
Abstract
Protein S-sulfenylation, which results from oxidation of free thiols on cysteine residues, has recently emerged as an important post-translational modification that regulates the structure and function of proteins involved in a variety of physiological and pathological processes. By altering the size and physiochemical properties of modified cysteine residues, sulfenylation can impact the cellular function of proteins in several different ways. Thus, the ability to rapidly and accurately identify putative sulfenylation sites in proteins will provide important insights into redox-dependent regulation of protein function in a variety of cellular contexts. Though bottom-up proteomic approaches, such as tandem mass spectrometry (MS/MS), provide a wealth of information about global changes in the sulfenylation state of proteins, MS/MS-based experiments are often labor-intensive, costly and technically challenging. Therefore, to complement existing proteomic approaches, researchers have developed a series of computational tools to identify putative sulfenylation sites on proteins. However, existing methods often suffer from low accuracy, specificity, and/or sensitivity. In this study, we developed SVM-SulfoSite, a novel sulfenylation prediction tool that uses support vector machines (SVM) to identify key determinants of sulfenylation among five feature classes: binary code, physiochemical properties, k-space amino acid pairs, amino acid composition and high-quality physiochemical indices. Using 10-fold cross-validation, SVM-SulfoSite achieved 95% sensitivity and 83% specificity, with an overall accuracy of 89% and Matthew’s correlation coefficient (MCC) of 0.79. Likewise, using an independent test set of experimentally identified sulfenylation sites, our method achieved scores of 74%, 62%, 80% and 0.42 for accuracy, sensitivity, specificity and MCC, with an area under the receiver operator characteristic (ROC) curve of 0.81. Moreover, in side-by-side comparisons, SVM-SulfoSite performed as well as or better than existing sulfenylation prediction tools. Together, these results suggest that our method represents a robust and complementary technique for advanced exploration of protein S-sulfenylation.
Similar content being viewed by others

Introduction
Redox-dependent signalling plays a critical role in physiological processes such as aging and the immune response, as well as in a number of pervasive diseases, including cancer, Alzheimer’s disease, cardiovascular disease and diabetes1,2,3. For instance, protein S-sulfenylation (Fig. 1), which typically occurs in the cell via H2O2–dependent conversion of free thiols (-SH) on cysteine residues to sulfenic acid (-SOH), has emerged as a key post-translation modification (PTM) that can regulate the function of target proteins4,5. For example, sulfenylation can modulate the enzymatic activity, binding affinity, stability and/or subcellular localization of cellular proteins2,5,6,7,8. While sulfenylation is readily reversible (either by reduction back to the free thiol or via disulfide formation followed by reduction by cellular enzymes, such as glutaredoxin and thioredoxin), further oxidation by various peroxides (ROOH) or H2O2 can convert sulfenic acid to sulfinic and sulfonic acid moieties that are largely irreversible inside the cell9.

In the presence of an oxidizing agent (ROOH), the thiolate anion of a redox-sensitive Cys (Cys-S−) is reversibly oxidized to form a sulfenic acid (SOH). When in close proximity to another reactive Cys (SH), either in the same protein molecule or in another protein, SOH leads to disulfide bond (S-S) formation. In addition to Cys residues in proteins, SOH can also react with the cellular antioxidant, glutathione (g-Glu-Cys-Gly; GSH) to form a mixed S-S bond (PSSG). Aside from altering the chemical properties of the Cys residue and the tertiary structure of the protein, S-S bonds are also believed to prevent terminal oxidation to sulfinic (SO2H) and sulfonic (SO3H) acid. Disulfide bonds can be reduced back to the thiol by cellular antioxidant enzymes, such as glutaredoxin (Grx) or thioredoxin (Trx).
To identify sulfenylation sites on proteins, several chemo-proteomic methods have been developed10,11,12,13,14,15,16. For instance, Yang et al. recently used selective labelling for cysteine sulfenic acid together with tandem mass spectrometry (MS/MS)-based detection to identify more than 1,000 sulfenylation sites in ~700 cellular proteins4.Though such chemo-proteomic methods provide a wealth of information about the redox regulation of cellular proteins in situ, these methods are time-consuming, expensive and require a high level of technical expertise. Moreover, though MS-based methods provide significant depth of coverage of the ‘sulfenylome’, they can be biased towards abundant proteins. Finally, other redox reactions that occur following lysis, such as adventitious oxidation by ambient oxygen, can generate SOH sites that do not necessarily occur under physiological conditions. Therefore, to complement global proteomic analysis of sulfenylation site identification, several computational approaches have been developed to predict sulfenylation sites in silico17,18,19,20,21,22. The first method, developed by Bui and colleagues in 2015, uses maximal dependence decomposition (MDD) and support vector machines (SVM) to identify sulfenylation motifs based on amino acid composition and solvent accessibility. Using 5-fold cross-validation, the resulting method, known as MDD-SOH, achieved efficiency scores of 68%, 70%, 70%, and 0.27 for accuracy (ACC), sensitivity (SN), specificity (SP), and Matthew’s correlation coefficient (MCC), respectively17. Subsequently, Xu et al. used SVM to identify key parameters for sulfenylation from among 14 types of physicochemical properties18. The resulting sulfenylation site prediction tool, termed iSulf-Cys, performed similarly to MDD-SOH based on 10-fold cross-validation, with a slight increase in MCC18. Similarly, other prediction tools based on strategies including position specific scoring matrices (PSSM) and SVM have offered modest improvements in performance19,20,21. For instance, Sakka et al. developed the PRotEin S-sulfenylation server (PRESS), which uses SVM to identify structural features that correlate with sulfenylation20. Using 10-fold cross-validation, PRESS achieved ACC, SN and SP scores of 77%, 80% and 74%, respectively. Most recently, two prediction tools, S-SulfPred and SulCysSite, were reported that also show significant improvements in one or more performance metric22,23. For instance, Hasan et al. developed a predictor named SulCysSite that uses a random forest-based strategy to identify key parameters related to sulfenylation from among four features. Using this approach, the authors observed SN, SP and MCC scores of 62%, 81%, and 0.45, respectively, upon 10-fold cross-validation23. Likewise, Jia and Zuo developed a method named S-SulfPred, which combines a synthetic minority oversampling technique (SMOTE) and an under-sampling strategy with an SVM implementation to predict sulfenylation sites based on three feature types22. Upon 10-fold cross-validation, S-SulfPred achieved impressive scores of 88%, 78%, 91% and 0.64 in terms of ACC, SN, SP and MCC, respectively.
Despite the significant progress that has been made over the past two years, there is still room for improvement in the performance of existing sulfenylation site prediction tools. For instance, many of the existing sulfenylation site prediction tools exhibit relatively low MCC scores compared to prediction tools for other posttranslational modifications, such as phosphorylation24, hydroxylation25 and glycosylation26. While this is likely a function of the size of datasets available for training and/or the mode of modification (i.e., enzyme-mediated modification for phosphorylation, hydroxylation and glycosylation versus predominantly non-enzymatic modification for sulfenylation), it is also likely that other factors contribute to the relatively low MCC scores. Moreover, in addition to the primary amino acid sequence, some methods, such as PRESS, require structural data about the protein-of-interest. As a consequence, the number of proteins that can be analysed is greatly reduced. Here, we describe the development and evaluation of SVM-SulfoSite, a novel sulfenylation site prediction tool designed to identify putative sulfenylation sites using only the primary amino acid sequence as input. SVM-SulfoSite combines multiple features, including physiochemical properties, amino acid composition and high-quality indices, with novel classifier algorithms and an SVM-based machine learning strategy to predict sulfenylation sites in proteins. Based on evaluation using both 10-fold cross-validation and an independent dataset, SVM-SulfoSite compares favourably to existing sulfenylation site prediction tools with regard to accuracy, sensitivity, specificity and MCC. Therefore, SVM-SulfoSite represents a robust, accessible sulfenylation prediction tool that promises to provide additional insights into the regulation and biological consequences of protein S-sulfenylation.
Results and Discussion
Model development
To develop a robust sulfenylation site prediction tool that is able to identify putative sulfenylation sites using only the primary amino acid sequence as input, we first compiled training and independent test sets similar to those described by Xu et al.18. After discarding sequences that exhibited ≥40% identity, 1,045 positive sites and 7,126 negative sites on 778 human proteins remained. All sites had been experimentally verified previously4. We then randomly selected 145 positive sites and 268 negative sites for the independent test set and used the remaining sites for model training and development. The training set consisted of 900 positive sites and 6,858 negative sites. Each 21-residue fragment contained a central cysteine (either sulfenylated or not sulfenylated) flanked by 10 residues on either side. Due to the disproportionate number of negative sites, the positive and negative sets were imbalanced. Therefore, we used oversampling to balance the positive and negative classes27. This approach, which randomly selects data points from the minority class until it is equivalent to the majority class, has been used for bioinformatics analysis in several biological contexts28,29. After balancing the positive and negative training sets, all fragment sequences were converted into vectors for analysis. To this end, we selected five features—binary encoding (BE), 14 types of physicochemical amino acid properties (AAindex), k-spaced amino acid pairs (KSAAP), amino acid composition (AAC) and high-quality indices (HQI)—and generated a unique vector for each feature. Finally, to build our predictor, a SVM-based machine learning strategy was employed to identify those parameters that correlated with sulfenylation in the training set. After each round of parameter optimization, the resulting model was assessed using 10-fold cross-validation until no further improvement in model performance was observed. The resulting model, which we termed SVM-SulfoSite, exhibited robust performance with respect to several standard scoring metrics, as described below.
Model evaluation
To assess the performance of SVM-SulfoSite, we first used 10-fold cross-validation30,31,32,33. To this end, the training set was partitioned into 10 subsets. We then used 9 of the subsets to train the model and the remaining subset for testing. We repeated this technique ten times, with the final result representing the average performance of 10 models. As can be seen in Table 1, SVM-SulfoSite exhibited high efficiency scores for ACC, SN and SP, leading to a strong MCC score of 0.79. Though the scores were relatively high across all metrics, SVM-SulfoSite performed particularly well with respect to SN (95%) and ACC (89%). Likewise, the area under the ROC curve was a robust 0.97 (Fig. 2). To gain further insights into the relative impact of each feature on the overall performance of our method, we also conducted 10-fold cross-validation using only one feature at a time (Table 1 and Fig. 2). While each of the individual features contributed substantially to the overall score for most metrics, KSAAP appears to have the largest impact on model performance across all metrics except SP. Interestingly, while BE and AAC both appear to have a relatively strong effect on sensitivity, AAC does not impact specificity to the same extent as the other features. Together, these data suggest that both the frequency (AAC) and the relative spacing of amino acids around the modified cysteine residue (KSAAP) may play an important role in correctly identifying positive sites of sulfenylation (i.e., SN) while the former is less important for correctly predicting negative sites (i.e., SP).

Receiver operator characteristic (ROC) curves for each of five features used to develop our method as well as that for the final method utilizing all features (SVM-SulfoSite) for the 10-fold cross-validation. The area under the curve (AUC) for each feature is given in parentheses.
In addition to 10-fold cross-validation, we also evaluated the performance of our method using an independent test set that had been kept hidden from the model during training. Though the efficiency scores were not as high as those observed using 10-fold cross-validation, SVM-SulfoSite still performed quite well using the independent test set (Table 2). Interestingly, while very little difference was observed between the SP scores obtained using the independent set and 10-fold cross-validation (80% and 83%,respectively), we noticed a marked decrease in SN using the independent set (62% for the independent test set versus 95% for 10-fold cross-validation). Likewise, there was a modest decrease in ACC between the two evaluation methods (74% for the independent set and 89% for 10-fold cross-validation). Together, these changes decreased the MCC score from 0.79 to 0.42. Likewise, the AUC dropped from 0.97 to 0.81. The observed differences between the performance metrics using 10-fold cross-validation and our independent set may be a consequence of the oversampling strategy used to balance the positive and negative datasets during training. Among the individual features, KSAAP scores decreased most dramatically when SVM-SulfoSite was evaluated using the independent test set, followed by BE. On the other hand, AAindex and HQI exhibited very similar scores regardless of the evaluation method used. It is interesting to note that both of these features rely on physiochemical properties of amino acids. This is consistent with the notion that sulfenylation is typically a non-enzymatic PTM that may be particularly sensitive to the surrounding physiochemical environment of the modified cysteine7.
Comparison with existing methods
Next, we compared the performance of SVM-SulfoSite with that of other recently developed sulfenylation site predictors using both 10-fold cross-validation and independent test sets. Based on 10-fold cross-validation, SVM-SulfoSite performed as well or better than all existing methods in each category (with the exception of SP, where SVM-SulfoSite was outperformed by S-SulfPred) (Table 3). The largest improvements were observed for MCC, where SVM-SulfoSite scored ~2.1-fold higher than the average MCC across all methods (ranging from a 23% improvement versus S-SulfPred to a 2.9-fold improvement over MDD-SOH). This was followed by SN, where SVM-SulfoSite exhibited an average increase of 36% (ranging from 19 to 61% over PRESS and SOHPRED, respectively) and ACC (average improvement of 19%, ranging from 1 to 35% improvement over S-SulfPred and iSulf-Cys, respectively). Finally, the smallest gains were observed for SP, where SVM-SulfoSite exhibited an average improvement of 11% (ranging from a 9% decrease versus S-SulfPred to a 30% increase compared to iSulf-Cys).
Interestingly, SP was one of the categories where we observed the greatest gains when the methods were compared using an independent test set (Table 4). In fact, the average increase in SP observed for SVM-SulfoSite using the independent dataset (18%) was ~65% greater than that observed using 10-fold cross-validation (11%). The gains in SP ranged from a 13% increase (which was observed for several methods, including S-SulfPred) to a 21% increase over iSulf-Cys. Similarly, SVM-SulfoSite outperformed all existing methods with respect to ACC using the independent set. Indeed, the average improvement in ACC was 6%, ranging from 3 to 10% versus S-SulfPred and iSulf-Cys, respectively. In contrast, SVM-SulfoSite exhibited lower SN scores than the other methods, with an average decrease of 14% (ranging from 10% versus iSulf-Cys to 18% compared to SulCysSite). Nonetheless, the improvements observed in the other areas led to an average increase in MCC of 33% for SVM-SulfoSite when assessed using the independent test set (ranging from a 2% decrease versus S-SulfPred to a 56% increase versus PRESS). Together, these data suggest that SVM-SulfoSite is able to distinguish between sulfenylated and non-sulfenylated cysteine residues in an efficient manner. Importantly, our predictor performs as well as or better than existing methods in all performance metrics except SN, where it exhibits modest decreases in performance when using an independent dataset. In particular, SVM-SulfoSite performs very well with respect to SP, where it outperforms existing methods in side-by-side comparisons using an independent dataset. These data suggest that SVM-SulfoSite is able to distinguish between true negative and false positive sites more efficiently than existing tools.
Conclusion
Reversible sulfenylation of proteins is an important PTM involved in the regulation of a number cellular processes that contribute to health and disease, yet experimental identification of sulfenylation sites remains challenging. This is due to several factors, including the highly reactive nature of the sulfenic acid moiety and the fact that sulfenylation is generally believed to be a non-enzymatic process7. Therefore, several computational methods have recently been developed to predict putative sulfenylation sites in silico. Despite these strides, there is still room for improvement - particularly with respect to overall predictor performance. Here, we developed a novel sulfenylation prediction tool, termed SVM-SulfoSite, that uses a SVM-based strategy to identify parameters that correlate with sulfenylation among five distinct feature sets. Based on analysis using both 10-fold cross-validation and an independent dataset, SVM-SulfoSite performed as well or better than existing methods with respect to MCC (Tables 3 and 4), which is often viewed as a surrogate for overall performance since it integrates information about the true positive, false positive, true negative and false negative rates34,35. For all previously developed sulfenylation prediction methods, the MCC is <0.65 based on 10-fold cross-validation (Table 3). In contrast, SVM-SulfoSite achieved an MCC of 0.79 when assessed using 10-fold cross-validation. Similarly, when performance was assessed using an independent test set, only S-SulfPred and SVM-SulfoSite achieved an MCC >0.42 (Table 4).
In particular, SVM-SulfoSite consistently ranked among the best methods (i.e., either first or second) with respect to ACC, SP and MCC regardless of the evaluation method employed. Interestingly, though SVM-SulfoSite exhibited the highest SN score when assessed by 10-fold cross-validation (95%), its performance in this area decreased substantially when evaluated using an independent test set (62%). This may be a consequence of the oversampling strategy that we employed to account for incongruities between the number of positive and negative sites in the training set. Indeed, due to the sub-stoichiometric nature of many PTMs inside the cell, a major challenge in PTM predictor development is how to handle imbalanced datasets. Whereas under-sampling is often used to balance training sets during the development of prediction tools for well-studied PTMs such as phosphorylation sites24,36, due to the relatively small number of experimentally-verified sulfenylation sites available for analysis, we chose instead to resolve this issue using an oversampling strategy so that no information about negative sites was lost during balancing. On the other hand, this strategy likely improved the performance of SVM-SulfoSite with respect to SP. Indeed, since negative sites were retained during training, our method is able to identify negative sites more efficiently than other methods, as reflected in the highest SP scores using both 10-fold cross-validation and an independent test set. Similarly, the ability to better distinguish between positive and negative sites may have also contributed to the improved fidelity of our method, which is supported by increased scores for ACC and MCC. In conclusion, we have developed and evaluated a novel and robust sulfenylation site prediction tool, termed SVM-SulfoSite, that is able to efficiently identify sulfenylation sites in proteins using only the primary amino acid sequence as input. As a complementary approach to experimental strategies, such as chemo-proteomic and biochemical analyses, SVM-SulfoSite has the potential to provide new insights into the characteristics and functional consequences of protein sulfenylation. To facilitate its use by the biomedical research community, the method has been posted on GitHub (https://github.com/HussamAlbarakati/SVM-Sulfosite) and we are currently developing a webserver that will allow biological researchers to use this method to predict putative sulfenylation sites in proteins-of-interest.
Materials and Methods
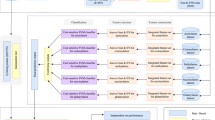
Our method was developed using a four-step procedure (Fig. 3). In the first step, data were collected and pre-processed to remove overlapping sequences. Next, sequences containing a central cysteine residue were identified and features (defined below) were extracted using binary encoding (BE), 14 types of physiochemical amino acid properties (AAindex), k-space amino acid pairs (KSAAP), amino acid composition (AAC) and high-quality indices (HQI). Following feature extraction, oversampling techniques were used to balance the training dataset. Finally, the model was evaluated using both 10-fold cross-validation and an independent dataset.

Schematic showing the workflow used to develop our method. KSAAP: k-space amino acid pairs; HQI: high-quality indices; BE: Binary encoding; AAC: Amino acid composition; AAindex: physiochemical amino acid properties; SVM: support vector machines; NCBI: National Center for Biotechnology Information.
Binary encoding (BE)
To convert each amino acid in a sequence into a unique numerical vector, we used an orthogonal binary scheme. According to this strategy, 21 dimensional vectors were used to represent each of the 20 canonical amino acid plus a 21st position, corresponding to non-existent residues at the extreme N- or C-terminus of a protein (denoted as X). For example, alanine (A) was encoded as “100000000000000000000”, while cysteine (C) was encoded as “010000000000000000000” and so on. “Non-existent” X residues were encoded as “000000000000000000001”. In all windows evaluated, the central residue was cysteine (C), which was removed from window sites. Therefore, the total BE feature vector was 420.
Amino acid index (AAindex) property
Each amino acid has several specific physicochemical and biological properties that directly or indirectly influence protein properties. Dissimilar mixtures of those properties have different impacts to the construction and function of proteins. AAindex37 is a database that holds numerous physicochemical and biological properties of amino acids. Several mixtures of physicochemical properties have been employed that effectively convert sequences of peptides into mathematical expressions18,38,39,40. In this study, we used 14 physicochemical properties: hydrophobicity, solvent accessibility, polarity, polarizability, accessibility, PK-N, PK-C, melting point, molecular weight, optical rotation, net charge index of side chains, entropy of formation, heat capacity and absolute entropy. In each case, the feature properties for non-existent residues, X, were represented as 0. Likewise, as above, the central residue was always Cys, which was not considered for window sites. Therefore, the total AAindex feature vector was 280.
K-space amino acid pairs (KSAAP)
Each KSAAP can be denoted as fr{k}fs(r, s = 1, 2, …, 20), where fr and fs represent any two residues from among the 20 canonical amino acids. If k = 0, fr{k}fs positions for a dipeptide and total feature vector is 40041. In this study, we also considered X, which represents non-existent residues such that fr{k}fs(r, s = 1, 2, …, 21). Since we defined kmax = 3, the total KSAAP feature vector was thus 21 * 21 * 3 = 1,323.
Amino acid composition (AAC)
AAC represents the frequency of each amino acid in a protein fragment. The fraction of 20 canonical amino acids can be calculated according to equation 1:
where Ra is the number of a given amino acid in a fragment and R denotes the length of the fragment42. The total AAC feature vector was 20.
High quality indices (HQI)
High quality physicochemical indices were similar to those used to develop the hydroxylation site prediction tools, predHydroxy and RF-Hydroxysite25,36. Briefly, 8 indices describing various physicochemical properties corresponding to 8 groups were generated by grouping 544 associated amino acid properties in the AAIndex database using fuzzy clustering43. The 8 indices are (BLAM930101)44, (BIOV880101)45, (MAXF760101)46, (TSAJ990101)47, (NAKH920108)48, (CEDJ970104)49, (LIFS790101)50 and (MIYS990104)51. We extracted HQIs for each amino acid residue neighbouring the cysteine residue (regardless of whether it was modified or unmodified). The central residue was always C, which was not considered for window sites. Thus, the total HQI feature vector was 160.
Oversampling to balance training data
Class imbalance, which happens when the sample sizes in the data classes are unequally dispersed52, has been identified as one of the most difficult issues in the machine-learning field53. Previous studies have described various approaches to address data imbalance27,28,29. A popular approach is random under-sampling, which decreases the number of sample points from the majority class such that the majority class is balanced with the minority class. However, the disadvantage of using this method is that it can dispose of conceivably valuable data that could be essential for classifiers27. Therefore, in this study, we utilized an alternative oversampling approach that randomly replicates data points from the minority class to balance with the majority class. The primary benefit of using this strategy is that there is no loss of information, as is seen when using an under-sampling approach28,29.
Model learning and testing
To train our algorithm, we utilized a SVM-based machine learning strategy. SVMs are supervised machine learning algorithms used for many classification problems. SVM has been widely used in many bioinformatics problems, such as SUMOylation site prediction54 and protein fold recognition55. Briefly, SVM constructs a hyperplane in a high dimension space that separates two classes of attribute vectors using the largest distance margin. In this study, we used a radial basis function (RBF) kernel, which facilitates the classification of data that is not linearly separable56. This approach was initially performed on the training dataset using 2,203 features. The fidelity of the model parameters was assessed after each round of analysis based on 10-fold cross-validation and optimized in an iterative manner until no further improvement in model performance was observed. Two parameters, penalty (C = 0.1) and kernel width (kw = 0.005), were chosen based on the best predictive performance of our method.
To evaluate the performance of our method, we assessed four commonly used metrics: accuracy (ACC), sensitivity (SN), specificity (SP) and the Matthew’s correlation coefficient (MCC). These metrics, which have been used in previous studies to evaluate performance of other predictors24,25, are defined below:
True positives (TP) indicate the number of correctly classified sulfenylation sites, true negatives (TN) represent the number of correctly classified negative sites, false positives (FP) denote the number of negative sites incorrectly classified as sulfenylation sites and false negatives (FN) indicate the number of actual sulfenylation sites incorrectly classified as negative sites. In addition, we also assessed the area under the receiver-operator characteristic (ROC) curve as an indicator of model performance. The ROC curve, which illustrates the trade-off between specificity and sensitivity57, yields a solitary execution measure called the area under curve (AUC) score, where the AUC for an arbitrary classifier is 0.5 and an AUC of 1.0 represents a perfect classifier58.
Data availability
The data, the source code, and other materials for the work are available in the GitHub repository, @[https://github.com/HussamAlbarakati/SVM-Sulfosite].
References
Poole, L. B. & Schoneich, C. Introduction: What we do and do not know regarding redox processes of thiols in signaling pathways. Free Radic Biol Med 80, 145–147 (2015).
Roos, G. & Messens, J. Protein sulfenic acid formation: from cellular damage to redox regulation. Free Radic Biol Med 51, 314–326 (2011).
Chung, H. S., Wang, S. B., Venkatraman, V., Murray, C. I. & Van Eyk, J. E. Cysteine oxidative posttranslational modifications: emerging regulation in the cardiovascular system. Circ Res 112, 382–392 (2013).
Yang, J., Gupta, V., Carroll, K. S. & Liebler, D. C. Site-specific mapping and quantification of protein S-sulphenylation in cells. Nat Commun 5, 4776 (2014).
Poole, L. B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic Biol Med 80, 148–157 (2015).
Leonard, S. E. & Carroll, K. S. Chemical ‘omics’ approaches for understanding protein cysteine oxidation in biology. Curr Opin Chem Biol 15, 88–102 (2011).
Poole, L. B. & Nelson, K. J. Discovering mechanisms of signaling-mediated cysteine oxidation. Curr Opin Chem Biol 12, 18–24 (2008).
Wani, R. et al. Isoform-specific regulation of Akt by PDGF-induced reactive oxygen species. Proc Natl Acad Sci USA 108, 10550–10555 (2011).
Lindahl, M., Mata-Cabana, A. & Kieselbach, T. The disulfide proteome and other reactive cysteine proteomes: analysis and functional significance. Antioxid. Redox. Signaling, 14, 2581−2642 (2011).
Weerapana, E. et al. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468, 790–795 (2010).
Wang, C., Weerapana, E., Blewett, M. M. & Cravatt, B. F. A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles. Nat Methods 11, 79–85 (2014).
Szychowski, J. et al. Cleavable biotin probes for labeling of biomolecules via azide-alkyne cycloaddition. J Am Chem Soc 132, 18351–18360 (2010).
Zheng, T., Jiang, H. & Wu, P. Single-stranded DNA as a cleavable linker for bioorthogonal click chemistry-based proteomics. Bioconjug Chem 24, 859–864 (2013).
Qian, Y. et al. An isotopically tagged azobenzene-based cleavable linker for quantitative proteomics. Chembiochem 14, 1410–1414 (2013).
Furdui, C. M. & Poole, L. B. Chemical approaches to detect and analyze protein sulfenic acids. Mass Spectrom Rev 33, 126–146 (2014).
Qian, J. et al. A simple and effective strategy for labeling cysteine sulfenic acid in proteins by utilization of beta-ketoesters as cleavable probes. Chem Commun (Camb) 48, 4091–4093 (2012).
Bui, V. M., Lu, C. T., Ho, T. T. & Lee, T. Y. MDD-SOH: exploiting maximal dependence decomposition to identify S-sulfenylation sites with substrate motifs. Bioinformatics 32, 165–172 (2016).
Xu, Y., Ding, J. & Wu, L. Y. iSulf-Cys: Prediction of S-sulfenylation Sites in Proteins with Physicochemical Properties of Amino Acids. PLoS One 11, e0154237 (2016).
Bui, V. M. et al. SOHSite: incorporating evolutionary information and physicochemical properties to identify protein S-sulfenylation sites. BMC Genomics 17(Suppl 1), 9 (2016).
Sakka, M. et al. PRESS: PRotEin S-Sulfenylation server. Bioinformatics 32, 2710–2712 (2016).
Wang, X., Yan, R., Li, J. & Song, J. SOHPRED: a new bioinformatics tool for the characterization and prediction of human S-sulfenylation sites. Mol Biosyst 12, 2849–2858 (2016).
Lei, G. -C., Tang, J. & Du, P. -F. Predicting S-sulfenylation Sites Using Physicochemical Properties Differences. Letters in Organic Chemistry 14, 665–672 (2017).
Hasan, M. M., Guo, D. & Kurata, H. Computational identification of protein S-sulfenylation sites by incorporating the multiple sequence features information. Mol Biosyst 13, 2545–2550 (2017).
Ismail, H. D., Jones, A., Kim, J. H., Newman, R. H. & Kc, D. B. RF-Phos: A Novel General Phosphorylation Site Prediction Tool Based on Random Forest. Biomed Res Int 2016, 3281590 (2016).
Ismail, H. D., Newman, R. H. & Kc, D. B. RF-Hydroxysite: a random forest based predictor for hydroxylation sites. Mol Biosyst 12, 2427–2435 (2016).
Hassan, H., Badr, A. & Abdelhalim, M. B. Prediction of O-glycosylation Sites Using Random Forest and GA-Tuned PSO Technique. Bioinform Biol Insights 9, 103–109 (2015).
KrishnaVeni, C. & Sobha Rani, T. On the classification of imbalanced datasets. IJCST 2, 145–148 (2011).
Guo, X., Yin, Y., Dong, C., Yang, G. & Zhou, G. On the class imbalance problem. in Natural Computation, 2008. ICNC'08. Fourth International Conference on, Vol. 4 192–201 (IEEE, 2008).
Kotsiantis, S., Kanellopoulos, D. & Pintelas, P. Handling imbalanced datasets: A review. GESTS International Transactions on Computer Science and Engineering 30, 25–36 (2006).
Chou, K. C. & Zhang, C. T. Prediction of protein structural classes. Crit Rev Biochem Mol Biol 30, 275–349 (1995).
Pan, Z. et al. Systematic analysis of the in situ crosstalk of tyrosine modifications reveals no additional natural selection on multiply modified residues. Sci Rep 4, 7331 (2014).
Xu, H. D., Shi, S. P., Wen, P. P. & Qiu, J. D. SuccFind: a novel succinylation sites online prediction tool via enhanced characteristic strategy. Bioinformatics 31, 3748–3750 (2015).
Liu, B. et al. iDNA-Prot|dis: identifying DNA-binding proteins by incorporating amino acid distance-pairs and reduced alphabet profile into the general pseudo amino acid composition. PLoS One 9, e106691 (2014).
Boughorbel, S., Jarray, F. & El-Anbari, M. Optimal classifier for imbalanced data using Matthews Correlation Coefficient metric. PLoS One 12, e0177678 (2017).
Powers, D. M. W. Evaluation: from precision, recall and F-measure to ROC, informedness, markedness and correlation. J. Mach. Learn. Technol. 2, 37–63 (2011).
Shi, S. P., Chen, X., Xu, H. D. & Qiu, J. D. PredHydroxy: computational prediction of protein hydroxylation site locations based on the primary structure. Mol Biosyst 11, 819–825 (2015).
Kawashima, S. et al. AAindex: amino acid index database, progress report 2008. Nucleic Acids Res 36, D202–205 (2008).
Zhao, X. et al. Position-specific analysis and prediction of protein pupylation sites based on multiple features. Biomed Res Int 2013, 109549 (2013).
Zheng, L. L. et al. Prediction of protein modification sites of pyrrolidone carboxylic acid using mRMR feature selection and analysis. PLoS One 6, e28221 (2011).
Xu, Y., Ding, Y. X., Ding, J., Wu, L. Y. & Xue, Y. Mal-Lys: prediction of lysine malonylation sites in proteins integrated sequence-based features with mRMR feature selection. Sci Rep 6, 38318 (2016).
Hasan, M. M. et al. Computational Identification of Protein Pupylation Sites by Using Profile-Based Composition of k-Spaced Amino Acid Pairs. PLoS One 10, e0129635 (2015).
Cao, D. S., Xu, Q. S. & Liang, Y. Z. propy: a tool to generate various modes of Chou’s PseAAC. Bioinformatics 29, 960–962 (2013).
Saha, I., Maulik, U., Bandyopadhyay, S. & Plewczynski, D. Fuzzy clustering of physicochemical and biochemical properties of amino acids. Amino Acids 43, 583–594 (2012).
Blaber, M., Zhang, X. J. & Matthews, B. W. Structural basis of amino acid alpha helix propensity. Science 260, 1637–1640 (1993).
Biou, V., Gibrat, J. F., Levin, J. M., Robson, B. & Garnier, J. Secondary structure prediction: combination of three different methods. Protein Eng 2, 185–191 (1988).
Maxfield, F. R. & Scheraga, H. A. Status of empirical methods for the prediction of protein backbone topography. Biochemistry 15, 5138–5153 (1976).
Tsai, J., Taylor, R., Chothia, C. & Gerstein, M. The packing density in proteins: standard radii and volumes. J Mol Biol 290, 253–266 (1999).
Nakashima, H. & Nishikawa, K. The amino acid composition is different between the cytoplasmic and extracellular sides in membrane proteins. FEBS Lett 303, 141–146 (1992).
Cedano, J., Aloy, P., Perez-Pons, J. A. & Querol, E. Relation between amino acid composition and cellular location of proteins. J Mol Biol 266, 594–600 (1997).
Lifson, S. & Sander, C. Antiparallel and parallel beta-strands differ in amino acid residue preferences. Nature 282, 109–111 (1979).
Miyazawa, S. & Jernigan, R. L. Self-consistent estimation of inter-residue protein contact energies based on an equilibrium mixture approximation of residues. Proteins 34, 49–68 (1999).
Daskalaki, S., Kopanas, I. & Avouris, N. Evaluation of classifiers for an uneven class distribution problem. Applied artificial intelligence 20, 381–417 (2006).
He, H. & Garcia, E. A. Learning from imbalanced data. IEEE Transactions on knowledge and data engineering 21, 1263–1284 (2009).
Yavuz, A. S. & Sezerman, O. U. Predicting sumoylation sites using support vector machines based on various sequence features, conformational flexibility and disorder. BMC Genomics 15(Suppl 9), S18 (2014).
Yan, R. X., Si, J. N., Wang, C. & Zhang, Z. DescFold: a web server for protein fold recognition. BMC Bioinformatics 10, 416 (2009).
Chang, C.-C. & Lin, C.-J. LIBSVM: a library for support vector machines. ACM transactions on intelligent systems and technology (TIST) 2, 27 (2011).
Fawcett, T. An introduction to ROC analysis. Pattern recognition letters 27, 861–874 (2006).
Hanley, J. A. & McNeil, B. J. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 143, 29–36 (1982).
Acknowledgements
This work was supported by National Science Foundation (NSF) grant nos. 1647884 and 1564606 (to DBK), DBI-0939454 (to RHN and DBK) and MCB-1714405 (to LMH) as well as National Institutes of Health grant 5SC2GM113784 (to RHN) and R33 CA 177461 (to LBP).
Author information
Authors and Affiliations
Contributions
D.B.K., R.H.N., L.M.H., L.B.P. conceived and designed the study. H.J.A. performed the experiments. H.J.A. and E.W.M. analyzed the results. All the authors wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
AL-barakati, H.J., McConnell, E.W., Hicks, L.M. et al. SVM-SulfoSite: A support vector machine based predictor for sulfenylation sites. Sci Rep 8, 11288 (2018). https://doi.org/10.1038/s41598-018-29126-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-29126-x
This article is cited by
-
IHEC_RAAC: a online platform for identifying human enzyme classes via reduced amino acid cluster strategy
Amino Acids (2021)
-
piEnPred: a bi-layered discriminative model for enhancers and their subtypes via novel cascade multi-level subset feature selection algorithm
Frontiers of Computer Science (2021)
-
SIMLIN: a bioinformatics tool for prediction of S-sulphenylation in the human proteome based on multi-stage ensemble-learning models
BMC Bioinformatics (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
