
Abstract
Liver homeostasis requires the presence of both parenchymal and non-parenchymal cells (NPCs). However, systems biology studies of the liver have primarily focused on hepatocytes. Using an organotypic three-dimensional (3D) hepatic culture, we report the first transcriptomic study of liver sinusoidal endothelial cells (LSECs) and Kupffer cells (KCs) cultured with hepatocytes. Through computational pathway and interaction network analyses, we demonstrate that hepatocytes, LSECs and KCs have distinct expression profiles and functional characteristics. Our results show that LSECs in the presence of KCs exhibit decreased expression of focal adhesion kinase (FAK) signaling, a pathway linked to LSEC dedifferentiation. We report the novel result that peroxisome proliferator-activated receptor alpha (PPARα) is transcribed in LSECs. The expression of downstream processes corroborates active PPARα signaling in LSECs. We uncover transcriptional evidence in LSECs for a feedback mechanism between PPARα and farnesoid X-activated receptor (FXR) that maintains bile acid homeostasis; previously, this feedback was known occur only in HepG2 cells. We demonstrate that KCs in 3D liver models display expression patterns consistent with an anti-inflammatory phenotype when compared to monocultures. These results highlight the distinct roles of LSECs and KCs in maintaining liver function and emphasize the need for additional mechanistic studies of NPCs in addition to hepatocytes in liver-mimetic microenvironments.
Similar content being viewed by others


Introduction
The liver participates in vital functions related to metabolism, detoxification, and mediation of the complex defense mechanisms within the body1. Hepatocytes, the primary cell type of the liver1, are responsible for several hepatic functions, including lipid metabolism, glucose homeostasis, and biotransformation of xenobiotics1. Hepatocytes are the most widely-studied type of cell in the liver1. In recent years, other types of non-parenchymal hepatic cells have received more attention, such as liver sinusoidal endothelial cells (LSECs) and Kupffer cells (KCs), especially to study the roles they play in the onset of hepatic fibrosis2, non-alcoholic fatty liver disease3 and cholestasis4. The liver has a stratified architecture where the Space of Disse ensures the separation of the hepatocytes from the LSECs1. This in vivo architecture and intercellular communications have been recognized as important for proper functioning of hepatic cells1.
Deciphering cell-type-specific signaling is challenging to achieve in vivo. Engineered hepatic tissues have been developed to overcome this challenge5,6,7,8,9,10,11,12,13. Early in vitro models of the liver included the hepatocyte monolayer (HM)5,6,7 and the collagen sandwich (CS)5,6,7 cultures comprised of primary rat hepatic parenchymal cells. HMs lose morphological characteristics in less than one week5,6,7. In contrast, CSs maintain their polarized morphology and hepatic functions, e.g., albumin and urea production, for up to six weeks5,6,7. Hepatocytes co-cultured with NPCs have been shown to better maintain morphology and function than HM and CS cultures8. We have previously reported 3D organotypic liver models9,10,12 that consist of hepatocytes separated from hepatic NPCs by a detachable, biopolymeric membrane that mimics the Space of Disse12. This in vitro system recapitulated the stratified sinusoidal architecture found in vivo, thus enabling intercellular communications arising from cell-cell contact and paracrine signaling. Each cell type maintained phenotype up to 16 days. Additionally, the 3D liver models elicited proliferation of each cell type, while still maintaining cellular ratios similar to those found in vivo12. Traditional 2D in vitro cultures9 did not exhibit these properties.
Genomewide transcriptional analysis has been used to better understand the functions of hepatic cell types. However, gene expression studies on the liver have largely focused on understanding transcriptional programs in hepatocytes14,15,16,17. HMs, compared to CSs, were shown to rapidly lose liver-specific functions, such as cytochrome P450 (CYP) activity and cholesterol metabolism, within 48 hours14,15,17. In addition, cultures derived from hepatic cell lines exhibited decreased expression of CYPs compared to those derived from primary hepatocytes15,16. A study of primary rat hepatocytes co-cultured with one of three fibroblasts cell lines sought to elucidate cell-cell interactions at a transcriptional level18. However, fibroblasts are an inadequate substitute for hepatic NPCs. Moreover, co-cultures do not emulate the stratification found in vivo in the liver. Previous analyses of LSEC transcriptomes19,20,21,22,23,24,25 have limitations since they have focused on comparing LSECs to other endothelial cells or have investigated perturbations after partial hepatectomy and viral infection. Transcriptional studies on KCs26,27,28,29 have identified specific genes and cues involved in macrophage functional specialization or have monitored these cells during liver regeneration. None of these studies have considered how hepatoctyes and NPCs may influence each other transcriptionally in a healthy environment.
In this study, we conduct a genome-wide transcriptomic analysis of two organotypic 3D liver models12 with the goal of uncovering the functional activities of NPCs. To the best of our knowledge, this is the first genome-wide gene expression study of hepatic NPCs cultured in an in vitro 3D liver model that includes multiple cell types. Through extensive bioinformatic analyses, we elucidate changes in perturbed pathways that occur within the 3D liver models as a result of the presence or absence of KCs. Based on these analyses of our transcriptional data, we show that the inclusion of KCs in the 3D liver models encourages LSECs to maintain a differentiated phenotype. Taken together, the results suggest that signaling between hepatocytes and NPCs results in cellular and organ level functions that indicate a healthy hepatic environment. We stress that our results are based on analyses of transcriptional data and protein interaction networks. Further mechanistic experiments are required in order to confirm them.
Results
We measured expression profiles of hepatocytes, LSECs, and KCs in five different culture systems: 3D models containing all three cell types, 3D models containing only hepatocytes and LSECs, collagen sandwiches, hepatocyte monolayers, and KC monolayers. We note that the 3D liver models maintained phenotype through day 16. However, to be consistent with our previous work12, all samples from the 3D liver models were taken at day 12. KCs in monocultures were assessed at day 3. Henceforth, we refer to 3D liver models that are comprised of hepatocytes (H) and LSECs (L) as 3DHL and those that also include KCs (K) as 3DHLK.
Each cell type has a distinct gene expression profile
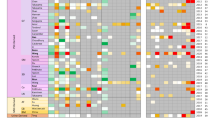
We first sought to confirm the purity of our samples to ensure that no cross-contamination had occurred during the cell separation processes (See “Materials and Methods”). We performed hierarchical clustering of the transcriptomic samples using the probe-set-level expression data (Fig. 1). From this clustering, we observed the replicates for each cell-type and culture condition (e.g., LSECs from the 3DHLK model or the LSECs from the 3DHL model) clustered together. We also found the NPC samples clustered separately from the hepatocytes. Within the cluster containing the NPCs, the KCs from the 3DHLK model clustered distinctly from the LSECs in either 3D liver model. We evaluated the robustness of this clustering to different linkage and distance measures. The NPCs clustered separately from hepatocytes. Within the NPCs, the LSECs clustered separately from the KCs. KC monocultures clustered separately from all samples in the liver models, thus having the most distinct expression profiles. This separation was consistent across different linkage methods (single, complete, and average) and distance measures (Euclidean, Manhattan, and correlation)30.

Hierarchical clustering of samples across cell types and culture conditions. Each column displays the expression values for one probe set. Each colored bar on to the left indicates the replicates of one cell type in one culture condition. From top to bottom, the samples are: hepatocytes in 3DHL models, HM, CS, and 3DHLK models; LSECs in 3DHL and 3DHLK models; KCs in 3DHKL models and monocultures of fresh KCs. We measured the expression profiles for the first seven groups on day 12 and for the last group on day 3.
We computed the average Euclidean distance among all hepatocyte samples, among all LSEC samples, between the KC samples in 3DHLK models, and across all pairs of cell types. The average distance from an LSEC sample to a KC sample was 81.59, which is at about 1.6 times the average distance among LSEC samples (55.02) or among KC samples (56.53). Hepatocytes were farther from both KCs and LSECs than KCs and LSECs were from each other.
These trends demonstrated that each cell type had a distinct and different expression pattern, and that the culture systems induced unique expression patterns in each cell type. Further, these data indicated that no contamination occurred during the cell separation process.
LSECs in the 3DHLK model displayed expression patterns indicative of a differentiated phenotype
We used Gene Set Enrichment Analysis (GSEA)31 to identify gene sets exhibiting up- or down-regulation in the LSECs in the 3DHLK model compared to the LSECs in the 3DHL models (see “Materials and Methods”). GSEA identified 77 gene sets as enriched (q-value ≤ 0.01; Supplementary Table S1), of which 61 sets were up-regulated in LSECs in the 3DHLK model and 16 were down-regulated. Since GSEA considers each gene set individually, it may return statistically significant functions that annotate similar sets of genes. These redundant functions do not provide additional biological insights. In such cases, we discuss all related gene sets together. For each gene set, we examine genes in its leading edge (see “Materials and Methods”) to further study their relationships to hepatic phenotypes. We summarize our results in this and the next five sections.
Two phenotypic characteristics of differentiated LSECs are the presence of open (100–150 nm) fenestrae and their endocytotic activity2. GSEA identified several down-regulated gene sets related to these characteristics, specifically, the FAK Pathway (q-value 1.16 × 10−3, rank 37), Actin Cytoskeleton (q-value 7.10 × 10−3, rank 69), and Actin Filament Based Process (q-value 7.13 × 10−3, rank 70). These results suggest that FAK and actin signaling are decreased in the LSECs from the 3DHLK model when compared to the same cell type in the 3DHL model.
We focus on the FAK Pathway where GSEA identified 29 genes in its leading edge. The FAK pathway initiates with signals from FAK, Talin 1, and two integrins (Itga5 and Itgb1); the latter three participate in the leading edge of this gene set. This pathway includes three regulators of cell migration: Rho-kinase (Rock2)32, Ras homolog family member A (RhoA)33 and Ras-related C3 botulinum toxin substrate 1 (Rac1)34. All three proteins have been shown to control LSEC fenestration in rats. Specifically, the inhibition of Rock2 resulted in an increase in the number of fenestrae. Likewise, Rac1 has been associated with the contraction of fenestrae in rat LSECs34. FAK signaling has also been shown to regulate endocytosis35. Specifically, FAK-mediated activation of Endophilin A2 (Sh3gl1) inhibits endocytosis35. We observed decreased expression of Sh3gl1 in the LSECs from the 3DHLK model. Taken together, these transcriptional trends suggest that LSECs cultured in the 3DHLK liver model may have more and larger fenestrae and exhibit increased endocytotic activity compared to LSECs in the 3DHL model.
Nitric oxide (NO) is critical for the maintenance of LSEC function and differentiation36. When we compared LSECs to hepatocytes in the 3DHLK model (Supplementary Table S1), we observed up-regulation of ion channel transport, acetylcholine binding, and G protein coupled receptor (GPCR) ligand binding genes. To preclude the possibility that this up-regulation may be caused by a down-regulation of these processes in hepatocytes in 3DHLK models, we applied GSEA to compare these hepatocytes to the same cell type in 3DHL model (Supplementary Table S1). We observed that none of these gene sets were significant at 0.01 level in this comparison. Nicotinic cholinergic receptor alpha 3 and alpha 6 receptors and Muscarinic cholinergic receptor 1 participated in the leading edge of functions related to ion channel activity, neurotransmitter receptor activity, and GPCR activity. Upon activation by acetylcholine, these cholinergic receptors open ion channels, enabling changes in intracellular calcium ion levels37,38. Increased calcium levels have been shown to activate NO synthase in endothelial cells in a Calmodulin-dependent manner39. We observed that Calmodulin-dependent protein kinase IV (Camk4) was up-regulated in LSECs compared to hepatocytes, suggesting an increase in NO synthesis in LSECs. Two other well known markers of LSEC differentiation, Protein Tyrosine Phosphatase, Receptor Type C (CD45)40 and Von Willebrand Factor (VWF)41, were up-regulated in LSECs in 3DHLK models compared to 3DHL models. Taken together, these transcriptional results suggest LSECs in the 3DHLK model maintain more phenotypic characteristics of a differentiated cell type.
A functional linkage network summarizes differences between LSECs in 3DHLK and in 3DHL systems
Several of the enriched gene sets identified by GSEA shared leading edge genes. In addition, previous methods have demonstrated that genes annotated to one function may interact with those genes from another function in an orchestrated manner42,43,44. We applied the Markov chain Monte Carlo Biological Process Network (MCMC-BPN) algorithm45 to identify a non-redundant set of gene sets that connect to each other via interactions between perturbed genes (See “Materials and Methods”). We applied MCMC-BPN to 77 gene sets with a q-value at most 0.01, as determined by GSEA. Figure 2a displays the biological process network (BPN) formed by all links with a probability greater than or equal to 0.45. This threshold corresponds to the top 2.5% of possible functional links. This BPN contains 36 links among 34 of the top 77 gene sets. The BPN contains three connected components, two of which consist of only gene sets down-regulated in LSECs in the 3DHLK model, with the third including only up-regulated gene sets.

Results from enrichment and network analysis comparing expression profiles of LSECs in 3DHLK models to 3DHL models. (a) Network computed by the MCMC-BPN algorithm. Each node is an enriched gene set reported by GSEA. Each link connects two gene sets. Red nodes are up-regulated and blue nodes are down-regulated in LSEC from the 3DHLK model. (b) Protein-protein and regulatory interactions up-stream of Ppara in the PPAR Signaling Pathway. Proteins highlighted in pink were up-regulated in LSECs in the 3DHLK model. (c) Protein interactions underlying the link between the PPAR Signaling Pathway (purple nodes) and the Terpenoid Backbone Biosynthesis Pathway (green nodes) in the BPN displayed in (a). Nodes with a saturated color are in the leading edge. Note that Hmgcs2 is annotated to both pathways. (d) Reactions involved in cholesterol biosynthesis. Pink proteins belong to the leading edge of either the Terpenoid Backbone Biosynthesis or the Steroid Biosynthesis pathway.
In the following sections, we discuss several of the top enriched functions identified by GSEA and the links between them in the BPN. Many of these up-regulated gene sets were related to the metabolism of fatty acids, bile acids and cholesterol. We first present leading edge genes from the Peroxisome Proliferator-activated Receptor (PPAR) signaling pathway. Next, we explore the link between PPAR signaling and cholesterol metabolism revealed by the BPN. Finally, we discuss how bile acid biosynthesis and fatty acid metabolism relate to these two gene sets.
LSECs in the 3DHLK model exhibited increased PPAR signaling
LSECs in the 3DHLK model show up-regulation of genes in the PPAR Signaling Pathway (rank 1, q-value < 10−5) compared to those in the 3DHL model (Supplementary Table S1). GSEA assigned 26 of the 63 genes from this gene set to its leading edge, including Ppara (SNR 0.21, rank 206), a primary transcription factor, and its co-activators Rxra and Rxrg (SNR 0.18, rank 346; SNR 0.15, rank 586, resp.)46,47,48. PPARs are a group of three nuclear hormone receptors: PPARα, PPARδ and PPARγ. (We use ‘Ppara’ to name the gene and ‘PPARα’ for the protein product.) Only Ppara was up-regulated in LSECs in the 3DHLK model; Ppard and Pparg showed no significant difference in expression levels in LSECs between the two 3D cultures. The expression of Ppara in LSECs was elevated in comparison to its level in hepatocytes in the 3DHLK model (SNR: 0.14, rank 3866). Note that this gene was somewhat down-regulated in hepatocytes in the 3DHLK model compared to the 3DHL model (SNR: −0.11, rank 11745).
Up-stream of PPARα in the signaling pathway lie members of both the Fatty Acid Transport Protein (FATP)49 and Fatty Acid Binding Protein (FABP) families of proteins50. Consistent with the liver specific expression patterns reported in the literature51,52,53, we observed up-regulation of FATPs (Slc27a2 and Slc27a5, SNR: 0.14, rank 684; SNR: 0.26, rank 101, resp.) and FABPs (Fabp1/4/6, SNR: 0.49, rank 11; SNR: 0.12, rank 909; SNR: 0.10, rank: 1323, resp.), all of which participated in the leading edge (Fig. 2b). FATPs are transmembrane proteins involved in the uptake of long and very-long chain fatty acids into the cell51,52. FABPs act as an intracellular buffer by ushering ligands from FATPs to PPARα54. A murine study showed a physical interaction between the FABP1 and PPARα proteins in vivo in the liver and in cultured primary hepatocytes50. Note that the predominantly enterocyte specific FATP Slc27a455 was highly down-regulated in LSECs.
Upon activation by ligands such as fatty acids and their derivatives, eicosinoids, and synthetic compounds, PPARα translocates from the cytoplasm to the nucleus46. In the nucleus, PPARα dimerizes with the co-activator retinoid X receptors (RXRα and RXRγ) and transcribes genes involved in lipid metabolism, gluconeogenesis and cell survival56. Both RXRα and RXRγ were up-regulated in LSECs in the 3DHLK model. These LSECs also showed increased expression of down-stream targets of PPARα. These targets are involved in cholesterol metabolism57 and bile acid biosynthesis (e.g., Cyp7a1 and Cyp8b1)58,59,60, fatty acid oxidation61,62 and amino acid degradation (e.g., Acadm and Ehhadh)63. We discuss the relevance of cholesterol metabolism, bile acid biosynthesis and fatty acid oxidation in the following sections.
LSECs in the 3DHLK model regulate cholesterol metabolism
The MCMC-BPN analysis identified a link between PPAR Signaling Pathway and Terpenoid Backbone Biosynthesis gene set (link probability 0.90; Fig. 2a). Figure 2c displays the set of interactions representing this link; each interaction connects one gene in the PPAR Signaling Pathway to one gene in the Terpenoid Backbone Biosynthesis gene set. Sixteen out of the 18 interactions connected genes in the leading edges of these two gene sets. Six of these 16 interactions reflected the transcriptional regulation of 3-Hydroxy-3-Methylglutaryl-CoA Reductase (Hmgcr), 3-Hydroxy-3-Methylglutaryl-CoA Synthase 1 (Hmgcs1), and 3-Hydroxy-3-Methylglutaryl-CoA Synthase 2 (Hmgcs2) by PPARα and by RXRα64. The MCMC-BPN analysis also identified a link between Terpenoid Backbone Biosynthesis and both Cholesterol Biosynthesis and Steroid Biosynthesis (link probabilities 0.56 and 0.47, respectively). The Terpenoid Backbone Biosynthesis and Cholesterol Biosynthesis gene sets share seven genes that correspond to the set of reactions responsible for converting Acetyl-CoA to Farnesyl diphosphate65. Additionally, the Cholesterol Biosynthesis and Steroid Biosynthesis gene sets encompass genes responsible for the conversion of Farnesyl diphosphate to cholesterol66. Together, these three gene sets, all of which are up-regulated in LSECs in the 3DHLK model, annotate the set of reactions in cholesterol biosynthesis that convert Acetyl-CoA to cholesterol (Fig. 2d)67,68,69. These links in the BPN indicate that the PPAR Signaling Pathway may be activated and in turn induce the expression of Cholesterol biosynthesis genes in LSECs in the 3DHLK model.
Further support for this observation comes from the significant up-regulation of the Reactome Cholesterol Biosynthesis gene set (rank 1, q-value < 10−5) in LSECs in the 3DHLK model versus LSECs in the 3DHL model (Supplementary Table S1 and Fig. 2a). However, cholesterol biosynthesis in the liver has mainly been attributed to hepatocytes1 and the involvement of LSECs has not been well documented. To further understand our results, we compared the expression of these genes in LSECs to hepatocytes in the 3DHLK model. We noted that the Cholesterol Biosynthesis gene set is not significantly perturbed when we compare Hepatocytes in the 3DHLK model to the same cell type in the 3DHL model (q-value 1). Cholesterol homeostasis occurs due to a combination of synthesis, transcytosis and metabolism69,70,71,72. Certain signaling events related to cholesterol homeostasis are triggered by cytoplasmic cholesterol concentrations (Fig. 3a). When cytoplasmic cholesterol levels are low, sterol regulatory element binding proteins (SREBPs) transcribe genes involved in de novo cholesterol biosynthesis72. In addition, activated SREBPs transcribe scavenger receptor B1 (Scarb1; SB-1), low-density lipoprotein receptor (LDLR), and proprotein convertase subtilisin/kexin type 9 (PCSK9), genes involved in the uptake of cholesterol72. Both LDLR and SB-1, which are responsible for the uptake of exogenous cholesterol72, show increased expression in LSECs (SB-1, SNR: 0.18, rank: 3121; LDLR, SNR: 0.49, rank: 7) suggesting that LSECs may participate in transcytosis of cholesterol. SB-1, the high density lipoprotein receptor, is known to be expressed abundantly in LSECs70. Interestingly, the SREBP transcriptional target PCSK9 plays a role in the clathrin-mediated endocytosis of LDLR72. In contrast, when cholesterol levels are high within a cell, the transcription factor liver X receptors (LXRs) are activated72. LXRs transcribe genes involved in the efflux of cholesterol, including ApoE, and the ATP-binding cassette transporters ABCA and ABCG family of transporters72. Though we observe down-regulation of ABCA1 and ABCG1 (SNR: −0.09, rank: 9463; SNR: −0.17, rank: 10834, resp.) in LSECs, the liver-specific ABCG5 displays increased expression (SNR: 0.192, rank: 2795). Another method for maintaining cholesterol homeostasis involves HMGCR, the rate-limiting step in cholesterol biosynthesis73. HMGCR may be post-translationally degraded by 24,25-dihydrolanosterol and squalene73. 24-Dehydrocholesterol Reductase (Dhcr24) and Farnesyl-Diphosphate Farnesyltransferase 1 (Fdft1), the two enzymes responsible for the synthesis of 24,25-dihydrolanosterol and squalene, respectively, exhibit increased expression in LSECs compared to hepatocytes (SNR: 2036, rank: 0.23; SNR: 0.39, rank: 232, resp.). Altogether, these trends in the transcriptional data suggest that LSECs in the 3DHLK models participate in cholesterol transcytosis as well as in the biosynthesis of this compound.

LSEC and KC signaling pathways in 3DHLK systems. (a) Pathway diagram of transcytosis of cholesterol in LSECs. (b) FXR-dependent feedback mechanisms involved in regulation of bile acid biosynthesis. Interactions represent both the positive feedback mechanism through Ppara and the negative feedback mechanism through SHP. Proteins highlighted in pink are up-regulated in LSECs in the 3DHLK model. (c) Fatty Acid Degradation and beta-oxidation pathway. Genes highlighted in pink were up-regulated in the LSECs in the 3DHLK model. (d) Diagram of the FC-ε RI Signaling Pathway. Proteins shown in blue are down-regulated in Kupffer cells from the 3DHLK model.
LSECs in the 3DHLK model up-regulated genes involved in bile acid biosynthesis
The LSECs from the 3DHLK model, compared to those in the 3DHL cultures, showed up-regulation of genes associated with the Primary Bile Acid (BA) Biosynthesis gene set (q-value 4.93 × 10−3, rank 55, Supplementary Table S1 and Fig. 2a). As in the case of cholesterol biosynthesis, we compared the expression levels of these genes in LSECs to hepatocytes from the 3DHLK model, since only hepatocytes have been shown to produce BAs58,74,75. Using GSEA, we found no enrichment of gene sets related to BA biosynthesis (q-value > 0.01), suggesting similar expression levels among these genes in both LSECs and hepatocytes and supporting the possibility of bile acid biosynthesis in LSECs.
Synthesis of BAs is the primary means of cholesterol catabolism58,76 and can occur via the classical (neutral) or alternate pathways (Fig. 3b)58. In the initial step of BA formation in the classical pathway, Cytochrome P450, Family 7, Subfamily A, Polypeptide 1 (Cyp7a1) hydroxylates the C-7 carbon of cholesterol, and is the rate-limiting enzyme for determining the BA pool58. Cytochrome P450, Family 8, Subfamily B, Polypeptide 1 (Cyp8b1) leads to the generation of cholic acid (CA)58. Following the alternate pathway, Cytochrome P450, Family 27, Subfamily A, Polypeptide 1 (Cyp27a1) and Aldo-keto Reductase Family 1, Member D1 (Akr1d1) control the population of chenodeoxycholic acid (CDCA)76. We observed the up-regulation of Cyp7a1, Cyp8b1, Cyp27a1, and Akr1d1 in the leading edge of the Primary Bile Acid Biosynthesis gene set from the KEGG database when we compared LSECs from the 3DHLK model to those from 3DHL cultures. These results suggest that higher levels of CA and CDCA may be synthesized in LSECs in the presence of hepatocytes and KCs. CDCA has been found to induce FXR activity in humans and mice76,77, resulting in increased expression of PPARA78. These transcription-based results suggest that a feedback mechanism among PPARα, FXR, and BA biosynthesis occurs in LSECs when they are cultured with hepatocytes and KCs.
LSECs in the 3DHLK model exhibited increased fatty acid metabolism
Six of the top 77 gene sets identified by GSEA were involved in fatty acid metabolism (Supplementary Table S1). These gene sets include Carboxylic Acid Metabolic Process, Organic Acid Metabolic Process and Monocarboxylic Acid Metabolic Process (rank 1, q-value < 10−5), Fatty Acid Oxidation (rank 14, q-value 3.34 × 10−5), Fatty Acid Metabolism (rank 17, q-value 1.36 × 10−4), and Fatty Acid Metabolic Process (rank 19, q-value 1.47 × 10−4). Our BPN included two gene sets related to fatty acids: Organic Acid Metabolic Process and Monocarboxylic Acid Metabolic Process. All six fatty acid related gene sets shared a core set of four genes in their leading edges: Cpt1a, Acadvl, Acadm, and Echs1, each being up-regulated in LSECs in the 3DHLK model compared to the 3DHL model (SNR ≥ 0.13; rank ≤ 833). Up-regulation of these gene sets suggests that LSECs in the presence of KCs may have increased fatty acid metabolism. In fact, LSECs are known to derive a large proportion of their energy (~45% of ATP) from fatty acid oxidation79.
Biosynthesis of cholesterol requires the availability of Acetyl-CoA80. Fatty acid oxidation and degradation provide one source of Acetyl-CoA81. These processes occur in either the peroxisome82 or the mitochondria83. For mitochondrial fatty acid oxidation, carnitine palmitoyltransferases (Cpt1a/Cpt2) first import fatty acids from the cytoplasm where their degradation occurs through β-oxidation (Fig. 3c)84,85,86,87. Acyl-CoA Dehydrogenase, Very Long Chain (Acadvl)88, Acyl-CoA Dehydrogenase, C-4 To C-12 Straight Chain (Acadm)88, Enoyl CoA Hydratase, Short Chain, 1 (Echs1)89, and Cpt1a90 participate in this process. Cpt1a regulates the rate-limiting step in rat hepatocytes91. All these genes highlighted in pink were up-regulated in the LSECs in the 3DHLK model. It is notable that Ppara, which is up-regulated in these LSECs, transcriptionally regulates Cpt1a and Acadm.
KCs display an anti-inflammatory phenotype in the 3DHLK model
KCs, like many macrophages, exhibit plasticity in their phenotype ranging from pro- to anti-inflammatory characteristics92,93. To better understand their phenotype in the 3DHLK liver model, we compared their gene expression profiles in the 3DHLK model on day 12 to KC monocultures on day 3. GSEA identified 21 significant functions in the 3DHLK model (q-value < 10−5, Supplementary Table S1). GSEA identified four functions as up-regulated (q-value < 10−5) in KCs from the 3DHLK liver model. The remaining 17 gene sets were down-regulated. Of those functions showing increased expression in the KCs from the 3DHLK cultures, three were related to drug metabolism and phase II conjugation, and one involved genes found up-regulated in hepatocytes in response to LSECs94. The down-regulated gene sets related to inflammation and immune response.
We focus on gene sets that we can map to pathways, specifically the Fcε receptor signaling pathway in the KEGG database (Fig. 3d). The Fcε receptor is involved in allergic and immune responses, and is responsible for the release of histamines, leukotrienes, and cytokines after activation95. The Fcε receptor, specifically the Fcεr1γ subunit, is one mechanism responsible for the M2-to-M1 transition of macrophages96. The gene set for this signaling pathway contains 70 genes, 21 of which participate in the leading edge. In order to initiate Fcε signaling, immunoglobulin E (IgE) binds to Fcε receptors, which activate Lyn or Fyn, both Src Family Tyrosine Kinases97,98. Once activated, Lyn or Fyn phosphorylates the Fcεr1γ-chain subunit of Fcε receptor, resulting in the recruitment and subsequent activation of Syk by Lyn or Fyn97,98. We observed the down-regulation of Fcer1g, Fyn, Lyn, Syk, Gab2 and PI3K (PIK3R1/PIK3CD) in KCs in the 3DHLK models compared to monocultures. All seven of these genes were members of the leading edge of the Fcε-receptor signaling pathway. Additional characteristics of M1 macrophages include expression of pro-inflammatory cytokines such as tumor necrosis factor (TNF), IL6, IL12, and nitric oxide synthase 2, inducible (iNOS)92. TNF, which is transcriptionally up-regulated by Fcε receptor signaling, IL6, and iNOS exhibited decreased expression in KCs in the 3D model compared to those in the monoculture. In contrast, macrophages with an anti-inflammatory phenotype express IL10 protein, IL1 receptor agonist, and IL1 decoy receptor99, which were up-regulated in the 3D cultures. These trends in the transcriptional data support the conclusion that KCs in 3DHLK cultures show a more M2-like phenotype in comparison to freshly isolated KCs.
Discussion
Hepatocytes are the most widely studied cell type in the liver. More recently, scientists have begun to understand the critical roles played by NPCs, especially LSECs and KCs, in modulating the function of healthy livers. In this paper, we have sought to discover transcriptional perturbations as a result of heterotypic communications among hepatic cell types in an organotypic liver model that maintained the phenotypes of each cell type for up to 16 days12. This model permits the separation of cell types thus enabling us to measure the transcriptome in each cell type individually. Thereby, we created the first-ever transcriptional dataset in hepatocytes and in two types of NPCs (LSECs and KCs) in an in vitro liver model. We noted that each cell type had a distinct expression profile and that each culture system induced unique expression patterns in each cell type. These results indicate that no contamination occurred during the cell separation process. We have already reported on the analysis of the hepatocyte transcriptome12. Here, we focused our detailed analysis on the LSECs and KCs since previous transcriptional studies on these NPCs in an in vitro context have been limited. We related our results, which are based on transcriptional data, to published in vivo or in vitro experiments.
Just like hepatocytes, LSECs require communication with other hepatic cells for normal physiological phenotype and function9,94. Phenotypic characteristics of LSECs include the presence of open fenestrae, which are associated with the endocytic capabilities of LSECs2. Maintaining this phenotype in vitro is challenging since LSECs rapidly dedifferentiate within days36,100. We have demonstrated earlier that fenestrae found in healthy LSECs disappear in the presence of activated KCs101. To the best of our knowledge, the current work is the first to provide clear transcriptional evidence that suggests that LSECs maintain a differentiated phenotype when cultured in a tri-cellular, organotypic hepatic environment.
Nitric oxide (NO) is one of the key factors that are involved in the maintenance of LSEC phenotype36. Endothelial NO synthase (eNOS; Nos2) is a primary contributor to the production of NO in LESCs2. LSECs in the 3DHLK model show increased levels of eNOS gene expression compared to those in the 3DHL model. There are several ways in which eNOS may be activated in LSECs; our data contains evidence to support two such mechanisms. First, Nicotinic and Muscarinic receptor activation leads to active eNOS in endothelial cells39 via increased cytosolic calcium ion levels as a result of opened ion channels37,38. We observed up-regulation of ion channel, neurotransmitter receptor, and GPCR activity, all functions involving the Nicotinic and Muscarinic receptors. Second, vascular endothelial growth factor A (Vegfa) promotes the maintenance of fenestrae by inducing eNOS activity and subsequent production of NO2,36. In culture and in vivo, Vegfa has been shown to be sufficient in maintaining LSECs fenestration102. Since KCs are known to express Vegfa103, it is possible that LSECs may be exposed to additional Vegfa originating from the KCs in the 3DHLK model. Further mechanistic experiments will need to be performed to identify the direct cause of this amplified eNOS expression. Nevertheless, our results are consistent with LSECs in the 3DHLK model producing increased levels of NO, thereby exhibiting a fenestrated phenotype.
In vivo, a decrease in the numbers and diameter of fenestrae on LSECs is often associated with fibrosis2. In addition, increased focal adhesions have been correlated with a loss of fenestrae in vitro101. LSECs in the 3DHLK model exhibited decreased FAK signaling. Three participants in FAK signaling, Rock232, RhoA33 and Rac134, have all been associated with negatively regulating LSEC fenestration. We observe decreased expression of Rock2 and Rac1 in LSECs cultured in the presence of KCs, i.e., in the 3DHLK model. Rock2 has been found to negatively regulate eNOS activity in cirrhosis104. The decreased expression of Rock2 may provide an additional explanation for the observed increased expression of eNOS in LSECs cultured in the presence of KCs., thus providing additional evidence for LSEC fenestration in the 3DHLK model. Taken together, these results support the conclusion that the 3DHLK liver model provides a non-fibrotic environment for LSECs.
PPARα activity has extensively been studied in hepatocytes56,105,106,107 with respect to its role in lipid metabolism56,106, insulin sensitivity56, and its target genes107. Altered PPARα signaling has also been linked to fatty liver disease108, obesity109, and diabetes. However, considerably less information is present for its gene expression or protein activity in LSECs. Several non-hepatic ECs have exhibited Ppara expression110,111, but a recent study in female murine liver ECs failed to detect significant Ppara mRNA112. In contrast, we observed that LSECs in the 3DHLK model express Ppara, and at higher levels than LSECs in the 3DHL model and hepatocytes from both 3D cultures. In the liver, PPARα is known to transcriptionally control gene sets related to fatty acid metabolism113, bile acid biosynthesis58,59,60, amino acid degradation63, and cholesterol metabolism57, phenomena that are mirrored in our LSEC-specific data. Our novel results indicate that PPARα may have a pivotal role in LSECs as a regulator of many biological processes that are critical to proper hepatic function.
PPARα transcriptionally up-regulates Cyp7a1, Cyp8b1 and Cyp27a158,59,60, which are responsible for maintaining the pool of bile acids (BAs)58,76. Studies have shown that the bile acid chenodeoxycholic acid (CDCA) induces FXR activity in humans and mice76,77. In human HepG2 cells, activation of FXR by CDCA has been associated with increased expression of PPARa78. We found transcriptional support for this positive feedback mechanism between PPARα and FXR in LSECs in the 3DHLK model (Fig. 3b). Our analysis confirms the up-regulation of Cyp7a1, Cyp8b1 and Cyp27a1 in LSECs from the 3DHLK model, which may be the result of increased PPARα activity as suggested by the functional link between PPAR Signaling and Terpenoid Backbone Biosynthesis in the BPN (Fig. 2a,c). Conversely, FXR is known to activate Nuclear Receptor Subfamily 0, Group B, Member 2 (SHP; Nr0b2), another nuclear receptor, which in turn inhibits Cyp7a158 (Fig. 3b). FXR and SHP showed increased expression in LSECs in the 3DHLK model. Taken together, these transcriptional results indicate that LSECs may balance the positive and negative feedback among PPARα, FXR, and BA biosynthesis, mechanisms which were previously observed in HepG2 cells.
Bile acid biosynthesis is a well known function of the liver58. We have previously shown that 3DHL models synthesize and metabolize bile acids74. Here, we find transcriptional support for altered bile acid metabolism in LSECs cultured in the organotypic liver models. Perturbed bile acid metabolism in LSECs may stem from the uptake of BAs originating at hepatocytes in the 3DHLK model. Solute Carrier Family 10 Member 1 (known as Slc10a1 or NTCP), a membrane transporter responsible for the influx of extracellular BAs, has been shown to be functionally present in hepatocytes114. In our data, this transporter shows higher expression levels in LSECs compared to hepatocytes from the same liver model (3DHLK or 3DHL). Moreover, Slc10a1 also shows a marked increase in expression in LSECs in the presence of KCs, i.e., in 3DHLK models. If LSECs in the 3DHLK model uptake BAs, then exogenous CDCA may activate the FXR receptor and cause transcriptional up-regulation of bile acid biosynthesis enzymes through the feedback mechanisms discussed earlier (Fig. 3b). Further mechanistic experiments will need to be performed to understand the precise role of Slc10a1 and exogenous BAs in the feedback loop between PPARα and FXR.
KCs, like other macrophages, display a continuum of phenotypes from proinflammatory (M1) to anti-inflammatory (M2)92,93,115. Under proinflammatory conditions, activated KCs release cytokines (e.g., Tnf-α, IL1, and IL6), chemokines, and reactive oxygen species93. In contrast, M2 KCs express IL10, IL1 receptor agonist, and IL1 decoy receptor99. Activated KCs have been implicated in liver fibrosis in non-alcoholic steatohepatitis mouse models116, whereas anti-inflammatory KCs promote tissue repair and suppress inflammation117. KCs in 3D cultures show decreased expression of proinflammatory cytokines, indicating a more M2-like phenotype and suggesting that the 3DHLK model provides a healthy (non-fibrotic) environment for culturing hepatic cells.
It is known that, in addition to proper in vivo architecture, intercellular communications are essential for the maintenance of phenotype and function of hepatic cells9,94. Despite the importance of these heterotypic signals originating in NPCs, transcriptional studies of the liver have mainly focused on hepatocytes5,6,7,9,14,15,16. Here, we provide the first genome-wide transcriptional study of hepatic NPCs in an organotypic liver model. Through an extensive functional enrichment and network analysis of these transcriptional data, we have shown that LSECs in the 3DHLK model maintain their differentiation and that KCs present with an anti-inflammatory phenotype. These results demonstrate the 3DHLK model represents a more in vivo-like environment for LSECs and KCs than 3DHL models. Our computational analysis has uncovered previously unknown events in LSECs, including the expression of PPARα and genes that participate in the processes it regulates. We also hypothesize that the feedback mechanisms that connect PPARα, FXR, and BA biosynthesis, previously identified in HepG2 cells, may also occur in LSECs. We note that our analyses are based on transcriptional data. Hence, further mechanistic experiments are required to confirm our results. Nevertheless, our work calls attention to the potential role LSECs may play in the pathogenesis of diseases such as obesity and diabetes. These discoveries emphasize the need for further study of the functions and activities of hepatic non-parenchymal cells both in vitro and in vivo.
Materials and Methods
Materials
Dulbecco’s modified Eagle’s medium (DMEM), phosphate-buffered saline (PBS), Hank’s buffered salt solution, Earle’s balanced salt solution, penicillin, streptomycin, human plasma fibronectin, and trypsin-ethylenediaminetetraacetic acid were purchased from Invitrogen Life Technologies, Carlsbad, CA. Type IV collagenase, HEPES (4-[2-hydroxyethyl] piperazine-1-ethanesulfonic acid), glucagon, calcium chloride, hydrocortisone, sodium dodecyl sulfate, hydrogen peroxide, glutaraldehyde, dicumarol, 3-methylcholanthrene, calf thymus DNA, chitosan, and hyaluronic acid (HA) was purchased from Sigma-Aldrich, St. Louis, MO. All other chemicals were purchased from Fisher Scientific, Waltham, MA, unless otherwise noted.
Assembly of detachable polymeric Space of Disse
Detachable polyelectrolyte multilayers (PEMs) were assembled using chitosan (cationic) and HA (anionic) as previously described12. Briefly, chitosan was dissolved in 1% v/v acetic acid and HA in 18 MΩ cm water (Hydro, Durham, NC). Their concentrations were 5 mM. The pH of the polyelectrolyte (PE) solutions was maintained at 4.0 and 5.0 for chitosan and HA, respectively. PEMs were assembled on hydrophobic poly-tetrafluoroethylene (PTFE; McMaster-Carr, Elmhurst, IL) substrates using a robotic deposition system (NanoStrata, Tallahassee, FL). Water contact angle values (KSV Instruments, Helsinki, Finland) on clean PTFE substrates ranged from 111.9 ± 4.2 (n = 15). A detailed study was performed to find the optimal number of bilayers (BLs), deposition times, optical properties and the Young’s modulus for PEMs12. 12.5 BLs with a deposition time of 40 min have previously been reported to provide stable detachable PEMs with properties similar to the Space of Disse12.
Isolation and culture of hepatocytes and LSECs and KCs
Primary hepatocytes were isolated from female Lewis rats (Harlan Laboratories, Indianapolis, IN; weighing 175–199 g) utilizing a two-step in situ collagenase perfusion method5,6,9,10,118. Hepatocyte yields ranged from 150–200 × 106 cells and their viability was determined to be 90%–95% based on trypan blue exclusion. LSECs, from the same isolation, were obtained using differential adhesion and were cultured on fibronectin coated flasks9. Cryopreserved primary rat KCs (Invitrogen Life Technologies) were maintained in DMEM supplemented with 10% (v/v) heat-inactivated fetal bovine serum, 100 U/mL penicillin, 100 μg/mL streptomycin, 10 μg/mL insulin, and 100 μM β-mercaptoethanol. Animal care and surgical procedures including the procedure for liver excision from rats were conducted in accordance with the guidelines and regulations set up by the Virginia Tech’s Institutional Animal Care and Use Committee. All cell culture work was approved by Virginia Tech’s Institutional Biosafety Committee.
Assembly of 3D multicellular hepatic cultures
Hepatocytes were initially cultured as monolayers up to 72 h in 12-well tissue culture polystyrene plates (BD Biosciences, San Jose, CA) coated with rat-tail type 1 collagen (BD Biosciences)9,10. For the 3D liver models, UV-sterilized detachable PEMs (12.5 BL) were placed above the layer of hepatocytes. The PEMs were hydrated in the presence of hepatocyte culture medium for 1 h. Thereafter, 25,000 LSECs were seeded on the PEM. In cultures containing KCs, initially 50,000 cells were seeded on the PEM to obtain an initial ratio of 10:1 hepatocytes:KCs to emulate healthy livers119. All multicellular cultures were maintained in hepatocyte culture medium.
Gene chip hybridization and microarray data quality control
Hepatocytes, LSECs and KCs were separated as previously reported12. Briefly, 3D liver models were exposed sequentially to DynabeadsR (CELLectionTM Kit; Invitrogen Life Technologies) coated with SE-1 and CD163 antibodies120. SE-1 and CD163 are LSEC- and KC-specific antibodies, respectively. LSEC and KC fractions were collected using a magnet (DynaMagTM; Invitrogen Life Technologies). Total RNA was extracted from each cell type from HM, CS, and 3DHL and 3DHLK on day 12, and KC monolayers on day 3 using an RNeasy mini kit (Qiagen, Germantown, MD). The samples were checked for RNA degradation based on the RNA Integrity Number121. Each cell type and model combination was performed in triplicates, except KC samples from 3DHLK on day 12 which had only two samples that passed the RNA quality metric. Samples were labeled according to the Affymetrix Standard Target labeling process, and hybridized to the GeneChip Rat Genome 230 2.0 (Affymetrix, Santa Clara, CA). Complementary RNA synthesis, hybridization, and GeneChip scanning were performed at the Virginia Bioinformatics Institute Core Laboratory.
Gene expression analysis
We processed the Affymetrix microarrays using the Robust MultiChip Average (RMA) method122 in R. RMA performs background correction, quantile normalization and probe summarization. Each of the microarrays met the quality standards recommended by Simpleaffy123. As a second quality control, we performed hierarchical clustering on the normalized data to show consistency between replicates (Fig. 1). We performed hierarchical clustering on our gene expression data using complete linkage and Euclidian distance30. We used a data-driven approach to analyze the gene expression data further, which we describe in the next two subsections.
Functional enrichment
To compare the functional differences between LSECs in the 3DHLK and 3DHL models at day 12, we performed functional enrichment on the normalized gene expression data using the Gene Set Enrichment Analysis (GSEA) package31. GSEA summarizes probe-level data into gene-level data, computes differential expression of each gene using the signal-to-noise ratio (SNR), and calculates the enrichment of each functional gene set. To obtain the null distribution of p-values, GSEA permutes gene sets 5000 times. GSEA estimates the false discovery rate (q-value) using the method of Benjamini and Hochberg124. We considered gene sets with an FDR q-value ≤ 0.01 as significant. For each enriched gene set, GSEA also computed the leading edge, defined as the subset of genes annotated to the gene set that contributed to its enrichment. We note that for the KCs in the 3DHLK model, we simulated a third sample to be the mean of the other two samples.
We used gene sets from the Molecular Signature Database (MSigDB) v4.031 and Netpath125, a curated resource for signal transduction pathways. To enable the detection of perturbed biological processes and pathways, we included C2:CP and C5 gene sets in the analysis. The C2:CP collection contained pathways taken from databases such as Biocarta (a resource that is no longer available independently), KEGG126, and Reactome127, publications in PubMed, and knowledge of domain experts. C5 gene sets corresponded to annotations to Gene Ontology (GO) terms128. NetPath provides three gene sets for each pathway: (1) the genes directly involved in the pathway, (2) the genes transcriptionally up-regulated as a result of activating the pathway, and (3) the genes transcriptionally down-regulated by the pathway. Since very small or very large gene sets may be hard to interpret, we filtered out gene sets with less than 10 or more than 500 member genes, leaving 2501 gene sets for functional enrichment analysis.
Biological process networks
We sought to better understand the relationship among the significantly enriched gene sets reported by GSEA by integrating the gene expression data with protein interaction networks. Our goal was to compute a network where nodes were gene sets and where links connected pairs of gene sets, with each link being supported by multiple interactions between genes annotated to each set. To this end, we used a previously-published algorithm: Markov chain Monte Carlo Biological Process Networks (MCMC-BPN)45 to link the significantly enriched gene sets reported by GSEA. MCMC-BPN computes a network whose nodes are gene sets and whose links connect gene sets (e.g., Fig. 2a). The algorithm uses a Markov Chain Monte Carlo process to select as few links between genes as possible that explain as many interactions as possible between perturbed genes annotated to different gene sets. In our earlier work, we have demonstrated that MCMC-BPN produces sparser and less redundant networks than competitive methods without sacrificing coverage of the protein interaction network45.
MCMC-BPN takes as input a protein interaction network, a collection of gene sets, and a set of perturbed genes. We created these inputs as follows:
-
(a)
We downloaded a rat functional interaction network from STRING129 on January 27, 2015. Each interaction (u,v) in this network has a score computed by STRING. To obtain a reliable network, we removed every interaction with a score less than 500. Further, we retained only those interactions (u,v) such that both u and v were present on the DNA microarray. The final network contained 7,994 nodes and 70,052 undirected interactions.
-
(b)
We used the gene sets deemed significant by GSEA (q-value ≤ 0.01).
-
(c)
We defined a gene to be perturbed if that gene participated in the leading edge of at least one significant gene set in the previous step. The final input contained all such genes.
We ran MCMC-BPN with a burn-in period of 10,000,000 steps followed by 100,000,000 MCMC steps.
Literature analysis
Until this stage, our analysis was driven completely by gene expression data, functional annotation of genes, and gene interaction networks. We examined the BPNs computed, manually selected gene sets or groups of gene sets from these networks, and used the published literature on these gene sets in the liver for discussion in the paper.
Data availability
The microarray datasets generated during the current study are available in the Gene Expression Omnibus repository at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE74424.
References
Arias, I. et al. The liver: biology and pathobiology. (John Wiley & Sons, 2011).
DeLeve, L. D. Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology 61, 1740–1746, https://doi.org/10.1002/hep.27376 (2015).
McMahan, R. H., Porsche, C. E., Edwards, M. G. & Rosen, H. R. Free Fatty Acids Differentially Downregulate Chemokines in Liver Sinusoidal Endothelial Cells: Insights into Non-Alcoholic Fatty Liver Disease. Plos One 11, https://doi.org/10.1371/journal.pone.0159217 (2016).
Sato, K. et al. Pathogenesis of Kupffer Cells in Cholestatic Liver Injury. American Journal of Pathology 186, 2238–2247, https://doi.org/10.1016/j.ajpath.2016.06.003 (2016).
Dunn, J. C., Yarmush, M. L., Koebe, H. G. & Tompkins, R. G. Hepatocyte function and extracellular matrix geometry: long-term culture in a sandwich configuration. The FASEB Journal 3, 174–177 (1989).
Dunn, J., Tompkins, R. & Yarmush, M. Long-term in vitro function of adult hepatocytes in a collagen sandwich configuration. Biotechnol. Prog. 7, 237–245, https://doi.org/10.1021/bp00009a007 (1991).
Moghe, P. V. et al. Culture matrix configuration and composition in the maintenance of hepatocyte polarity and function. Biomaterials 17, 373–385 (1996).
Bhatia, S. N., Balis, U. J., Yarmush, M. L. & Toner, M. Effect of cell-cell interactions in preservation of cellular phenotype: cocultivation of hepatocytes and nonparenchymal cells. FASEB J 13, 1883–1900 (1999).
Kim, Y. & Rajagopalan, P. 3D Hepatic Cultures Simultaneously Maintain Primary Hepatocyte and Liver Sinusoidal Endothelial Cell Phenotypes. PLoS ONE 5, e15456, https://doi.org/10.1371/journal.pone.0015456 (2010).
Kim, Y., Larkin, A., Davis, R. & Rajagopalan, P. The Design of In Vitro Liver Sinusoid Mimics Using Chitosan–Hyaluronic Acid Polyelectrolyte Multilayers. Tissue Engineering Part A 16, 2731–2741, https://doi.org/10.1089/ten.tea.2009.0695 (2010).
Shan, J. et al. In Molecular pathology of liver diseases 321–342 (Springer, 2011).
Larkin, A., Rodrigues, R., Murali, T. M. & Rajagopalan, P. Designing a Multicellular Organotypic 3D Liver Model with a Detachable, Nanoscale Polymeric Space of Disse. Tissue Engineering Part C: Methods 19, 875–884, https://doi.org/10.1089/ten.tec.2012.0700 (2013).
Lee, P. J., Hung, P. J. & Lee, L. P. An artificial liver sinusoid with a microfluidic endothelial‐like barrier for primary hepatocyte culture. Biotechnology and bioengineering 97, 1340–1346 (2007).
Baker, T. et al. Temporal Gene Expression Analysis of Monolayer Cultured Rat Hepatocytes. Chem. Res. Toxicol. 14, 1218–1231, https://doi.org/10.1021/tx015518a (2001).
Boess, F. et al. Gene Expression in Two Hepatic Cell Lines, Cultured Primary Hepatocytes, and Liver Slices Compared to the in Vivo Liver Gene Expression in Rats: Possible Implications for Toxicogenomics Use of in Vitro Systems. Toxicological Sciences 73, 386–402, https://doi.org/10.1093/toxsci/kfg064 (2003).
Olsavsky, K. et al. Gene expression profiling and differentiation assessment in primary human hepatocyte cultures, established hepatoma cell lines, and human liver tissues. Toxicology and Applied Pharmacology 222, 42–56, https://doi.org/10.1016/j.taap.2007.03.032 (2007).
Kim, Y., Lasher, C., Milford, L., Murali, T. M. & Rajagopalan, P. A Comparative Study of Genome-Wide Transcriptional Profiles of Primary Hepatocytes in Collagen Sandwich and Monolayer Cultures. Tissue Engineering Part C: Methods 16, 1449–1460, https://doi.org/10.1089/ten.tec.2010.0012 (2010).
Khetani, S., Szulgit, G., Del Rio, J., Barlow, C. & Bhatia, S. Exploring interactions between rat hepatocytes and nonparenchymal cells using gene expression profiling. Hepatology (Baltimore, Md.) 40, 545–554, https://doi.org/10.1002/hep.20351 (2004).
Hu, J. et al. Endothelial cell-derived angiopoietin-2 controls liver regeneration as a spatiotemporal rheostat. Science 343, 416–419, https://doi.org/10.1126/science.1244880 (2014).
Geraud, C. et al. Liver sinusoidal endothelium: a microenvironment-dependent differentiation program in rat including the novel junctional protein liver endothelial differentiation-associated protein-1. Hepatology (2010).
Rowe, I. A. et al. Paracrine signals from liver sinusoidal endothelium regulate hepatitis C virus replication. Hepatology 59, 375–384, https://doi.org/10.1002/hep.26571 (2014).
Xu, C.-s et al. Analysis of time-course gene expression profiles of sinusoidal endothelial cells during liver regeneration in rats. Molecular and cellular biochemistry 350, 215–227, https://doi.org/10.1007/s11010-010-0701-5 (2011).
Ginsberg, M. et al. Efficient direct reprogramming of mature amniotic cells into endothelial cells by ETS factors and TGFbeta suppression. Cell 151, 559–575, https://doi.org/10.1016/j.cell.2012.09.032 (2012).
Kern, M. et al. Virally infected mouse liver endothelial cells trigger CD8+ T-cell immunity. Gastroenterology 138, 336–346, https://doi.org/10.1053/j.gastro.2009.08.057 (2010).
Nonaka, H., Sugano, S. & Miyajima, A. Serial analysis of gene expression in sinusoidal endothelial cells from normal and injured mouse liver. Biochem Biophys Res Commun 324, 15–24, https://doi.org/10.1016/j.bbrc.2004.09.014 (2004).
Okabe, Y. & Medzhitov, R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell 157, 832–844, https://doi.org/10.1016/j.cell.2014.04.016 (2014).
Eijgelaar, W. J., Horrevoets, A. J., Bijnens, A. P., Daemen, M. J. & Verhaegh, W. F. Equivalence testing in microarray analysis: similarities in the transcriptome of human atherosclerotic and nonatherosclerotic macrophages. Physiol Genomics 41, 212–223, https://doi.org/10.1152/physiolgenomics.00193.2009 (2010).
Zigmond, E. et al. Infiltrating monocyte-derived macrophages and resident kupffer cells display different ontogeny and functions in acute liver injury. Journal of immunology 193, 344–353, https://doi.org/10.4049/jimmunol.1400574 (2014).
Xu, C. et al. Characterization of transcriptional profiling of Kupffer cells during liver regeneration in rats. Cell Biol Int 36, 721–732, https://doi.org/10.1042/CBI20110104 (2012).
Hastie, T., Tibshirani, R. & Friedman, J. H. The elements of statistical learning: data mining, inference, and prediction. 2nd edn, (Springer, 2009).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102, 15545–15550, https://doi.org/10.1073/pnas.0506580102 (2005).
Venkatraman, L. & Tucker-Kellogg, L. The CD47-binding peptide of thrombospondin-1 induces defenestration of liver sinusoidal endothelial cells. Liver Int 33, 1386–1397, https://doi.org/10.1111/liv.12231 (2013).
Yokomori, H. et al. Rho modulates hepatic sinusoidal endothelial fenestrae via regulation of the actin cytoskeleton in rat endothelial cells. Lab Invest 84, 857–864, https://doi.org/10.1038/labinvest.3700114 (2004).
Yokomori, H. et al. Caveolin-1 and Rac regulate endothelial capillary-like tubular formation and fenestral contraction in sinusoidal endothelial cells. Liver Int 29, 266–276, https://doi.org/10.1111/j.1478-3231.2008.01891.x (2009).
Wu, X., Gan, B., Yoo, Y. & Guan, J.-L. FAK-mediated src phosphorylation of endophilin A2 inhibits endocytosis of MT1-MMP and promotes ECM degradation. Developmental cell 9, 185–196 (2005).
DeLeve, L. D., Wang, X., Hu, L., McCuskey, M. K. & McCuskey, R. S. Rat liver sinusoidal endothelial cell phenotype is maintained by paracrine and autocrine regulation. Am J Physiol Gastrointest Liver Physiol 287, G757–763, https://doi.org/10.1152/ajpgi.00017.2004 (2004).
Felder, C. C. Muscarinic acetylcholine receptors: signal transduction through multiple effectors. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 9, 619–625 (1995).
Vernino, S., Amador, M., Luetje, C. W., Patrick, J. & Dani, J. A. Calcium modulation and high calcium permeability of neuronal nicotinic acetylcholine receptors. Neuron 8, 127–134 (1992).
Busse, R. & Mulsch, A. Calcium-dependent nitric oxide synthesis in endothelial cytosol is mediated by calmodulin. FEBS letters 265, 133–136 (1990).
DeLeve, L. D. Liver sinusoidal endothelial cells and liver regeneration. J Clin Invest 123, 1861–1866, https://doi.org/10.1172/JCI66025 (2013).
Katz, S. C., Pillarisetty, V. G., Bleier, J. I., Shah, A. B. & DeMatteo, R. P. Liver sinusoidal endothelial cells are insufficient to activate T cells. J Immunol 173, 230–235 (2004).
Lasher, C. D., Rajagopalan, P. & Murali, T. M. Discovering networks of perturbed biological processes in hepatocyte cultures. PLoS One 6, e15247, https://doi.org/10.1371/journal.pone.0015247 (2011).
Dotan-Cohen, D., Letovsky, S., Melkman, A. A. & Kasif, S. Biological process linkage networks. PLoS One 4, e5313, https://doi.org/10.1371/journal.pone.0005313 (2009).
Li, Y., Agarwal, P. & Rajagopalan, D. A global pathway crosstalk network. Bioinformatics 24, 1442–1447, https://doi.org/10.1093/bioinformatics/btn200 (2008).
Lasher, C. D., Rajagopalan, P. & Murali, T. M. Summarizing cellular responses as biological process networks. BMC Syst Biol 7, 68, https://doi.org/10.1186/1752-0509-7-68 (2013).
Schupp, M. & Lazar, M. Endogenous ligands for nuclear receptors: digging deeper. The Journal of biological chemistry 285, 40409–40415, https://doi.org/10.1074/jbc.r110.182451 (2010).
Berger, J. P., Akiyama, T. E. & Meinke, P. T. PPARs: therapeutic targets for metabolic disease. Trends in pharmacological sciences 26, 244–251, https://doi.org/10.1016/j.tips.2005.03.003 (2005).
Straus, D. S. & Glass, C. K. Anti-inflammatory actions of PPAR ligands: new insights on cellular and molecular mechanisms. Trends in immunology 28, 551–558, https://doi.org/10.1016/j.it.2007.09.003 (2007).
Stahl, A. A current review of fatty acid transport proteins (SLC27). Pflugers Archiv: European journal of physiology 447, 722–727, https://doi.org/10.1007/s00424-003-1106-z (2004).
Hostetler, H. et al. L-FABP directly interacts with PPARalpha in cultured primary hepatocytes. Journal of lipid research 50, 1663–1675, https://doi.org/10.1194/jlr.m900058-jlr200 (2009).
Hirsch, D., Stahl, A. & Lodish, H. F. A family of fatty acid transporters conserved from mycobacterium to man. Proceedings of the National Academy of Sciences of the United States of America 95, 8625–8629 (1998).
Stahl, A. A current review of fatty acid transport proteins (SLC27). Pflügers Archiv 447, 722–727, https://doi.org/10.1007/s00424-003-1106-z (2004).
Ockner, R. K., Manning, J. A. & Kane, J. P. Fatty acid binding protein. Isolation from rat liver, characterization, and immunochemical quantification. The Journal of biological chemistry 257, 7872–7878, doi:citeulike-article-id:12725170 (1982).
Stahl, A., Gimeno, R. E., Tartaglia, L. A. & Lodish, H. F. Fatty acid transport proteins: a current view of a growing family. Trends in endocrinology and metabolism: TEM 12, 266–273 (2001).
Anderson, C. M. & Stahl, A. SLC27 fatty acid transport proteins. Molecular aspects of medicine 34, 516–528, https://doi.org/10.1016/j.mam.2012.07.010 (2013).
Ferré, P. The Biology of Peroxisome Proliferator-Activated Receptors: Relationship with Lipid Metabolism and Insulin Sensitivity. Diabetes 53, S43–S50, https://doi.org/10.2337/diabetes.53.2007.s43 (2004).
Marrapodi, M. & Chiang, J. Y. Peroxisome proliferator-activated receptor alpha (PPARalpha) and agonist inhibit cholesterol 7alpha-hydroxylase gene (CYP7A1) transcription. J Lipid Res 41, 514–520 (2000).
Chiang, J. Y. Regulation of bile acid synthesis. Frontiers in bioscience: a journal and virtual library 3 (1998).
Hunt, M. C. et al. The peroxisome proliferator-activated receptor alpha (PPARalpha) regulates bile acid biosynthesis. J Biol Chem 275, 28947–28953, https://doi.org/10.1074/jbc.M002782200 (2000).
Wang, J. et al. Human sterol 12α-hydroxylase (CYP8B1) is mainly expressed in hepatocytes in a homogenous pattern. Histochemistry and Cell Biology 123, 441–446, https://doi.org/10.1007/s00418-005-0779-0 (2005).
Keller, H. et al. Fatty acids and retinoids control lipid metabolism through activation of peroxisome proliferator-activated receptor-retinoid X receptor heterodimers. Proc Natl Acad Sci USA 90, 2160–2164 (1993).
Erol, E. et al. Liver fatty acid binding protein is required for high rates of hepatic fatty acid oxidation but not for the action of PPARalpha in fasting mice. FASEB journal 18, 347–349, https://doi.org/10.1096/fj.03-0330fje (2004).
Kersten, S. et al. The peroxisome proliferator-activated receptor alpha regulates amino acid metabolism. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 15, 1971–1978, https://doi.org/10.1096/fj.01-0147com (2001).
Vallett, S. M., Sanchez, H. B., Rosenfeld, J. M. & Osborne, T. F. A direct role for sterol regulatory element binding protein in activation of 3-hydroxy-3-methylglutaryl coenzyme A reductase gene. J Biol Chem 271, 12247–12253 (1996).
Wright, L. D. Biosynthesis of isoprenoid compounds. Annual review of biochemistry 30, 525–548 (1961).
Edwards, P. A. & Ericsson, J. Sterols and isoprenoids: signaling molecules derived from the cholesterol biosynthetic pathway. Annual review of biochemistry 68, 157–185, https://doi.org/10.1146/annurev.biochem.68.1.157 (1999).
Jassal, B. Cholesterol biosynthesis. Reactome - a curated knowledgebase of biological pathways, (2007).
Liscum, L. In Biochemistry of Lipids, Lipoproteins and Membranes, 4th Edition(eds J. E. Vance & Dennis Vance) (Elsevier Science, 2002).
Sharpe, L. & Brown, A. Controlling cholesterol synthesis beyond 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR). Journal of Biological Chemistry, jbc. R113, 479808, https://doi.org/10.1074/jbc.r113.479808 (2013).
Ganesan, L. P. et al. Scavenger receptor B1, the HDL receptor, is expressed abundantly in liver sinusoidal endothelial cells. Sci Rep 6, 20646, https://doi.org/10.1038/srep20646 (2016).
Herrnberger, L. et al. Formation of fenestrae in murine liver sinusoids depends on plasmalemma vesicle-associated protein and is required for lipoprotein passage. PLoS One 9, e115005, https://doi.org/10.1371/journal.pone.0115005 (2014).
Sharpe, L. J., Cook, E. C., Zelcer, N. & Brown, A. J. The UPS and downs of cholesterol homeostasis. Trends Biochem Sci 39, 527–535, https://doi.org/10.1016/j.tibs.2014.08.008 (2014).
Sharpe, L. J. & Brown, A. J. Controlling cholesterol synthesis beyond 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR). J Biol Chem 288, 18707–18715, https://doi.org/10.1074/jbc.R113.479808 (2013).
Detzel, C. J., Kim, Y. & Rajagopalan, P. Engineered three-dimensional liver mimics recapitulate critical rat-specific bile acid pathways. Tissue Eng Part A 17, 677–689, https://doi.org/10.1089/ten.TEA.2010.0423 (2011).
Eloranta, J. J. & Kullak-Ublick, G. A. Coordinate transcriptional regulation of bile acid homeostasis and drug metabolism. Archives of biochemistry and biophysics 433, 397–412, https://doi.org/10.1016/j.abb.2004.09.019 (2005).
Thomas, C., Pellicciari, R., Pruzanski, M., Auwerx, J. & Schoonjans, K. Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov 7, 678–693, https://doi.org/10.1038/nrd2619 (2008).
Parks, D. J. et al. Bile acids: natural ligands for an orphan nuclear receptor. Science 284, 1365–1368 (1999).
Pineda Torra, I. et al. Bile acids induce the expression of the human peroxisome proliferator-activated receptor alpha gene via activation of the farnesoid X receptor. Molecular endocrinology 17, 259–272, https://doi.org/10.1210/me.2002-0120 (2003).
Spolarics, Z., Lang, C. H., Bagby, G. J. & Spitzer, J. J. Glutamine and fatty acid oxidation are the main sources of energy for Kupffer and endothelial cells. The American journal of physiology 261, G185–190 (1991).
Russell, D. W. Cholesterol biosynthesis and metabolism. Cardiovascular drugs and therapy / sponsored by the International Society of Cardiovascular Pharmacotherapy 6, 103–110 (1992).
Schulz, H. Beta oxidation of fatty acids. Biochimica et biophysica acta 1081, 109–120 (1991).
Lazarow, P. B. Rat liver peroxisomes catalyze the beta oxidation of fatty acids. J Biol Chem 253, 1522–1528 (1978).
Mannaerts, G. P., Debeer, L. J., Thomas, J. & De Schepper, P. J. Mitochondrial and peroxisomal fatty acid oxidation in liver homogenates and isolated hepatocytes from control and clofibrate-treated rats. J Biol Chem 254, 4585–4595 (1979).
McGarry, J. D. & Brown, N. F. The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. European journal of biochemistry/FEBS 244, 1–14 (1997).
Rinaldo, P., Matern, D. & Bennett, M. Fatty Acid Oxidation Disorders. Annual Review of Physiology 64, 477–502, https://doi.org/10.1146/annurev.physiol.64.082201.154705 (2002).
Berg, J., Tymoczko, J. & Stryer, L. Biochemistry, 5th edition. (W H Freeman, 2002).
Gillespie, M. E. Mitochondrial Fatty Acid Beta-Oxidation. Reactome - a curated knowledgebase of biological pathways 06, https://doi.org/10.3180/react_1473.1 (2003).
Hashimoto, T. et al. Peroxisomal and mitochondrial fatty acid beta-oxidation in mice nullizygous for both peroxisome proliferator-activated receptor alpha and peroxisomal fatty acyl-CoA oxidase. Genotype correlation with fatty liver phenotype. J Biol Chem 274, 19228–19236 (1999).
Janssen, U., Davis, E. M., Le Beau, M. M. & Stoffel, W. Human mitochondrial enoyl-CoA hydratase gene (ECHS1): structural organization and assignment to chromosome 10q26.2-q26.3. Genomics 40, 470–475, https://doi.org/10.1006/geno.1996.4597 (1997).
McGarry, J. D. & Foster, D. W. Regulation of hepatic fatty acid oxidation and ketone body production. Annual review of biochemistry 49, 395–420, https://doi.org/10.1146/annurev.bi.49.070180.002143 (1980).
Louet, J. F. et al. Long-chain fatty acids regulate liver carnitine palmitoyltransferase I gene (L-CPT I) expression through a peroxisome-proliferator-activated receptor alpha (PPARalpha)-independent pathway. Biochem J 354, 189–197 (2001).
Dixon, L. J., Barnes, M., Tang, H., Pritchard, M. T. & Nagy, L. E. Kupffer cells in the liver. Compr Physiol 3, 785–797, https://doi.org/10.1002/cphy.c120026 (2013).
Sica, A., Invernizzi, P. & Mantovani, A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology 59, 2034–2042, https://doi.org/10.1002/hep.26754 (2014).
March, S., Hui, E. E., Underhill, G. H., Khetani, S. & Bhatia, S. N. Microenvironmental regulation of the sinusoidal endothelial cell phenotype in vitro. Hepatology 50, 920–928, https://doi.org/10.1002/hep.23085 (2009).
Ravetch, J. V. & Kinet, J. P. Fc receptors. Annu Rev Immunol 9, 457–492, https://doi.org/10.1146/annurev.iy.09.040191.002325 (1991).
Dominguez-Soto, A. et al. Intravenous immunoglobulin promotes antitumor responses by modulating macrophage polarization. J Immunol 193, 5181–5189, https://doi.org/10.4049/jimmunol.1303375 (2014).
Turner, H. & Kinet, J. P. Signalling through the high-affinity IgE receptor Fc epsilonRI. Nature 402, B24–30 (1999).
DeFranco, A. L. Transmembrane signaling by antigen receptors of B and T lymphocytes. Current opinion in cell biology 7, 163–175 (1995).
Mantovani, A., Sozzani, S., Locati, M., Allavena, P. & Sica, A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends in immunology 23, 549–555 (2002).
Sellaro, T. L., Ravindra, A. K., Stolz, D. B. & Badylak, S. F. Maintenance of hepatic sinusoidal endothelial cell phenotype in vitro using organ-specific extracellular matrix scaffolds. Tissue Eng 13, 2301–2310, https://doi.org/10.1089/ten.2006.0437 (2007).
Ford, A. J., Jain, G. & Rajagopalan, P. Designing a fibrotic microenvironment to investigate changes in human liver sinusoidal endothelial cell function. Acta Biomater 24, 220–227, https://doi.org/10.1016/j.actbio.2015.06.028 (2015).
Xie, G. et al. Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats. Gastroenterology 142, 918–927 e916, https://doi.org/10.1053/j.gastro.2011.12.017 (2012).
Ishikawa, K. et al. Expressions of vascular endothelial growth factor in nonparenchymal as well as parenchymal cells in rat liver after necrosis. Biochem Biophys Res Commun 254, 587–593, https://doi.org/10.1006/bbrc.1998.9984 (1999).
Anegawa, G. et al. Defective endothelial nitric oxide synthase signaling is mediated by rho-kinase activation in rats with secondary biliary cirrhosis. Hepatology 47, 966–977, https://doi.org/10.1002/hep.22089 (2008).
Braissant, O., Foufelle, F., Scotto, C., Dauça, M. & Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 137, 354–366 (1996).
Latruffe, N. & Vamecq, J. Peroxisome proliferators and peroxisome proliferator activated receptors (PPARs) as regulators of lipid metabolism. Biochimie 79, 81–94 (1997).
Mandard, S., Muller, M. & Kersten, S. Peroxisome proliferator-activated receptor alpha target genes. Cellular and molecular life sciences: CMLS 61, 393–416, https://doi.org/10.1007/s00018-003-3216-3 (2004).
Rao, M. S. & Reddy, J. K. PPARalpha in the pathogenesis of fatty liver disease. Hepatology 40, 783–786, https://doi.org/10.1002/hep.20453 (2004).
Evans, R. M., Barish, G. D. & Wang, Y. X. PPARs and the complex journey to obesity. Nat Med 10, 355–361, https://doi.org/10.1038/nm1025 (2004).
Jackson, S. M. et al. Peroxisome proliferator-activated receptor activators target human endothelial cells to inhibit leukocyte-endothelial cell interaction. Arteriosclerosis, thrombosis, and vascular biology 19, 2094–2104 (1999).
Marx, N., Sukhova, G., Collins, T., Libby, P. & Plutzky, J. PPARα Activators Inhibit Cytokine-Induced Vascular Cell Adhesion Molecule-1 Expression in Human Endothelial Cells. Circulation 99, 3125–3131, https://doi.org/10.1161/01.cir.99.24.3125 (1999).
Li, Z., Kruijt, K., van der Sluis, R., Van Berkel, T. & Hoekstra, M. Nuclear receptor atlas of female mouse liver parenchymal, endothelial, and Kupffer cells. Physiological Genomics 45, 268–275, https://doi.org/10.1152/physiolgenomics.00151.2012 (2013).
Minnich, A., Tian, N., Byan, L. & Bilder, G. A potent PPARα agonist stimulates mitochondrial fatty acid β-oxidation in liver and skeletal muscle. American Journal of Physiology-Endocrinology And Metabolism 280, E270–E279 (2001).
Meier, P. J. Molecular mechanisms of hepatic bile salt transport from sinusoidal blood into bile. The American journal of physiology 269, G801–812 (1995).
Zhou, D. et al. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell Signal 26, 192–197, https://doi.org/10.1016/j.cellsig.2013.11.004 (2014).
Tomita, K. et al. Tumour necrosis factor alpha signalling through activation of Kupffer cells plays an essential role in liver fibrosis of non-alcoholic steatohepatitis in mice. Gut 55, 415–424, https://doi.org/10.1136/gut.2005.071118 (2006).
Murray, P. J. & Wynn, T. A. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 11, 723–737, https://doi.org/10.1038/nri3073 (2011).
Rajagopalan, P. et al. Polyelectrolyte Nano-scaffolds for the Design of Layered Cellular Architectures. Tissue Engineering 12, 1553–1563, https://doi.org/10.1089/ten.2006.12.1553 (2006).
Sunman, J., Hawke, R., LeCluyse, E. & Kashuba, A. Kupffer cell-mediated IL-2 suppression of CYP3A activity in human hepatocytes. Drug metabolism and disposition: the biological fate of chemicals 32, 359–363, https://doi.org/10.1124/dmd.32.3.359 (2004).
Tokairin, T. et al. A highly specific isolation of rat sinusoidal endothelial cells by the immunomagnetic bead method using SE-1 monoclonal antibody. J Hepatol 36, 725–733 (2002).
Schroeder, A. et al. The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol 7, 3, https://doi.org/10.1186/1471-2199-7-3 (2006).
Irizarry, R. et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics (Oxford, England) 4, 249–264, https://doi.org/10.1093/biostatistics/4.2.249 (2003).
Wilson, C. & Miller, C. Simpleaffy: a BioConductor package for Affymetrix Quality Control and data analysis. Bioinformatics 21, 3683–3685, https://doi.org/10.1093/bioinformatics/bti605 (2005).
Benjamini, Y. & Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society. Series B (Methodological) 57, 289–300, https://doi.org/10.2307/2346101 (1995).
Kandasamy, K. et al. NetPath: a public resource of curated signal transduction pathways. Genome biology 11, R3, https://doi.org/10.1186/gb-2010-11-1-r3 (2010).
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M. & Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic acids research 40, D109–D114, https://doi.org/10.1093/nar/gkr988 (2012).
Croft, D. et al. Reactome: a database of reactions, pathways and biological processes. Nucleic acids research 39, D691–D697, https://doi.org/10.1093/nar/gkq1018 (2011).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics 25, 25–29, https://doi.org/10.1038/75556 (2000).
Franceschini, A. et al. STRINGv9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res 41, D808–815, https://doi.org/10.1093/nar/gks1094 (2013).
Acknowledgements
Grants from the National Science Foundation (DMR-090750, CBET-0933225, DBI-1062380, DMR-1410341, and CBET-1510920), the National Institutes of Health (F32-ES024062), and the U.S. Environmental Protection Agency (RD-83499801) supported this research.
Author information
Authors and Affiliations
Contributions
T.M.M. and P.R. designed the study. A.L. and L.V. performed the experiments. A.N.T. and R.R. performed the computational analysis. A.N.T., T.M.M. and P.R. analyzed the results and wrote the paper.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tegge, A.N., Rodrigues, R.R., Larkin, A.L. et al. Transcriptomic Analysis of Hepatic Cells in Multicellular Organotypic Liver Models. Sci Rep 8, 11306 (2018). https://doi.org/10.1038/s41598-018-29455-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-29455-x
This article is cited by
-
Comparative transcriptomic and phenotypic analysis of induced pluripotent stem cell hepatocyte-like cells and primary human hepatocytes
Cell and Tissue Research (2024)
-
Liver sinusoidal endothelial cells are implicated in multiple fibrotic mechanisms
Molecular Biology Reports (2021)
-
Current progress in hepatic tissue regeneration by tissue engineering
Journal of Translational Medicine (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
