
Abstract
Quantitatively benchmarking similarities and differences between the in vivo central nervous system and in vitro neuronal cultures can qualify discrepancies in functional responses and establish the utility of in vitro platforms. In this work, extracellular electrophysiology responses of cortical neurons in awake, freely-moving animals were compared to in vitro cultures of dissociated cortical neurons. After exposure to two well-characterized drugs, atropine and ketamine, a number of key points were observed: (1) significant differences in spontaneous firing activity for in vivo and in vitro systems, (2) similar response trends in single-unit spiking activity after exposure to atropine, and (3) greater sensitivity to the effects of ketamine in vitro. While in vitro cultures of dissociated cortical neurons may be appropriate for many types of pharmacological studies, we demonstrate that for some drugs, such as ketamine, this system may not fully capture the responses observed in vivo. Understanding the functionality associated with neuronal cultures will enhance the relevance of electrophysiology data sets and more accurately frame their conclusions. Comparing in vivo and in vitro rodent systems will provide the critical framework necessary for developing and interpreting in vitro systems using human cells that strive to more closely recapitulate human in vivo function and response.
Similar content being viewed by others

Introduction
Efforts to develop human cell-based microphysiological systems that recapitulate organ function are aimed at reducing the extensive use of experimental animal models, which often inaccurately predict human specific responses1,2. Fully validated systems hold promise for evaluating new drugs, characterizing toxicants, and aiding in the elucidation of disease mechanisms. A system that faithfully captures important brain function would be particularly relevant; neurological diseases and injuries are significant causes of morbidity and mortality but are often intractable to development of new and effective therapeutics when relying solely on animal models.
Neuronal cultures are frequently used to evaluate the effects of drugs or toxicants on cellular responses, taking advantage of multi-electrode arrays (MEAs) to provide a means to non-invasively interrogate neuronal cell health and function3,4,5,6,7. However, little to no information is available on the extent to which these in vitro systems reflect what is observed in vivo, despite calls from the research community to examine this relationship more closely4,8. While organotypic brain slices that preserve cellular architecture and circuitry are often superior to dissociated neurons as in vitro models, cell cultures are widely used as diagnostic screening platforms9,10. Furthermore, neuronal cultures, in contrast to brain slices cultured on MEAs, are more readily adapted to human-relevant systems by utilizing either human primary or stem-cell derived neurons. As such, a direct comparison of dissociated neuronal cultures to whole brain provides important and necessary information to frame the relevance of in vitro data sets. To that end, we chose to compare the prefrontal cortex in vivo to dissociated cortical neurons in vitro. The prefrontal cortex was chosen due to ease of reproducibly recording single-unit activity from neuronal probes in this brain region in vivo and its overall relevance in evaluating neuronal responses to chemical exposure11,12,13,14. The dissociated cortical neuron culture contains the representative neuronal cell types found in the prefrontal cortex15.
While the cellular milieu and network inputs differ significantly between neurons in an isolated culture compared to neurons in their native brain environment, many responses, particularly response trends, are expected to be similar. To verify these expectations, however, a systematic and quantitative comparison of functional responses to chemical stimuli between these systems is necessary. Herein, both experimental systems were exposed to two different drugs known to induce dual responses (i.e. both increases and decreases in single neuron firing rates): atropine and ketamine. These well-characterized drugs elicit effects both in vivo and in vitro and are known to work through interaction with different receptor populations involved in cortical function. Atropine is a muscarinic receptor antagonist affecting both the central and parasympathetic nervous systems16. Muscarinic receptors regulate acetylcholine release and neuronal spiking activity and are located on both excitatory and inhibitory cortical neurons17. Ketamine is an N-methyl-D-aspartate (NMDA) receptor antagonist that blocks receptor activity. It can elicit an increase or decrease in firing rate depending on the dosage18,19,20, cell type21, cell state12 and through modulation of other glutamatergic receptors21. One important consideration when directly comparing in vitro to in vivo responses is the possible effect of metabolism or modification of the parental drug molecule in vivo (which would not be similarly metabolized in a simple in vitro system). While no neuroactive metabolites have been reported for atropine, metabolites for ketamine (i.e. (R,S)-norketamine or (2R,6R)-hydroxynorketamine) have been shown to elicit neuronal responses22,23. These secondary effects may influence the overall outcome in an in vivo system. In this study, the administered doses of the drugs were normalized using reported region-specific brain accumulation values for the parent compound to allow for direct comparisons between the systems22,23.
Conducting direct comparisons between in vivo and in vitro experimental systems provides a more informed framework in which to interpret functional data and responses to chemical stimuli and has important implications for increasing the significance of drug development and toxicology studies. Our study illustrates that the relevance of results obtained from in vitro cultures likely depends on many factors including indirect neuronal effects that may be a result of neuroactive metabolites or the interaction of the parent compound with non-neuronal targets.
Results
Spontaneous single-unit activity
Neuronal electrophysiology was compared between rodent cortical neurons cultured on planar MEAs (in vitro) and awake, freely moving rats using implantable MEA probes (in vivo). Key similarities and differences for single-unit activity between these two neuronal systems were assessed by comparing both baseline electrophysiological activity and response to two classes of well-characterized neuroactive chemicals. Spontaneous (baseline) single-unit extracellular neuronal spiking activity in both in vitro and in vivo systems was compared by quantifying features typically observed for both systems24,25,26,27,28. Most notably, in vitro firing events were typically in bursts, whereas in vivo firing events were predominantly composed of single spikes (Fig. 1a,b; Supplementary Fig. S1). We also noted greater variability within the recorded features among experimental replicates (firing rate, interspike interval, and reduced single spike activity outside of bursts) in the in vitro system (Fig. 1c–e).

Baseline activity of neurons recorded from in vivo and in vitro multielectrode arrays (MEAs). Three representative electrodes with neuronal activity are shown for in vitro and in vivo recordings in (a,b), respectively. Comparisons of firing features are shown including: (c), interspike interval (ISI) (0.061 ± 0.001 sec in vitro [n = 247] and 0.079 ± 0.002 sec in vivo [n = 96]), (d), firing rate (2.0 ± 0.2 Hz in vitro [n = 474]) and 1.06 ± 0.09 Hz in vivo [n = 362] and (e), percent of spikes out of bursts (73 ± 1% in vitro [n = 322] and 93.9 ± 0.9% in vivo [n = 345]). Data are shown as mean ± s.e.m. Asterisks (*) indicate significance in Mann-Whitney U test. *p < 0.01, ***p < 0.001.
Drug effects on spontaneous single-unit activity
Upon exposure to atropine, single-unit responses for firing rate and non-burst activity were similar between the systems (Supplementary Fig. S2a,b). Importantly, the expected dual response was observed in both neuronal systems, whereby isolated single units exhibiting simultaneously increasing or decreasing responses to atropine were observed both in vitro (Fig. 2a,b) and in vivo (Fig. 2c,d) after exposure to 1 µM atropine. This dose of atropine caused no statistically significant differences in response between the two systems, as characterized by both the ratio of cells showing each type of response (Fig. 2e) and by changes in features such as firing rate (Fig. 2f) and interspike interval (Fig. 2h). Responses to the 5 µM dose differed markedly (Fig. 2e) with respect to cells showing a decrease in activity (~10% in vivo vs. >60% in vitro), although the overall change in firing rate for these cells does not change with dose (Fig. 2f). For both concentrations evaluated, only the 5 µM exposure demonstrated statistically significant differences in the single-spike activity between the two systems (Fig. 2g). Cells in vitro appeared more likely to increase in firing rate after atropine exposure, although the response was not statistically significant compared to the in vivo response from similar exposures (Fig. 2i). Changes in single-spike activity also showed a similar but statistically insignificant trend (Fig. 2j).

In vitro and in vivo responses to atropine challenge. Dual response examples for both in vitro and in vivo systems are shown in (a–d). Increases and decreases in spiking activity on a single channel when exposed in vitro are shown in (a,b), respectively. In vivo responses are shown in (c) (increase) and (d) (decrease). The proportions of single units showing increase, decrease, or no change in firing activity after exposure are shown in (e). A difference in distribution between in vitro and in vivo single-unit response types for atropine was noted: 1 µM, Χ2 (2, n = 94) = 1.92, p = 0.38 and 5 µM, Χ2 (2, n = 354) = 83.77, p < 0.0001. For the cells in (e) that showed decreased single unit activity, (f) presents the decreases in firing rate in vitro (1 µM n = 14; 5 µM n = 13) and in vivo (1 µM n = 35; 5 µM n = 90). (g) Shows the change in non-burst firing activity for this same group of cells that showed decrease in firing rate to atropine: in vitro (1 µM n = 14; 5 µM n = 10), in vivo (1 µM n = 16; 5 µM n = 35). (h) Shows interspike interval (ISI) for all cells at each dosage of atropine: in vitro (0 µM n = 135; 1 µM n = 30; 5 µM n = 112), in vivo (0 µM n = 51; 1 µM n = 9; 5 µM n = 54). For the cells in (e) that showed increase in cell activity, (i) presents the increases in firing rate: in vitro (1 µM n = 4; 5 µM n = 6), in vivo (1 µM n = 26; 5 µM n = 121). (j) Shows the change in non-burst firing activity for this same group of cells that showed decrease in firing rate to atropine: in vitro (1 µM n = 4; 5 µM n = 12), in vivo (1 µM n = 5; 5 µM n = 45). Data are shown as mean ± s.e.m. Asterisks (*) in panel represent significance from Dunn’s post-hoc test. *p < 0.01, **p < 0.05, ***p < 0.001, ****p < 0.0001.
Next, we investigated the effects of ketamine in both systems. Studies have reported that at sub-anesthetic doses in vivo, ketamine induces both increases and decreases in cortical neuron firing rate. At higher, anesthetic doses, most neurons exhibit a reduced firing rate, while a small but significant subset of neurons still exhibits a firing rate increase20. In the current study, we observed ketamine’s dual response behavior both in vitro and in vivo (Fig. 3). Overall, cortical neurons in vitro demonstrated greater sensitivity to ketamine exposure, with approximately one order of magnitude decrease in the dose range in vitro required to elicit the same response as in vivo (Supplementary Fig. S3). The difference in sensitivity is further illustrated in Fig. 3e and Supplementary Fig. S2c,d, which examines single-unit responses for the two concentrations of ketamine (45, 90 µM) that elicited responses in both systems. Nearly all cells in vitro (>99%) showed a decrease in activity when exposed to either 45 or 90 µM ketamine. In vitro, this decrease is reflected as a near complete cessation of activity (Fig. 3f); this was not the case for ≤1 µm ketamine (Supplementary Fig. S3) where a subset of in vitro cells showed increased activity. In vivo, however, fewer cells demonstrated decreases in activity for both 45 µM and 90 µM ketamine (68.8% and 37.8%, respectively). This reduction corresponded to a 40–50% decrease in activity for these cells (Fig. 3f). Interspike intervals (ISI) for both systems did not show a significant change from baseline (0 µM ketamine, Fig. 3h), aside from the in vitro exposure to 90 µM ketamine, which showed a complete (but reversible, Supplementary Fig. S4) cessation in single-unit activity. Finally, Fig. 3g shows a trend towards less non-burst firing in vitro compared to in vivo, similar to the changes in firing rate seen in Fig. 3f. This does not indicate an increase in bursting, but instead indicates decreases in single spike activity overall when interpreted in the context of the firing rate decreases in Fig. 3f.

In vitro and in vivo responses to ketamine challenge. Dual response examples for both in vitro and in vivo systems are shown in (a–d). Increases and decreases in spiking activity during simultaneous exposure to 1 µM ketamine in vitro are shown in (a,b), respectively. In vivo responses are shown in (c) (increase) and (d) (decrease) to ~90 µM ketamine. Outcomes after ketamine exposure are shown in (e). In vitro, 3.1% of cells showed increased activity and 96.9% of cells showed decreased activity to 45 µM ketamine. 100% of cells showed decreased activity to 90 µM ketamine. In vivo, 31.2% of cells increased activity and 68.8% of cells decreased activity to 45 µM ketamine. For 90 µM ketamine, 56.1% of cells increased activity, 37.8% of cells decreased activity, and 6.1% of cells showed no response. A difference in distribution between in vitro and in vivo single-unit response types for ketamine was noted: 45 µM, Χ2 (2, n = 85) = 17.08, p < 0.0001, and 90 µM Χ2 (2, n = 229) = 43.96, p < 0.0001. For the cells in (e) that showed decreased single unit activity, (f) presents the decreases in firing rate: in vitro (45 µM n = 31; 90 µM n = 33), in vivo (45 µM n = 29; 90 µM n = 74), (g) presents the decreases in non-burst activity: in vitro (45 µM n = 31; 90 µM n = 33), in vivo (45 µM n = 21; 90 µM n = 59), and (h) presents the interspike intervals (ISI): in vitro (0 µM n = 90; 45 µM n = 5; 90 µM n = 1), in vivo (0 µM n = 100; 45 µM n = 12; 90 µM n = 109). Response to vehicle is reported as 0 µM. Data are shown as mean ± s.e.m. Asterisks (*) in panel represent significance from Dunn’s post-hoc test. *p < 0.01, ***p < 0.001, ****p < 0.0001.
Discussion
This study aimed to quantify the similarities and differences of cultured cortical neurons and cortical neurons in vivo by comparing spontaneous electrophysiological activity and changes in electrophysiology after exposure to atropine or ketamine. These experiments showed: (1) significant differences in spontaneous in vivo and in vitro firing activity, (2) similar response trends in both systems in single-unit spiking activity after exposure to atropine, (3) greater sensitivity to the effects of ketamine in neurons in vitro. It is important to be aware that several factors could influence the differences observed in this study. These factors include: the age of neurons (embryonic versus adult)29, the ratio of GABA to glutamatergic cells15, presence and composition of supporting glial cells30, and the isolated environment for the in vitro system which lacks input from other organs31,32. Each of these points will be discussed in greater detail below.
In vitro cultures typically rely on embryonic neurons, as cell viability after harvesting and long-term culturing is more robust in embryonic and postnatal rodent systems33,34 compared to adult35,36,37. Electrical properties for neurons cultured in vitro at different ages have been compared and found to be similar across age groups29 apart from bursting behavior, which was more common in embryonic than adult neurons29. This characteristic of embryonic neurons may contribute to the differences observed here for spontaneous baseline neuronal activity between the two systems. Cultured neurons demonstrated more burst-like features (i.e. fewer spikes out of bursts) compared to neurons in vivo. With these inherent differences between systems in mind, responses to chemical stimuli were evaluated relative to the spontaneous (baseline) activity of the given system. This evaluation allowed for a more direct comparison for responses between systems.
The composition and ratios of neuronal subspecies, as well as the level of their direct interaction and feedback ability, are considerably different between in vivo and in vitro systems. Previous work has shown that a NMDA antagonist similar to ketamine, MK-801, decreases the neuronal activity of pyramidal cortical cells in culture and anesthetized rats38 while increasing cell activity in pyramidal cortical cells in awake rats39. This difference was attributed to decreased GABAergic interneuron activity in the awake animal, resulting in disinhibition of cortical neurons and a subsequent increase in spiking activity27. In cultured neurons or in anesthetized neurons in vivo, this disinhibition of cortical neurons is ineffective due to low basal levels of extracellular glutamate, the additional factor required to induce pyramidal cell action potentials40,41. Additionally, rodent cortical neurons in culture show an increased sensitivity to the excitotoxic effects of glutamate42 and neuronal NMDA receptors show differing sensitivity depending on the developmental stage of the neurons, with a hypersensitive stage seen in the first two weeks of rat neonatal life43. These factors, as well as the differences in GABAergic neurons between systems, may have contributed to the differential responses observed for ketamine. Within our in vitro system a small percentage (~8%, Supplementary Fig. S5) of neurons are GABAergic, compared to ~20% in vivo15. Supporting glia composition also differed between systems. Neuroglia including astrocytes, oligodendrocytes, and microglia all play a role in maintaining and influencing neuronal health and function44,45. While our in vitro system contains some contaminating astrocytes (GFAP+, ~13%, Supplementary Fig. S5), biologically relevant ratios and other cell types are not present. Cortical regions in vivo contain a much larger percentage of glial support cells (~40%)46 which are known to help protect against deleterious glutamate effects and possess NMDA receptors themselves47.
While differences in composition and ratios of cell types between systems may have influenced the response to ketamine, atropine responses were more comparable. Expression of muscarinic receptors is low in the embryonic rat cerebral cortex (E18; 5% of adult levels) but receptor expression increases rapidly through the first 11 days postnatally30 before stabilizing at levels that will be maintained through adulthood48. Interestingly, Heacock et al. showed that despite low expression of muscarinic receptors in embryonic neurons, their sensitivity to the agonist carbachol was similar to that of adult neurons30. The authors attributed the comparable sensitivity to a change in effective turnover rate (or the addition of unused receptors to cerebral cortical neurons as rats age) to explain why the neurons respond similarly to muscarinic agonists despite the differences in the maturity of the cultured neurons. A similar argument could explain why our in vivo and in vitro systems responded similarly to atropine. Additionally, muscarinic receptor expression on supporting cell types such as microglia and oligodendrocytes does not change with age in the rodent model49,50. Therefore, the recognized differences in ratios of neuronal to supporting cells for both systems may have not affected overall response.
As isolated tissue mimetics, in vitro model systems lack the ability to metabolize drug compounds. Therefore, the metabolites found after drug administration in vivo, and their potential off-target effects, are likely not recapitulated in vitro. This represents a significant difference between the two systems, particularly the differing responses seen for ketamine, a drug known to be readily metabolized in vivo. Importantly, ketamine metabolites have known neuroactive properties, and can act on other receptors to counter ketamine effects21 or directly work on the same receptors as ketamine with less efficacy31. These neuroactive metabolites may have contributed to the differences observed between our experimental systems.
Neuromodulatory inputs from other regions are absent in the in vitro system; these inputs influence cortical neurons in vivo32. The cortex is ~25% of the rodent brain’s total volume51 but is comprised of many specialized subregions that are a result of these interregional inputs. The input for each individual cell in the cortex is influenced by the larger circuitry of the brain; this can also have additional, complex effects on the ultimate response a single neuron shows to systemically applied drugs.
Taken together, our results demonstrate that there are quantifiable differences and similarities in extracellular electrophysiology between in vivo and in vitro cortical systems both for spontaneous activity and in response to chemical challenge with atropine and ketamine. This study represents only a preliminary step towards a more thorough comparison between in vivo and in vitro responses, which would require testing additional representative drug compounds across multiple drug classification categories. These more extensive comparisons are necessary to correlate and benchmark the responses between the two systems to better interpret in vitro data in the context of the whole organism. While the complexity of the in vivo system is a key determinant of chemical response, directly demonstrating the ability of the relatively simple in vitro system to recapitulate the fundamental response observed in vivo is critically important. Using two different classes of drugs, we demonstrate the utility of cortical in vitro neuronal systems to recapitulate whole organ responses to certain classes of chemicals. While we show that changes in extracellular electrical activity in neuronal cultures may be appropriate for certain types of pharmacological studies, we also demonstrate that this system may not fully recapitulate the responses observed in vivo. Understanding the boundaries of a representative response with this in vitro system will provide more relevant data sets, and a framework to develop in vitro systems that more closely mimic in vivo function and response. As such, in addition to expanding the number and type of drug compounds to test, future work should be aimed at developing more complex cultures (i.e. additional cell types, three dimensional cultures) that capture more of the whole brain response for complex drugs of action like ketamine, while concomitantly verifying that functional responses pivot towards a more in vivo functional phenotype. However, until more complex cultures or alternative in vitro systems have been developed and validated, we hope to begin a larger conversation between in vitro and in vivo communities to understand the right questions we can correctly answer with in vitro platforms to directly contribute to human health and disease.
Methods
Chemicals
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. Ketamine HCl (100 mg/mL in sterile water) was purchased from ACE surgical (Brockton, MA).
Microelectrode device fabrication
The MEA device used for in vitro platform testing was microfabricated as previously described52,53. Briefly, MEAs (20–50 µm platinum electrodes spread evenly over 1.8 mm2) were fabricated on a glass substrate. Standard photolithography and wet or plasma etching were used to pattern metal constituting the 60 electrodes and electrode traces; polyimide was deposited and patterned as an insulating layer over the metal traces. A polystyrene cylinder was affixed over the MEA to enable cell culturing over the array. The well area was 113 mm2 and accommodated approximately 700 μL. ZIF connectors were added to the device for electrical connections and electrodes were plated with platinum black53. Impedance measurements were taken prior to seeding and ranged from ~50–250 kΩ at 1 kHz.
The MEA devices implanted in vivo were microfabricated and plated as described previously54,55. Briefly, the implantable MEAs featured two separate shanks (each 15 μm thick and 100 µm wide) spaced 2 mm apart medial-laterally. At the tip of each shank, 16 electrodes were evenly distributed over a length of 1.89 mm (dorsal-ventrally through medial prefrontal cortex). MEA devices were fabricated on silicon substrates using alternating stacks of metal (conductive layers) and polyimide (insulation layers). Standard photolithography and wet or plasma etching were used to pattern the 32 electrodes, trace metal, and device outlines. Polyimide was deposited and patterned, followed by deposition of platinum for the electrode metal. The electrode metal area was then patterned and etched creating electrodes 20 μm in diameter, followed by an insulating polyimide layer. Next, layers of gold (for the metal traces connecting the electrodes to the connector region) and polyimide were deposited to create a layered structure. After fabrication, devices were released from the silicon wafer and individually connected to 36-channel Nano Strip connectors (Omnetics Connector Corporation, Minneapolis, MN) using conductive epoxy. Electrodes were then plated as described above with final impedances ranging from 50–400 kΩ at 1 kHz and prepped for surgical implantation.
Cell seeding
MEAs were sterilized with 70% ethanol for 20 minutes, rinsed 5x with sterile water, and coated with 0.1 mg/mL poly-D-lysine in borate buffer prior to seeding. Primary embryonic rat cortical neurons (E18/19, Lonza, Walkersville, MD) were seeded at a density of 180 K cells/device (N = 4 seedings, n = 16 devices, [lot #170505 and #091104]). Cultures were maintained with bi-weekly exchanges of 50% media (Primary Neuron Basal Medium (PNBM) supplemented with 2 mM L-glutamine, 50 μg/mL gentamicin, 37 ng/mL amphotericin, and 2% NSF-1) in a humidified incubator (5% CO2, 37 °C). Custom device caps, made from a polytetrafluoroethylene (PTFE) housing and a fluorinated ethylene-propylene (FEP) membrane, were used to maintain sterility and to allow for gas exchange.
In Vivo Surgical Implantation
All animal experiments and surgical procedures were conducted following the guidelines and regulations set by Lawrence Livermore National Laboratory, including Institutional Animal Care and Use Committee approval (#240). Devices were surgically implanted into medial Prefrontal Cortex (2.52 mm anterior-posterior, 1.2–1.8 mm medial-lateral, 1.5 mm dorsal-ventral) of male Sprague-Dawley rats (6–8 weeks, ~200 g). The device, with a silica insertion tool temporarily attached to the probe with polyethylene glycol (PEG), was implanted using a microdrive positioner (Kopf Instruments, Tujunga, CA). The PEG holding the positioner was then dissolved with saline and the insertion tool was retracted with the microdrive, leaving the array implanted in the brain54,56. The array connector was then affixed to the skull with dental acrylic.
Chemical dosing
For in vitro administration, chemicals were either dissolved (atropine; powder, >99.9% purity) or diluted (ketamine) in primary neuron basal media (PNBM) to 4X the final desired in vitro concentration. Cultures used for chemical testing were between day in vitro (DIV) 19 and 78. For chemical exposures, 25% of the culture media was removed from the MEA well and replaced with an equal volume of 4X chemical working stock (0.0004–360 µM ketamine, 4–10 µM atropine), resulting in a final culture concentration that ranges from 0.0001–90 µM for ketamine and 1–5 µM for atropine.
For in vivo administration, chemicals were either dissolved (atropine powder) or diluted (ketamine solution) in 0.2–0.4 mL saline (0.9% NaCl in sterile water). Intraperitoneal (i.p) dosing concentrations were based on published studies that evaluated brain region specific accumulation of non-metabolized (radiolabeled) drug upon i.p. injection57,58. Atropine (10 mg/kg) was given i.p. to rats (400–550 g); this was equivalent to 1 µM atropine in cortex (and 50 mg/kg = 5.2 µM). For ketamine, a concentration of 45 µM in the cortex was achieved with 5 mg/kg i.p. administration (and 10 mg/kg i.p. for 90 µM, 25 mg/kg for 200 µM).
Electrophysiology recording and processing
Parallel in vivo and in vitro experiments began on the day of probe implant in vivo (Day In Vivo) or day of cell seeding on the MEAs (Day In Vitro). Time from probe implant or cell seeding were both referred to as DIV, with DIV 0 referring to the day of in vivo probe implantation or in vitro cell seeding. Electrophysiology recordings began as early as DIV 3 to acclimate the animal to the recording chamber and to monitor the health of the in vitro cultures. Chemical challenges were initiated on DIV 21, when neuroinflammation from the surgery was resolved in vivo54 and the cultures began to show stable neuronal activity in vitro24.
For in vitro electrophysiology measurements, devices were placed on a heated stage (37 °C) in a 5% CO2 controlled chamber during recordings. Electrophysiology measurements were acquired using a multi-channel recording system (AlphaLab SnR, Alpha Omega, Alpharetta, GA) and were sampled at a frequency of 22.3 kHz and bandpass filtered between 268 and 8036 Hz. Baseline and chemical exposure measurements were recorded for 20 minutes. Chemical exposure measurements were collected for 20 minutes immediately following chemical addition. After testing, cultures were given at least two days for recovery between experiments.
For in vivo measurements, drug application was randomized so that there were no more than two recordings from the same animal in the same week. Each rat (N = 3) was administered both concentrations of each drug three times over a period of 6–12 weeks with at least two days between experimental drug exposures. For each drug administration, animals were placed in an operant behavioral chamber (Med Associates, Inc., St. Albans, VT) and acclimated to the chamber for >15 minutes before recording was initiated. Electrophysiology was recorded using a multichannel freely moving animal system (Tucker-Davis Technologies, Alachua, FL) at 24,414.1 Hz with a band pass filter of 100–5000 Hz. Baseline was monitored to ensure proper connectivity of devices and noise thresholds (auto set to ±4.0 standard deviations above noise). Animals then were administered a vehicle injection of 0.2–0.4 mL saline (0.9% NaCl in sterile water, i.p.) and 20 min of baseline condition data was collected. Next, animals were injected with the chemical challenge dissolved in 0.2–0.4 mL saline (0.9% NaCl in sterile water, i.p.) and >20 min of chemical condition data were then collected.
Feature analysis and statistics
After collection, all in vivo and in vitro data were analyzed to extract single-units from the multiunit signal data using Offline Sorter™ (PLEXON, Dallas, TX) and further analysis carried out only on isolated single-units. For all isolated single units, spike and burst features were calculated. Each single unit was analyzed individually and considered a data point for the experiment since each cell could respond differently to both drugs; averaging responses across a device would confound results and interpretation. Electrophysiological data from each unit on the in vitro MEA device or in vivo probe was analyzed using parameters we have previously evaluated on cortical and hippocampal cultures seeded on MEA devices53. Bursts were defined as having a maximum beginning interspike interval of 0.1 sec, a maximum end interspike interval of 0.2 sec, a minimum interburst interval of 0.5 sec, a minimum burst duration of 0.05 sec, and a minimum of 10 spikes per burst53. Feature analysis was carried out with an in-house custom R package.
Calculated features were inspired by those described in the work of Charlesworth et al.24: interspike interval (ISI), overall firing rate and percent of spikes outside of bursts. For drug challenges, the difference in electrophysiological activity before and after challenge was calculated and reported as the change in mean frequency from baseline. Here, n indicates the number of isolated single units. Single units that showed a complete cessation of firing activity with drug challenge were defined at a -100% change in activity. Single units exhibiting greater than 1000% change in the feature were excluded. Data was considered non-normally distributed for statistical analysis and were analyzed using GraphPad Prism7. In vivo and in vitro baseline firing were compared with the non-parametric Mann-Whitney test (Fig. 1c–e). For drug challenges, a chi-square test was run on the proportions of single units showing an increase, decrease, or no change for both systems (Figs 2e and 3e). Differences between multiple groups for features (Figs 2f–j and 3f–h, Supplementary Figs S2 and S3) were evaluated by the non-parametric Kruskal-Wallis one-way analysis of variance (ANOVA), followed by Dunn’s post hoc test. A p value of <0.05 was considered significant. All error bars indicate standard error of the mean (SEM).
Code availability
The analysis code used to analyze data in this study is available upon request.
Immunocytochemistry
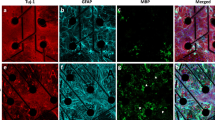
Cells were rinsed 4X with 1X PBS, fixed with 4% paraformaldehyde, washed with PBS (4X), and permeabilized using 10% saponin before blocking with 10% goat serum (1 hr at room temperature). Primary antibodies commonly used to identify cell populations were used: class III beta-tubulin for neurons (Tuj-1, chicken or mouse, Neuromics, Edina, MN, 1:200 dilution), GAD-67 for GABAergic neuron (mouse, Millipore, Burlington, MA, 1:200) and glial fibrillary acidic protein (GFAP) for astrocytes (rabbit, Millipore, Burlington, MA, 1:1000). After primary antibody incubation (overnight at 4 °C), cells were washed with PBS (4X) and stained with secondary antibodies (1 hr at 37 °C). Secondary antibodies included: goat anti-mouse linked to Alexa Fluor 488, goat anti-chicken linked to Alexa Fluor 647 and goat anti-rabbit linked to Alexa Fluor 594 (1:500 dilution, Life Technologies, Eugene, OR). After secondary antibody incubation, the cells were washed with PBS (4X), and incubated (20 min) with the nuclear stain, diamidino-2-phenylindole (DAPI, ThermoFisher, 300 nM), before imaging. Three fields of view (or regions) from each cell culture were imaged and acquired using a Leica inverted microscope controlled using Meta-morph imaging software (Molecular Devices, Sunnyvale, CA) for Supplemental Fig. 1d–f. A LSM700 confocal microscope (Carl Zeiss Microscopy, Thornwood, NY) was used for Supplemental Fig. 1a–c. For cell type quantification, positive cells (GFAP+, astrocyte marker and GAD-67+, GABAergic neuron marker) were counted and averaged from three to five fields of view of each culture (n = 3) and normalized to the average total nuclei count or Tuj-1 positive cells using ImageJ.
Histology
In vivo probe placement was verified after CO2 euthanasia of animals. Rats were perfused using a 10% formalin solution. Brains were removed, cryoprotected, and stored in 10% formalin for >3 days. Tissue was then sectioned coronally into 40–50 μm thick slices on a cryostat (Leica CM3050 S, Buffalo Grove, IL). The probe tract was identified visually under a microscope equipped with a camera. Approximate recording sites are shown in Supplementary Fig. 6.
Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
References
McGonigle, P. & Ruggeri, B. Animal models of human disease: Challenges in enabling translation. Biochemical Pharmacology 87, 162–171, https://doi.org/10.1016/j.bcp.2013.08.006 (2014).
Langley, G. R. et al. Towards a 21st-century roadmap for biomedical research and drug discovery: consensus report and recommendations. Drug Discov Today 22, 327–339, https://doi.org/10.1016/j.drudis.2016.10.011 (2017).
Gramowski, A., Schiffmann, D. & Gross, G. W. Quantification of acute neurotoxic effects of trimethyltin using neuronal networks cultured on microelectrode arrays. Neurotoxicology 21, 331–342 (2000).
Kreir, M. et al. Do in vitro assays in rat primary neurons predict drug-induced seizure liability in humans? Toxicology and Applied Pharmacology 346, 45–57, https://doi.org/10.1016/j.taap.2018.03.028 (2018).
Novellino, A. et al. Development of micro-electrode array based tests for neurotoxicity: assessment of interlaboratory reproducibility with neuroactive chemicals. Frontiers in neuroengineering 4, 4, https://doi.org/10.3389/fneng.2011.00004> (2011).
Han, Y. et al. The Effects of Acute GABA Treatment on the Functional Connectivity and Network Topology of Cortical Cultures. Neurochemical research 42, 1394–1402, https://doi.org/10.1007/s11064-017-2190-3 (2017).
Wallace, K., Strickland, J. D., Valdivia, P., Mundy, W. R. & Shafer, T. J. A multiplexed assay for determination of neurotoxicant effects on spontaneous network activity and viability from microelectrode arrays. NeuroToxicology 49, 79–85, https://doi.org/10.1016/j.neuro.2015.05.007 (2015).
Horvath, P. et al. Screening out irrelevant cell-based models of disease. Nature Reviews Drug Discovery 15, 751, https://doi.org/10.1038/nrd.2016.175 (2016).
Fang, Y. & Eglen, R. M. Three-Dimensional Cell Cultures in Drug Discovery and Development. Slas Discovery 22, 456–472, https://doi.org/10.1177/1087057117696795 (2017).
Langhans, S. A. Three-Dimensional in Vitro Cell Culture Models in Drug Discovery and Drug Repositioning. Frontiers in Pharmacology 9, https://doi.org/10.3389/fphar.2018.00006 (2018).
Roth, B. L., Sheffler, D. J. & Kroeze, W. K. Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nature Reviews Drug Discovery 3, 353, https://doi.org/10.1038/nrd1346 (2004).
Shapiro, D. A. et al. Aripiprazole, A Novel Atypical Antipsychotic Drug with a Unique and Robust Pharmacology. Neuropsychopharmacology 28, 1400, https://doi.org/10.1038/sj.npp.1300203 (2003).
Perry, J. L. et al. Prefrontal Cortex and Drug Abuse Vulnerability: Translation to Prevention and Treatment Interventions. Brain research reviews 65, 124–149, https://doi.org/10.1016/j.brainresrev.2010.09.001 (2011).
Goldstein, R. Z. & Volkow, N. D. Dysfunction of the prefrontal cortex in addiction: neuroimaging findings and clinical implications. Nature Reviews Neuroscience 12, 652, https://doi.org/10.1038/nrn3119 (2011).
Sahara, S., Yanagawa, Y., O’Leary, D. D. & Stevens, C. F. The fraction of cortical GABAergic neurons is constant from near the start of cortical neurogenesis to adulthood. J Neurosci 32, 4755–4761, https://doi.org/10.1523/jneurosci.6412-11.2012 (2012).
McKenna, T. M., Ashe, J. H., Hui, G. K. & Weinberger, N. M. Muscarinic agonists modulate spontaneous and evoked unit discharge in auditory cortex of cat. Synapse 2, 54–68, https://doi.org/10.1002/syn.890020109 (1988).
Groleau, M., Kang, J. I., Huppe-Gourgues, F. & Vaucher, E. Distribution and effects of the muscarinic receptor subtypes in the primary visual cortex. Front Synaptic Neurosci 7, 10, https://doi.org/10.3389/fnsyn.2015.00010 (2015).
Zorumski, C. F., Izumi, Y. & Mennerick, S. Ketamine: NMDA Receptors and Beyond. The Journal of Neuroscience 36, 11158–11164, https://doi.org/10.1523/jneurosci.1547-16.2016 (2016).
Tyler, M. W., Yourish, H. B., Ionescu, D. F. & Haggarty, S. J. Classics in Chemical Neuroscience: Ketamine. ACS Chemical Neuroscience 8, 1122–1134, https://doi.org/10.1021/acschemneuro.7b00074 (2017).
Patel, I. M. & Chapin, J. K. Ketamine Effects on Somatosensory Cortical Single Neurons and on Behavior in Rats. Anesthesia & Analgesia 70, 635–644 (1990).
Zanos, P. et al. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 533, 481–486, https://doi.org/10.1038/nature17998 (2016).
Suzuki, K., Nosyreva, E., Hunt, K. W., Kavalali, E. T. & Monteggia, L. M. Effects of a ketamine metabolite on synaptic NMDAR function. Nature 546, E1, https://doi.org/10.1038/nature22084 (2017).
Moaddel, R. et al. The distribution and clearance of (2S,6S)-hydroxynorketamine, an active ketamine metabolite, in Wistar rats. Pharmacology Research & Perspectives 3, e00157, https://doi.org/10.1002/prp2.157 (2015).
Charlesworth, P., Cotterill, E., Morton, A., Grant, S. G. & Eglen, S. J. Quantitative differences in developmental profiles of spontaneous activity in cortical and hippocampal cultures. Neural Development 10, 1, https://doi.org/10.1186/s13064-014-0028-0 (2015).
Smith, I. et al. Neuronal-glial populations form functional networks in a biocompatible 3D scaffold. Neurosci Lett 609, 198–202, https://doi.org/10.1016/j.neulet.2015.10.044 (2015).
Chiappalone, M., Bove, M., Vato, A., Tedesco, M. & Martinoia, S. Dissociated cortical networks show spontaneously correlated activity patterns during in vitro development. Brain Res 1093, 41–53, https://doi.org/10.1016/j.brainres.2006.03.049 (2006).
Homayoun, H. & Moghaddam, B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci 27, 11496–11500, https://doi.org/10.1523/jneurosci.2213-07.2007 (2007).
Fujisawa, S., Amarasingham, A., Harrison, M. T. & Buzsaki, G. Behavior-dependent short-term assembly dynamics in the medial prefrontal cortex. Nat Neurosci 11, 823–833, https://doi.org/10.1038/nn.2134 (2008).
Evans, M. S., Collings, M. A. & Brewer, G. J. Electrophysiology of embryonic, adult and aged rat hippocampal neurons in serum-free culture. J Neurosci Methods 79, 37–46 (1998).
Heacock, A. M., Fisher, S. K. & Agranoff, B. W. Enhanced coupling of neonatal muscarinic receptors in rat brain to phosphoinositide turnover. J Neurochem 48, 1904–1911 (1987).
Dinis-Oliveira, R. J. Metabolism and metabolomics of ketamine: a toxicological approach. Forensic Sciences Research 2, 2–10, https://doi.org/10.1080/20961790.2017.1285219 (2017).
Kirkby, L. A., Sack, Georgeann S., Firl, A. & Feller, M. B. A Role for Correlated Spontaneous Activity in the Assembly of Neural Circuits. Neuron 80, 1129–1144, https://doi.org/10.1016/j.neuron.2013.10.030.
Banker, G. A. & Cowan, W. M. Rat hippocampal neurons in dispersed cell culture. Brain Res 126, 397–342 (1977).
Ahlemeyer, B. & Baumgart-Vogt, E. Optimized protocols for the simultaneous preparation of primary neuronal cultures of the neocortex, hippocampus and cerebellum from individual newborn (P0.5) C57Bl/6J mice. J Neurosci Methods 149, 110–120, https://doi.org/10.1016/j.jneumeth.2005.05.022 (2005).
Brewer, G. J. Isolation and culture of adult rat hippocampal neurons. J Neurosci Methods 71, 143–155 (1997).
Kaneda, M., Nakamura, H. & Akaike, N. Mechanical and enzymatic isolation of mammalian CNS neurons. Neuroscience Research 5, 299–315, https://doi.org/10.1016/0168-0102(88)90032-6 (1988).
Brewer, G. J. & Torricelli, J. R. Isolation and culture of adult neurons and neurospheres. Nat Protoc 2, 1490–1498, https://doi.org/10.1038/nprot.2007.207 (2007).
Chen, L., Muhlhauser, M. & Yang, C. R. Glycine tranporter-1 blockade potentiates NMDA-mediated responses in rat prefrontal cortical neurons in vitro and in vivo. J Neurophysiol 89, 691–703, https://doi.org/10.1152/jn.00680.2002 (2003).
Jackson, M. E., Homayoun, H. & Moghaddam, B. NMDA receptor hypofunction produces concomitant firing rate potentiation and burst activity reduction in the prefrontal cortex. Proc Natl Acad Sci USA 101, 8467–8472, https://doi.org/10.1073/pnas.0308455101 (2004).
Amaral, A. I., Meisingset, T. W., Kotter, M. R. & Sonnewald, U. Metabolic aspects of neuron-oligodendrocyte-astrocyte interactions. Front Endocrinol (Lausanne) 4, 54, https://doi.org/10.3389/fendo.2013.00054 (2013).
Herculano-Houzel, S. The glia/neuron ratio: how it varies uniformly across brain structures and species and what that means for brain physiology and evolution. Glia 62, 1377–1391, https://doi.org/10.1002/glia.22683 (2014).
Choi, D. W., Maulucci-Gedde, M. & Kriegstein, A. R. Glutamate neurotoxicity in cortical cell culture. J Neurosci 7, 357–368 (1987).
Ikonomidou, C. et al. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 283, 70–74 (1999).
Clarke, L. E. & Barres, B. A. Emerging roles of astrocytes in neural circuit development. Nature Reviews Neuroscience 14, 311, https://doi.org/10.1038/nrn3484 (2013).
Salter, M. W. & Beggs, S. Sublime Microglia: Expanding Roles for the Guardians of the CNS. Cell 158, 15–24, https://doi.org/10.1016/j.cell.2014.06.008.
Ren, J. Q., Aika, Y., Heizmann, C. W. & Kosaka, T. Quantitative analysis of neurons and glial cells in the rat somatosensory cortex, with special reference to GABAergic neurons and parvalbumin-containing neurons. Experimental Brain Research 92, 1–14, https://doi.org/10.1007/bf00230378 (1992).
Verkhratsky, A. & Kirchhoff, F. NMDA Receptors in glia. Neuroscientist 13, 28–37, https://doi.org/10.1177/1073858406294270 (2007).
Tice, M. A. B., Hashemi, T., Taylor, L. A. & McQuade, R. D. Distribution of muscarinic receptor subtypes in rat brain from postnatal to old age. Developmental Brain Research 92, 70–76, https://doi.org/10.1016/0165-3806(95)01515-9 (1996).
Pannell, M. et al. The subpopulation of microglia expressing functional muscarinic acetylcholine receptors expands in stroke and Alzheimer’s disease. Brain Struct Funct 221, 1157–1172, https://doi.org/10.1007/s00429-014-0962-y (2016).
Molina-Holgado, E., Khorchid, A., Liu, H.-N. & Almazan, G. Regulation of muscarinic receptor function in developing oligodendrocytes by agonist exposure. British Journal of Pharmacology 138, 47–56, https://doi.org/10.1038/sj.bjp.0705002 (2003).
Mengler, L. et al. Brain maturation of the adolescent rat cortex and striatum: changes in volume and myelination. Neuroimage 84, 35–44, https://doi.org/10.1016/j.neuroimage.2013.08.034 (2014).
Enright, H. A. et al. Long-term non-invasive interrogation of human dorsal root ganglion neuronal cultures on an integrated microfluidic multielectrode array platform. Analyst 141, 5346–5357, https://doi.org/10.1039/c5an01728a (2016).
Soscia, D. et al. Controlled placement of multiple CNS cell populations to create complex neuronal cultures. PLoS One 12, e0188146, https://doi.org/10.1371/journal.pone.0188146 (2017).
Tooker, A. et al. In 2014 36th Annual International Conference of the IEEE Engineering in Medicine and Biology Society. 6830–6833 (2014).
Chung, J. E. et al. High-density, long-lasting, and multi-region electrophysiological recordings using polymer electrode arrays. bioRxiv, https://doi.org/10.1101/242693 (2018).
Felix, S. H. et al. Insertion of Flexible Neural Probes Using Rigid Stiffeners Attached with Biodissolvable Adhesive. J. Vis. Exp. (79), e50609, https://doi.org/10.3791/50609 (2013).
Cohen, M. L. & Trevor, A. J. On the Cerebral Accumulation of Ketamine and the Relationship Between Metabolism of the Drug and its Pharmacological Effects. J. Pharmacology and Experimental Therapeutics 189, 351 (1974).
Witter, A., Slangen, J. L. & Terpstra, G. K. Distribution of 3H-methylatropine in rat brain. Neuropharmacology 12, 835–841, https://doi.org/10.1016/0028-3908(73)90036-1 (1973).
Acknowledgements
We would like to thank David Soscia and Angela Tooker at LLNL for the use of their fabricated microelectrode arrays for in vitro and in vivo measurements, respectively. We would also like to thank Mona Hwang for assistance with cell maintenance and Doris Lam for the helpful manuscript discussion. This work was performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344 through LDRD awards 14-SI-001 and 17-SI-002. LLNL IM release #LLNL-JRNL-746078.
Author information
Authors and Affiliations
Contributions
A.M.B., H.A.E., K.K., N.O.F. and E.K.W. all contributed to experimental design. A.M.B., H.A.E., J.O. and E.A.K. performed all experiments. A.M.B., H.A.E. and A.P.S. analyzed the data. N.O.F. and E.K.W. supervised the project. The manuscript was written by A.M.B. and H.A.E. and edited by A.P.S., K.K. and N.O.F.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Belle, A.M., Enright, H.A., Sales, A.P. et al. Evaluation of in vitro neuronal platforms as surrogates for in vivo whole brain systems. Sci Rep 8, 10820 (2018). https://doi.org/10.1038/s41598-018-28950-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-28950-5
This article is cited by
-
Advances in 3D neuronal microphysiological systems: towards a functional nervous system on a chip
In Vitro Cellular & Developmental Biology - Animal (2021)
-
Enhanced LTP of population spikes in the dentate gyrus of mice haploinsufficient for neurobeachin
Scientific Reports (2020)
-
Functional and transcriptional characterization of complex neuronal co-cultures
Scientific Reports (2020)
-
Tissue-specific extracellular matrix accelerates the formation of neural networks and communities in a neuron-glia co-culture on a multi-electrode array
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
