
Abstract
We investigated the association between HPV infection and bacterial microbiota composition in the placenta, uterine cervix and mouth in thirty-nine women. HPV DNA genotyping of 24 types was conducted using Multimetrix®. Microbiota composition was characterized by 16S rRNA gene sequencing. HPV DNA was detected in 33% of placenta, 23% cervical and 33% oral samples. HPV16 was the most frequent type in all regions. HPV infection was associated with higher microbiota richness (p = 0.032) in the mouth but did not influence microbial diversity or richness in other samples. HPV infection was associated with higher abundance of Lactobacillaceae (p = 0.0036) and Ureaplasma (LDA score > 4.0, p < 0.05) in the placenta, Haemophilus (p = 0.00058) and Peptostreptococcus (p = 0.0069) genus in the cervix and Selenomonas spp. (p = 0.0032) in the mouth compared to HPV negative samples. These data suggest altered bacterial microbiota composition in HPV positive placenta, cervix and mouth. Whether the changes in bacterial microbiota predispose or result from HPV remains to be determined in future studies.
Similar content being viewed by others


Introduction
Human papilloma virus (HPV) is a known oncovirus and it has potential to cause carcinoma in the genital, anal and oropharyngeal regions1. HPV has long been regarded mainly as a sexually transmitted disease (STD) but recent studies have shown that HPV infection can be acquired by vertical transmission as well as via the placenta from mother to child2,3,4,5,6,7. Traditionally, HPV infection in the placenta has been considered to increase the risk of pregnancy complications8,9,10,11 but HPV DNA has also been discovered from placentas from healthy pregnancies3,5,12,13,14 and transabdominally obtained placental samples7,15,16.
In addition to viruses, humans have diverse bacterial microbiota which has been recognized to play an important role in human health. Recently published data suggest that even the placenta may harbor a unique microbiota which is mainly composed of Proteobacteria17,18. Parnell et al.19 have reported that the microbiota composition varies depending on the location within the placenta and that the placental microbiota may change according to maternal factors such as gestational diabetes mellitus20. However, not all published data support the notion of intrauterine colonization21,22.
Healthy vaginal microbiota is known to be predominantly dominated by lactobacilli. Shifts in vaginal microbiota balance may result in altered composition referred to as polybacterial dysbiosis and to disease such as bacterial vaginosis, both of which have been connected to vaginal HPV infection23,24. Individuals with HPV infection more frequently have higher diversity of cervico-vaginal microbiota and HPV clearance in turn has been associated with increased number of antigen-representing Langerhans cells25. A study by Gao et al.26 showed more L. gasseri and Gardnerella vaginalis in HPV infected women’s cervico-vaginal microbiota. HPV persistence is considered a critical event in malignant transformation. We have previously shown that bacterial vaginosis, even when asymptomatic, predicted HPV persistence23. Di Paola et al.27 propose that Atopobium spp. and G. vaginalis may serve as microbial markers for HPV persistence in Italian women while Adebamowo et al.28 showed that Mycoplasma hominis was associated with HPV persistence in Nigerian women. Furthermore, changes in the vaginal microbial composition, connected to lower levels of Lactobacillus spp., have been associated with more severe cervical intraepithelial neoplasia and higher diversity in microbiota might lead to more severe disease29.
Recent research has revealed that malignancies may arise in various human organs in reaction to bacterial inflammation or by bacteria’s own direct oncogenic mutations to human cells. Bacteria have been linked with conditions including liver cancer, esophageal cancer, pancreatic cancer, gall bladder cancer and colorectal cancer30,31. Furthermore, there are differences in the microbiota between normal oral mucosa and patients with oral squamous cell carcinoma32. It has even been suggested that the oral microbiota may have an effect on developing pancreatic cancer, possibly mediated by P. gingivalis. P. gingivalis has also been linked to chronic inflammation and atherosclerosis33,34. Accumulating evidence thus suggests that both HPV and bacterial dysbiosis might play a significant role in malignant transformation. Nonetheless, our knowledge about the interactions between HPV infection and the bacterial microbiota and its impact on human health is still rudimentary.
We aimed to investigate whether an existing HPV infection has influence on the bacterial microbiota composition in the placenta, the uterine cervix or the mouth. This could lead us to understand part of the mechanisms modifying HPV clearance and persistence leading to malignancy.
Results
HPV status of the samples
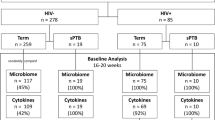
The HPV genotypes of all the samples including sex of the infant, delivery mode and gestational age are presented in Table 1. HPV DNA was detected in 33% of the placental, 23% cervical and 36% oral samples. HPV16 was the most frequent type found in all groups studied (54% of placenta, 22% of cervix and 54% of oral samples). Infection with multiple HPV types was found in three oral and five cervical samples but in none of the placental samples. Four women displayed HPV DNA both in the oral and cervical samples, seven in oral and placental samples and six in cervical and placental samples. Of these, two women in oral/cervix, two in oral/placenta and four in cervix/placenta samples had the same HPV genotype in the two different sites, either HPV6 or HPV16. Only two women had HPV detectable in all three locations. Interestingly, the HPV genotype found was the same in all sites, i.e. either HPV6 or HPV16.
Placenta microbiota and HPV
To study the impact of potential contaminants, pooled placental negative controls yielded 726 reads and the identified OTUs (n = 11) representing 6 different genera listed in Supplementary Table 1. Finally, a total of 19/39 placenta samples (49%) were included in the analysis after exclusion of the specimens with low number of reads (n < 1000 reads) followed by filtering the OTUs present in the negative controls. The most abundant phylum in the placenta microbiota was Firmicutes (58.3% vs. 66.6%) followed by Proteobacteria (21.1% vs. 15.7%), Actinobacteria (13.8% vs 10.0%) and Bacteroidetes (6.5% vs. 6.1%) in HPV negative and positive respectively (Fig. 1C). At family level, Staphylococcaceae (22.5% vs 29.1%), Enterococacceae (15.6% vs 13.8%), Veillonellaceae (8.8% vs 7.4%), Corynebacteriaceae (6.3% vs 0.8%) and Moraxellaceae (6.1% vs 0.1%) were the predominant groups in both HPV negative and positive samples, respectively. At genus level, Staphylococcus (22.9% vs 29.5%); unclassified Enterococcaceae genus (15.6% vs 13.7%), Corynebacterium (6.3% vs 0.1%), and Acinetobacter (6.0% vs 0.1%) were the most abundant in HPV negative and positive placenta samples.

The bacterial microbiota in placenta samples negative and positive for HPV DNA. No significant differences in microbial richness (A) or diversity (B) were detected between HPV negative (red) and positive (blue) placenta samples. The relative abundance of bacteria are presented on the phylum (C) and family levels (D). The relative abundance of the genus Lactobacillus was significantly higher in HPV positive (blue) as compared to HPV negative (red) samples (E). By LEfSe analysis (F), the genera Lactobacillus and Ureaplasma were significantly enriched in HPV positive (blue) placenta samples as compared to HPV negative (LDA score > 4.0, p > 0.05).
No differences in bacterial richness or diversity (as assessed by the Chao1 and Shannon indexes, respectively) were found between HPV positive (n = 6) and negative (n = 13) placenta groups (Fig. 1A,B). However, higher abundance of Lactobacillaceae (p = 0.0036) and Lactobacillus genus (p = 0.0023), were observed in HPV positive group compared to HPV negative placenta samples (Fig. 1,E). Interestingly, the relative abundance of Lactobacillus iners was higher in HPV negative group than in HPV positive (47.7% vs. 18.7% and p = 0.07). We also performed detailed analyses of the Lactobacillus species associated with HPV positivity in the placenta samples. In the HPV positive group, we detected L. zeae, L. reuteri and Unclassified Lactobacillus. To further investigate the composition, we selected LEfSe analyses at OTU level to reveal the Lactobacillus group in HPV positive placenta samples to be combined by L. iners, L. crispatus, L. jensenii, L. gasseri and L. reuteri. In addition, in HPV negative group L.iners was evident. LEfSe analysis showed that Ureaplasma and Lactobacillus genus were significantly enriched in HPV positive placenta samples compared to HPV negative (LDA score > 4.0, p < 0.05), (Fig. 1F).
Despite the lower number of HPV positive samples, comparing HPV positive samples between low-risk HPV types (LR-HPV, including HPV6, 11, 42, 43, 44 and 70) and high-risk HPV types (HR-HPV, including HPV16, 18, 26, 31, 33, 35, 39, 45, 51, 52, 53, 56, 58, 59, 66, 68, 73 and 82) genotypes we observed higher abundance of Staphylococcus (p = 0.012) and Lachnospira genus (p = 0.050) in LR-HPV genotype compared to those observed in HR-HPV genotype. Although not with significant statistical differences, a higher diversity and lower richness were observed in HR-HPV genotype compared to LR-HPV genotype. LEfSe analysis showed that Ureaplasma and Lactobacillus genus were significantly enriched in HR-HPV genotype placenta samples while Lachnospira genus was enriched in LR-HPV genotype.
Cervical microbiota and HPV
The most abundant phyla in the cervix were Firmicutes (91.0% vs. 90.7%) and Actinobacteria (7.3% vs. 5.1%), followed by Fusobacteria (1.2% vs. 0.1%), Bacteroidetes (0.2% vs. 3.3%) and Tenericutes and Proteobacteria in HPV negative and positive samples, respectively. No statistically significant differences in species richness or diversity were detected between HPV positive and negative cervical samples (Fig. 2A,B) nor were we able to point out statistical differences on the phylum level (Fig. 2C).

The bacterial microbiota in cervical samples negative and positive for HPV DNA. No significant differences in microbial richness (A) or diversity (B) were detected between HPV negative (red) and positive (blue) cervical samples. The relative abundance of bacteria are presented on the phylum (C) and family levels (D). By LEfSe analysis (E), unclassified Coriobacteriaceae were significantly enriched in HPV positive (blue) samples as compared to HPV negative (LDA score > 4.40, p < 0.05), while Haemophilus (LDA score > 4.18) were enriched in HPV negative (red) compared to HPV positive cervical specimens.
On the family level, the most abundant groups belonged to Lactobacillaceae (89.7% vs. 86.8% in HPV negative and positive samples, respectively). The HPV positive cervical samples mainly harboured L. crispatus and L. jensenii whereas in HPV negative cervix group L. iners and L. reuteri were more abundant. The relative abundance of Coriobacteriaceae (p = 0.083) was slightly increased in HPV positive cervical samples. On the other hand, Peptostreptococcaceae (p = 0.0065) and Enterococcaceae (p = 0.022) families were significantly increased in HPV negative compared to HPV positive cervical samples (Fig. 2D). The HPV positive cervix harbored more unclassified Coriobacteriaceae (p = 0.07) genus as compared to HPV negative cervical samples whereas Haemophilus (p = 0.00058) and Peptostreptococcus (p = 0.0069) were significantly increased in HPV negative group at genus level. At genus level, Lactobacillus was predominant in both HPV groups; negative and positive (89.7% in HPV negative and 86.7% in HPV positive). At species level, L. iners, mainly the OTU 133075, was the predominant Lactobacillus group in both HPV negative and positive samples (47.7% vs 18.6%, respectively, p-value = 0.07).
In LEfSe analysis, unclassified Coriobacteriaceae genus was significantly enriched in HPV positive group compared to HPV negative (LDA score > 4.40, p < 0.05), while Haemophilus (LDA score > 4.18) were enriched in HPV negative compared to HPV positive cervix (Fig. 2E). To further identify the species abundant in the HPV positive cervical samples group, we discovered the group mainly being composed of Atopobium vaginae.
Oral microbiota and HPV
The most abundant phyla in the mouth were Firmicutes (48.9% vs. 53.5%), Proteobacteria (24.6% vs. 18.6%), Actinobacteria (12.5% vs. 11.8%), Bacteroides (8.7% vs. 10.6%) followed by Fusobacteria (3.8% vs. 4.5%) and TM7 (0.6% vs. 1.0%) in HPV negative and positive samples, respectively. The predominant microbial families belonged to Streptococcaceae, Pasteurellaceae, Veillonellaceae, Micrococcaceae, Prevotellaceae, Neisseriaceae, Gemellaceae and Fusobacteriaceae. At genus level, Streptococcus, Haemophilus, Veillonella, Prevotella, unclassified Gemellaceae, Fusobacteriumand Actinomyces were the predominant bacterial genera in both groups.
At phylum level, the relative abundance of TM7 was increased (p = 0.026) in HPV positive oral samples compared to the HPV negative group by ANOVA test. Increased relative abundance of TM73 (p = 0.011) at family level as well as Selenomonas spp. (p = 0.0032), Megasphaera spp. (p = 0.026) and TM73 (p = 0.018) at species level was detected in HPV positive oral samples. Haemophilus spp. was higher (p = 0.019) in HPV negative compared to HPV positive.
HPV positive samples displayed higher richness (Chao1 index) of bacterial microbiota as compared to HPV negative samples (p = 0.0319) (Fig. 3A). No difference in diversity (Shannon index, Fig. 3B) was detected between HPV positive and HPV negative groups. Furthermore, the LEfSe algorithm showed that unclassified Bifidobacteriaceae and Finegoldia genera were significantly enriched in HPV positive samples as compared to HPV negative (LDA score > 3.36 and 3.32 respectively, p < 0.05), while Haemophilus genus was enriched in HPV negative compared to HPV positive (Fig. 3E).

The bacterial microbiota in oral samples negative and positive for HPV DNA. Increased bacterial richness as assessed by the Chao1 index (A) was detected in HPV positive oral samples (blue) as compared to HPV negative samples (red). No difference in bacterial diversity (B) was detected between HPV negative (red) and positive (blue) oral samples. The relative abundance of bacteria are presented on the phylum (C) and family levels (D). In LEfSe analysis, unclassified Bifidobacteriaceae and Finegoldia genera were significantly enriched in HPV positive (blue) samples as compared to HPV negative (red) (LDA score > 3.36 and 3.32 respectively, p < 0.05), while Haemophilus genus was enriched in HPV negative (red) compared to HPV positive (E).
Discussion
HPV infection is associated with altered bacterial microbiota in the placenta, uterine cervix and mouth. Distinct differences were detected in the relative abundance of specific microbial groups on family and genus levels while the overall microbial richness and diversity appeared to be associated with HPV infection with the exception of higher richness in HPV positive oral samples compared to HPV negative samples. HPV positive placenta samples showed increased abundance of Lactobacillus genus (p = 0.0023) and Ureaplasma (LDA score > 4.0, p < 0.05), Coriobacteriaceae (p = 0.083) in cervix and Selenomonas spp. (p = 0.0032), Megasphaera spp. (p = 0.026) and TM73 (p = 0.018) in the mouth. In cervical samples Peptostreptococcaceae (p = 0.0065) and Enterococcaceae (p = 0.0069) were significantly enriched in HPV negative samples.
It has recently been suggested that the fetus does not develop in a sterile environment based on discovery of a distinct microbiota in the placenta17,18. Still there has been controversy about whether the findings are genuine or a result from contamination21,35. Some studies have not found specific placental microbiota21,22. Several studies have also reported HPV DNA in the placenta, even in samples collected transabdominally and sometimes associated with adverse effects in pregnancies3,5,8,9,10,11,12,13,14,15,36,37.
In this study we attempted to meticulously rule out the possibility of false positive results of either with bacterial microbiota or HPV DNA. We took special care and performed several steps to ensure that our findings are relevant and not result from contamination. The reagents used for PCR amplification were sequenced as well as negative controls to rule out possible reagent contamination. With placenta samples, negative placental controls reached 726 reads and identified 11 OTUs. We considered >1000 reads as a limit for inclusion. A total of 19/39 placenta samples (49%) reached that and therefore were included in this study. At OTU picking we further excluded singletons and OTUs with a relative frequency below 0.01. Cyanobacteria, Chloroplasts and Rhizobiales and sequences that could not be classified to domain level at OTU picking were removed from the results to control any possible bias. The presence and localization of HPV infection of our placental samples (Finnish Family HPV Study samples) have been examined before by Sarkola et al.12 and Koskimaa et al.7 and co-workers. Sarkola and coworkers confirmed the PCR positive HPV results by localizing the HPV16 and HPV6 DNA in syncytiotrophoblasts with tyramide amplified in-situ hybridization.
We recognized placental microbiota composed of Firmicutes, Proteobacteria, Actinobacteria and Bacteroidetes, which is in line with recent previous studies17,19,20,38,39. With HPV positive samples, we saw a significant increase in the amount of Lactobacillus genus which was mainly composed by L. iners, L. crispatus, L. jensenii, L. gasseri and L. reuteri species. Lactobacillus is the most abundant phyla in the vaginal microbiota40 but has also been detected in placenta in previous studies without analyzing the presence of HPV19,38. The most abundant Lactobacillus species in cervix and placenta were L. iners (41.0 vs. 0.2, p = 0.0000018), L. crispatus (13.8 vs. 0.1, p = 0.0012), L. gasseri (120.3 vs. 0.06, p = 0.023) and L. jensenii (6.01 vs. 0.0035, p = 0.042, respectively) without taking into account the HPV status of the samples.
In the present study, Ureaplasma spp. (Ureaplasma parvum 99% identity) was enriched in HPV positive placenta sample group. Ureaplasma spp. has previously in several studies been associated with chrorioamnionitis, which jeopardizes normal pregnancy and might lead to preterm delivery38,41,42,43,44,45. However, our results did not support the view that HPV presence would explain pregnancy complications and risk pregnancies as all had normal pregnancy without any complication. Nevertheless, the growing evidence of bacteria and virus in the placenta warrants further investigation from the point of view of a potential causal role in pregnancy complications including chorioamnionitis and preterm delivery46.
HPV infection has previously been linked to higher richness in vaginal microbiota3,24,25,26,29,47 and may be regarded as a predictor of imbalanced vaginal flora, especially decreased amount of Lactobacillus23,27,47,48. In the present study, we did not detect differences in the amount of Lactobacillus but rather an increase of Peptostreptococcaceae, Enterococcaceae and Haemophilus in HPV negative cervical samples. Peptostreptococcus has previously been associated with high-grade invasive cervical carcinoma when encountered together with HPV infection29. Interestingly, Enterococcus has also previously been linked to HPV-16 positive cervical cancer biopsies49 in contrast to our present study. Still, these previous findings have been made in single studies only and Enterococcus has even been observed from asymptomatic and healthy female vaginal flora50. It is currently not clear whether the vaginal microbiota play an important role in acquiring and persistence of HPV infection51. We have also previously presented data that some of the women from Finnish Family HPV Study, who presented HPV16 positive, developed incident cervical intraepithelial neoplasia within 14 years of follow-up52. Whether Lactobacillus plays a protective role in HPV clearance remains also to be determined51,53,54. Moreover, LEfSe test showed higher abundance of unclassified Coriobacteriaceae genus in HPV positive group cervical samples which was mostly attributable to Atopobium vaginae. A.vaginae has previously been reported to be associated with HPV persistence in cervico-vaginal region27,51 and bacterial vaginosis55,56. Still, A.vaginae has also been suggested in some cases to be a part of healthy vaginal microbiota57,58,59,60. This would suggest that present HPV infection and the observed changes in cervical bacterial microbiota may independently or synergistically create an environment favoring HPV persistence, dysbiosis and neoplastic transformation.
In this study, we observed a number of women with multiple HPV infection simultaneously at different body sites. Other studies have also observed this phenomenon but the cause behind this is currently not known61,62,63,64. HPV infection has primarily been considered as a sexually transmitted disease but recent research has begun to challenge this notion and so far we also know that vertical transmission during delivery or later in life, horizontal transmission and even autoinoculation have been established as possible ways of acquiring HPV infection3,65. Based on the results of the current study, we are able to conclude that bacterial microbiota can be one of the factors influencing the composition of overall microbiota in different anatomical sites.
The mouth harbors one of the most diverse microbiota in the human body and the most abundant bacterial phyla in the oral cavity include Actionbacteria, Bacteroidetes, Firmicutes, Fusobacterium, Streptococcus, Veillonella and Proteobacteria40,66. In our study the presence of HPV infection increased the amount of Selenomonas spp., Megasphaera spp. and TM73 in the oral microbiota. Selenomonas is a part of oral normal microbiota and oral biofilm but is also associated in disease, especially Selenomonas noxia in (aggressive) periodontitis (gum disease) and increased caries activity67,68,69. Megasphaera spp. is a part of dental plaque, so called early colonizer and has been discovered in endodontic abscess specimens70. The microbiota changes we found in oral HPV positive samples have not been earlier connected with HPV infection but may affect oral health. The association between oral HPV infection and oral health has not been extensively investigated but our data suggests that the oral microbiota composition in the presence of present HPV infection may favor potential pathogens.
Our study has several limitations. In our material of 329 mothers and deliveries and 315 placentas, only 13 placenta samples were HPV positive. This small number of HPV positive samples most likely explains why differences in the overall richness in the microbiota were not found. Secondly, the amount of usable DNA residues from bacteria in placenta samples is low which renders drawing definitive conclusions of the overall composition of microbiota difficult.
To our knowledge, this is the first study to show that HPV infection is associated with altered bacterial microbiota composition in the placenta and the mouth. We used the same protocol to indicate microbiota from oral, cervical and placenta samples. The diversity and composition of the bacterial microbiota was characterized by sequencing of the 16S rRNA gene and because a single bacterium can harbor multiple sequences of the 16S rRNA genes we have better possibilities to detect them as a sign that there has truly been a bacterial colonization. Still we were only able to investigate a small proportion of the placenta samples and needed to exclude some samples because the low number of reads to strengthen the reliability of our data. This definitely has an impact on our results but it has been an issue also in recent published articles investigating the placenta microbiota18,19.
In summary, we report a novel finding of association between microbiota and HPV infection. Our data may be interpreted to corroborate the hypothesis of a distinct microbiota of placenta. These initial findings still need to be replicated in larger studies to either confirm our findings or to reveal new perspectives into this neglected field of viral bacterial interactions. Whether the changes in bacterial microbiota predispose or result from HPV and if it influences HPV persistence, remains also to be determined in future studies.
Methods
Samples collected in the Finnish Family HPV Study71 were utilized in this study. The original study was designed to evaluate the interactions of HPV infection in families. A total of 329 pregnant women in their third trimester were enrolled in the study together with their male spouses (n = 131) and offsprings to come (n = 313) with 6-year follow-up.
For the present substudy, DNA samples from the placenta, the uterine cervix and the mother’s mouth taken during the last trimester were included. The presence of HPV DNA has been examined earlier7,12,62. The subjects were selected according to the HPV status of the placenta and mode of delivery. Altogether 13 placentas with HPV DNA were available. Thirteen HPV DNA negative placentas from vaginal deliveries with 13 HPV DNA negative placentas from caesarean section deliveries were selected as controls. The placenta samples obtained after delivery included all the tissue layers from the maternal side of placenta and the HPV DNA was localized in syncytiotrophoblasts12. Furthermore, the oral and cervical samples of these women had been collected as mucosal scrapings with a brush (Cytobrush, MedScand, Malmö, Sweden) and immediately frozen −70 °C until used.
The clinical study and its amendments were found acceptable by the Ethical Committee of the Intermunicipal Hospital District of Southwest Finland (#3/1998, #2/2006, 45/180/2010). The methods were carried out in accordance with the relevant guidelines and regulations. Informed consent was obtained from all the subjects participating in the study.
DNA extraction
HPV DNA was extracted by the high salt-method72.
HPV detection
HPV testing was performed with nested PCR using MY09/MY11 as external primers and GP05+/GP06+ as internal primers followed by genotyping with Multimetrix® kit (Progen Biotechnik GmbH, Heidelberg, Germany), which detects 24 HPV types (low-risk HPV6, 11, 42, 43, 44, and 70; high-risk HPV16, 18, 26, 31, 33, 35, 39, 45, 51, 52, 53, 56, 58, 59, 66, 68, 73 and 82 types) as described in Koskimaa et al.7 and Syrjänen et al.62.
16S bacterial gene sequencing
Isolated DNA concentrations were measured using a Qubit® 2.0 Fluorometer (Life Technology, Carlsbad, CA, USA) and normalized to10 ng/μL. The V3-V4 region of 16S rDNA gene was amplified by PCR using Illumina adapter overhang nucleotide sequences following Illumina protocols. After 16S rDNA gene amplification, the multiplexing step was performed using Nextera XT Index Kit (Illumina, San Diego, CA, USA) and PCR product was checked in a Bioanalyzer DNA 1000 chip (Agilent Technologies, Santa Clara, CA, USA). Libraries were sequenced using a 2 × 300 pb paired-end run (MiSeq Reagent kit v3) on a MiSeq-Illumina platform (Lifesequencing sequencing service, Valencia, Spain). To rule out and control for possible contaminations, PCR amplification and libraries controls were also sequenced as negative controls.
Bioinformatics and statistical analysis
Quality control of the FASTQ files was performed using Fastx tool kit version 0.0134 to remove reads with quality less than Q20, once the sequences were clean based on quality scores, we trimmed traces of the 16S rRNA primers and sequencing adapters using cutadapt version 1.2.5. After primer removal, sequences with <300 nucleotides read length were trimmed using perl scripting. Sequences were mapping against the human genome BWA version 0.7.173 and filtered with samtools version 1.3.1–50. Clean FASTQ files were converted to FASTA files and UCHIME program version 4.2 was used to remove chimeras.
An open reference OTU picking method using 97% identity to the Greengenes 13_8 database was performed using QIIME pipeline (version 1.9.0)74. Singletons and OTUs with a relative frequency below 0.01 were removed. Sequences that could not be classified to domain level, or were classified as Cyanobacteria, Chloroplasts (as they likely represent cellulose and cotton material) and Rhizobiales (as potential environmental contaminants) were removed from the dataset.
Alpha diversity indices (Chao1: richness and Shannon: diversity) and beta diversity using UNIFRAC (phylogenetic) and Bray Curtis distance (non-phylogenetic) among samples were studied and PERMANOVA was used to test significance. Calypso software version 8.10 (http://cgenome.net/calypso/) was used with total sum normalization (TSS) for the statistical analysis, and also, Cumulative Sum Scaling normalization (CSS) for multivariate tests (Redundancy Analysis - RDA). Linear discriminant analysis effect size (LEfSe) was used to detect unique biomarkers (LDA score > 3.0) in relative abundance of bacterial taxonomy. P-values ≤ 0.05 were regarded as statistically significant.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Giuliano, A. R. et al. EUROGIN 2014 Roadmap: Differences in HPV infection natural history, transmission, and HPV-related cancer incidence by gender and anatomic site of infection HHS Public Access. Int J Cancer Int J Cancer June https://doi.org/10.1002/ijc.29082 (2015).
Chatzistamatiou, K., Sotiriadis, A. & Agorastos, T. Effect of mode of delivery on vertical human papillomavirus transmission - A meta-analysis. J. Obstet. Gynaecol. 36, 10–4 (2016).
Lee, S. M. et al. Risk of vertical transmission of human papillomavirus throughout pregnancy: a prospective study. PLoS One 8, e66368 (2013).
M., M. et al. Transmission of carcinogenic human papillomavirus types from mother to child: A meta-analysis of published studies. Eur. J. Cancer Prev. https://doi.org/10.1097/CEJ.0b013e3283592c46 (2013).
Park, H. et al. Rate of vertical transmission of human papillomavirus from mothers to infants: relationship between infection rate and mode of delivery. Virol. J. 9, 80 (2012).
Syrjänen, S. & Puranen, M. Human papillomavirus infections in children: the potential role of maternal transmission. Crit. Rev. Oral Biol. Med. 11, 259–74 (2000).
Koskimaa, H.-M. et al. Human papillomavirus genotypes present in the oral mucosa of newborns and their concordance with maternal cervical human papillomavirus genotypes. J. Pediatr. 160, 837–43 (2012).
Ambühl, L. M. M. et al. Human Papillomavirus Infection as a Possible Cause of Spontaneous Abortion and Spontaneous Preterm Delivery. Infect. Dis. Obstet. Gynecol. 2016, 3086036 (2016).
Slatter, T. L. et al. A clinicopathological study of episomal papillomavirus infection of the human placenta and pregnancy complications. Mod. Pathol. 28, 1369–82 (2015).
Hong, L. J., Oshiro, B. T. & Chan, P. J. HPV-16 exposed mouse embryos: a potential model for pregnancy wastage. Arch. Gynecol. Obstet. 287, 1093–1097 (2013).
Gomez, L. M. et al. Placental infection with human papillomavirus is associated with spontaneous preterm delivery. Hum. Reprod. 23, 709–715 (2008).
Sarkola, M. E., Grénman, S. E., Rintala, M. A. M., Syrjänen, K. J. & Syrjänen, S. M. Human papillomavirus in the placenta and umbilical cord blood. Acta Obstet. Gynecol. Scand. 87, 1181–1188 (2008).
Kim, Y. H. et al. Genotypic prevalence of human papillomavirus infection during normal pregnancy: A cross-sectional study. J. Obstet. Gynaecol. Res. 40, 200–207 (2014).
Skoczyński, M., Goździcka-Józefiak, A. & Kwaśniewska, A. Prevalence of human papillomavirus in spontaneously aborted products of conception. Acta Obstet. Gynecol. Scand. 90, 1402–5 (2011).
Weyn, C. et al. Evidence of human papillomavirus in the placenta. J. Infect. Dis. 203, 341–3 (2011).
Syrjänen, S. & Puranen, M. Human papillomavirus infections in children: the potential role of maternal transmission. Crit. Rev. Oral Biol. Med. 11, 259–74 (2000).
Aagaard, K. et al. The placenta harbors a unique microbiome. Sci. Transl. Med. 6, 237ra65 (2014).
Collado, M. C., Rautava, S., Aakko, J., Isolauri, E. & Salminen, S. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci. Rep. 6, 23129 (2016).
Parnell, L. A. et al. Microbial communities in placentas from term normal pregnancy exhibit spatially variable profiles. Sci. Rep. 7, 11200 (2017).
Zheng, J. et al. The Placental Microbiota Is Altered among Subjects with Gestational Diabetes Mellitus: A Pilot Study. Front. Physiol. 8, 675 (2017).
Perez-Muñoz, M. E., Arrieta, M.-C., Ramer-Tait, A. E. & Walter, J. A critical assessment of the ‘sterile womb’ and ‘in utero colonization’ hypotheses: implications for research on the pioneer infant microbiome. Microbiome, https://doi.org/10.1186/s40168-017-0268-4 (2017).
Lauder, A. P. et al. Comparison of placenta samples with contamination controls does not provide evidence for a distinct placenta microbiota. Microbiome 4, 29 (2016).
Kero, K., Rautava, J., Syrjänen, K., Grenman, S. & Syrjänen, S. Association of asymptomatic bacterial vaginosis with persistence of female genital human papillomavirus infection. Eur. J. Clin. Microbiol. Infect. Dis. https://doi.org/10.1007/s10096-017-3048-y (2017).
Van De Wijgert, J. H. H. M. et al. The vaginal microbiota: What have we learned after a decade of molecular characterization? PLoS One, https://doi.org/10.1371/journal.pone.0105998 (2014).
Shannon, B. et al. Association of HPV infection and clearance with cervicovaginal immunology and the vaginal microbiota. Mucosal Immunol. https://doi.org/10.1038/mi.2016.129 (2017).
Gao, W., Weng, J., Gao, Y. & Chen, X. Comparison of the vaginal microbiota diversity of women with and without human papillomavirus infection: a cross-sectional study. BMC Infect. Dis. https://doi.org/10.1186/1471-2334-13-271 (2013).
Di Paola, M. et al. Characterization of cervico-vaginal microbiota in women developing persistent high-risk Human Papillomavirus infection. Sci. Rep. https://doi.org/10.1038/s41598-017-09842-6 (2017).
Adebamowo, S. N. et al. Mycoplasma hominis and Mycoplasma genitalium in the Vaginal Microbiota and Persistent High-Risk Human PapillomavirusInfection. Front. Public Heal. https://doi.org/10.3389/fpubh.2017.00140 (2017).
Mitra, A. et al. Cervical intraepithelial neoplasia disease progression is associated with increased vaginal microbiome diversity. Sci. Rep. 5, 16865 (2015).
Schwabe, R. F. & Jobin, C. The microbiome and cancer. Nat. Rev. Cancer, https://doi.org/10.1038/nrc3610 (2013).
Ohtani, N. Microbiome and cancer. Semin. Immunopathol. 37, 65–72 (2015).
Wang, L. & Ganly, I. The oral microbiome and oral cancer. Clinics in Laboratory Medicine, https://doi.org/10.1016/j.cll.2014.08.004 (2014).
Michaud, D. S. & Izard, J. Microbiota, oral microbiome, and pancreatic cancer. Cancer J. https://doi.org/10.1097/PPO.0000000000000046 (2014).
Hayashi, C., Gudino, C. V., Gibson, F. C. & Genco, C. A. Pathogen-induced inflammation at sites distant from oral infection: Bacterial persistence and induction of cell-specific innate immune inflammatory pathways. Molecular Oral Microbiology, https://doi.org/10.1111/j.2041-1014.2010.00582.x (2010).
Wassenaar, T. M. & Panigrahi, P. Is a foetus developing in a sterile environment? Lett. Appl. Microbiol. 59, 572–9 (2014).
Chen, M. L. et al. Placenta Microbiology and histology and the risk for severe retinopathy of prematurity. Investig. Ophthalmol. Vis. Sci. https://doi.org/10.1167/iovs.11-7380 (2011).
Stout, M. J. et al. Identification of intracellular bacteria in the basal plate of the human placenta in term and preterm gestations. Am. J. Obstet. Gynecol. https://doi.org/10.1016/j.ajog.2013.01.018 (2013).
Doyle, R. M. et al. Bacterial communities found in placental tissues are associated with severe chorioamnionitis and adverse birth outcomes. PLoS One, https://doi.org/10.1371/journal.pone.0180167 (2017).
Pelzer, E., Gomez-Arango, L. F., Barrett, H. L. & Nitert, M. D. Review: Maternal health and the placental microbiome. Placenta https://doi.org/10.1016/j.placenta.2016.12.003 (2017).
Huse, S. M., Ye, Y., Zhou, Y. & Fodor, A. A. A core human microbiome as viewed through 16S rRNA sequence clusters. PLoS One 7, e34242 (2012).
Kikhney, J. et al. Is Ureaplasma spp. the leading causative agent of acute chorioamnionitis in women with preterm birth? Clin. Microbiol. Infect. https://doi.org/10.1016/j.cmi.2016.10.010 (2017).
Cox, C. et al. The common vaginal commensal bacterium Ureaplasma parvum is associated with chorioamnionitis in extreme preterm labor. J. Matern. Neonatal Med. 10.3109/14767058.2016.1140734 (2016).
Sweeney, E. L. et al. Placental Infection with Ureaplasma species is Associated with Histologic Chorioamnionitis and Adverse Outcomes in Moderately Preterm and Late-Preterm Infants. J. Infect. Dis. https://doi.org/10.1093/infdis/jiv587 (2016).
Sweeney, E. L., Dando, S. J., Kallapur, S. G. & Knox, C. L. The human Ureaplasma species as causative agents of chorioamnionitis. Clin. Microbiol. Rev. https://doi.org/10.1128/CMR.00091-16 (2017).
Kwak, D. W., Cho, H. Y., Kwon, J. Y., Park, Y. W. & Kim, Y. H. Usefulness of maternal serum C-reactive protein with vaginal Ureaplasma urealyticum as a marker for prediction of imminent preterm delivery and chorioamnionitis in patients with preterm labor or preterm premature rupture of membranes. J. Perinat. Med, https://doi.org/10.1515/jpm-2014-0142 (2015).
Prince, A. L. et al. The placental membrane microbiome is altered among subjects with spontaneous preterm birth with and without chorioamnionitis. Am. J. Obstet. Gynecol. 214, 627.e1–627.e16 (2016).
Dareng, E. O. et al. Prevalent high-risk HPV infection and vaginal microbiota in Nigerian women. Epidemiol. Infect. 144, 123–37 (2016).
Lee, J. E. et al. Association of the Vaginal Microbiota with Human Papillomavirus Infection in a Korean Twin Cohort. PLoS One 8, e63514 (2013).
Ma, Z. et al. Human papillomavirus type 16 exists in bacteria isolated from cervical cancer biopsies. J Int Med Res, https://doi.org/10.1177/147323000903700411 (2009).
Babu, G., Singaravelu, B. G., Srikumar, R., Reddy, S. V. & Kokan, A. Comparative Study on the Vaginal Flora and Incidence of Asymptomatic Vaginosis among Healthy Women and in Women with Infertility Problems of ReproductiveAge. J. Clin. DIAGNOSTIC Res. 11, DC18–DC22 (2017).
Mitra, A. et al. The vaginal microbiota, human papillomavirus infection and cervical intraepithelial neoplasia: what do we know and where are we going next? Microbiome 4, 58 (2016).
Koskimaa, H.-M. et al. Human papillomavirus 16 E2-, E6- and E7-specific T-cell responses in children and their mothers who developed incident cervical intraepithelial neoplasia during a 14-year follow-up of the Finnish Family HPV cohort. J. Transl. Med. 12, 44 (2014).
Brotman, R. M. et al. Interplay between the temporal dynamics of the vaginal microbiota and human papillomavirus detection. J. Infect. Dis. 210, 1723–33 (2014).
Verhoeven, V. et al. Probiotics enhance the clearance of human papillomavirus-related cervical lesions. Eur. J. Cancer Prev. https://doi.org/10.1097/CEJ.0b013e328355ed23 (2013).
Hay, P. Bacterial vaginosis. F1000Research 6, 1761 (2017).
Xia, Q. et al. Identification of vaginal bacteria diversity and it’s association with clinically diagnosed bacterial vaginosis by denaturing gradient gel electrophoresis and correspondence analysis. Infect. Genet. Evol. 44, 479–486 (2016).
Mendes-Soares, H. et al. Fine-scale analysis of 16S rRNA sequences reveals a high level of taxonomic diversity among vaginal Atopobium spp. Pathog. Dis. 73, (2015).
Jespers, V. et al. Quantification of bacterial species of the vaginal microbiome in different groups of women, using nucleic acid amplification tests. BMC Microbiol. 12, 83 (2012).
Shipitsyna, E. et al. Composition of the Vaginal Microbiota in Women of Reproductive Age – Sensitive and Specific Molecular Diagnosis of Bacterial Vaginosis Is Possible? PLoS One 8, e60670 (2013).
Vitali, B. et al. Vaginal microbiome and metabolome highlight specific signatures of bacterial vaginosis. Eur. J. Clin. Microbiol. Infect. Dis. 34, 2367–2376 (2015).
Steinau, M. et al. Prevalence of Cervical and Oral Human Papillomavirus Infections Among US Women. J. Infect. Dis. 209, 1739–1743 (2014).
Syrjänen, S. et al. Dynamics of human papillomavirus serology in women followed up for 36 months after pregnancy. J. Gen. Virol. https://doi.org/10.1099/vir.0.007823-0 (2009).
Paaso, A. E. et al. Lack of type-specific concordance between human papillomavirus (HPV) serology and HPV DNA detection in the uterine cervix and oral mucosa. J. Gen. Virol. 92, 2034–2046 (2011).
Termine, N. et al. Oral human papillomavirus infection in women with cervical HPV infection: new data from an Italian cohort and a metanalysis of the literature. Oral Oncol. 47, 244–50 (2011).
Liu, Z., Rashid, T. & Nyitray, A. G. Penises not required: a systematic review of the potential for human papillomavirus horizontal transmission that is non-sexual or does not include penile penetration. Sex. Health 13, 10–21 (2015).
Wade, W. G. The oral microbiome in health and disease. Pharmacol. Res. 69, 137–143 (2013).
Gonçalves, L. F. H. et al. Levels of Selenomonas species in generalized aggressive periodontitis. J. Periodontal Res. 47, 711–8 (2012).
Cruz, P., Mehretu, A. M., Buttner, M. P., Trice, T. & Howard, K. M. Development of a polymerase chain reaction assay for the rapid detection of the oral pathogenic bacterium, Selenomonas noxia. BMC Oral Health 15, 95 (2015).
Johansson, I., Witkowska, E., Kaveh, B., Lif Holgerson, P. & Tanner, A. C. R. The Microbiome in Populations with a Low and High Prevalence of Caries. J. Dent. Res. 95, 80–6 (2016).
George, N. et al. Oral microbiota species in acute apical endodontic abscesses. J. Oral Microbiol. https://doi.org/10.3402/jom.v8.30989 (2016).
Rintala, M. A. M. et al. Transmission of high-risk human papillomavirus (HPV) between parents and infant: A prospective study of HPV in families in Finland. J. Clin. Microbiol. https://doi.org/10.1128/JCM.43.1.376-381.2005 (2005).
Miller, S. A., Dykes, D. D. & Polesky, H. F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. https://doi.org/10.1093/nar/16.3.1215 (1988).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics, https://doi.org/10.1093/bioinformatics/btp698 (2010).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods, https://doi.org/10.1038/nmeth.f.303 (2010).
Acknowledgements
The authors thank all the past and present researchers of the Finnish Family HPV study and especially the families participating in this study. We also want to thank Ms. Tatjana Peskova for her skillful help in the laboratory.
Author information
Authors and Affiliations
Contributions
S.S. designed and was responsible of the original study; H.T. and M.C. analyzed the samples and data set; H.T., S.R., S.S., M.C., J.R. designed the present study, as well as wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tuominen, H., Rautava, S., Syrjänen, S. et al. HPV infection and bacterial microbiota in the placenta, uterine cervix and oral mucosa. Sci Rep 8, 9787 (2018). https://doi.org/10.1038/s41598-018-27980-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-27980-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
