
Abstract
Host-associated microbiota have been claimed to play a role in hosts’ responses to parasitic infections, often protecting the hosts from infection. We tested for such a role in the crustacean Daphnia and the parasitic bacterium Pasteuria ramosa, a widely used model system for host-parasite interactions. We first determined the infection phenotype (i.e., resistotype) of eight clonal D. magna genotypes against four strains of P. ramosa by attachment test, followed by 16 S rDNA amplicon sequencing to determine if their genotype or their parasite resistotype influences the composition of their microbiome. We then reciprocally transplanted the microbiota of two host genotypes with opposite resistotypes to four P. ramosa isolates, followed by a reassessment of their resistotype after transplantation. We found significant differences in microbiome composition and structure between Daphnia genotypes and between Daphnia resistotypes to specific P. ramosa strains. Reciprocal microbiota exchange or making the Daphnia hosts bacteria-free, however, did not influence the resistotypes of the hosts. Thus, in contrary to what has been observed in some taxa, our results suggest that D. magna susceptibility to P. ramosa is strongly dictated by the genetic differences of the hosts and is still dependent on Daphnia’s first line of immune defense against the esophageal attachment of P. ramosa, which appears to be uninfluenced by the host’s microbiota.
Similar content being viewed by others

Introduction
Until a few years ago, conventional wisdom stated that differences among host populations in resistance to pathogens are largely explained by host genetic differences1,2,3 with environmental factors playing a subordinate role. Recently, microbiota have been added to the list of non-genetic factors that contribute to host resistance against parasites4,5,6,7. The specificity of infection from a gut pathogen in honeybees, for instance, is purportedly influenced by microbiome composition, and hints at the protective role of the microbiome in counteracting parasite infection5,8. Contrastingly, reports that the microbiome can also mediate host susceptibility to diseases have been gaining a lot of attraction in the biomedical sciences9,10,11. The beneficial role of the microbiome to host health has been overtly emphasized in recent years, but their possible influence on host pathogen resistance comes from a handful of investigations from a few host taxa so far. Therefore, this emerging concept requires more evidence before a solid generalization across taxa can be made.
Next generation sequencing has allowed researchers to characterize the microbiome composition of many organisms, resulting in increasingly more complex patterns. Environmental conditions such as diet, can explain variation in microbiome composition between individuals12,13, but species-specificity of microbiome composition is still observed, and differences between groups of organisms can still be explained in part by their phylogenetic position14,15. Recently, the host genotype in mice was reported to influence gut microbial composition13,16, with some members of microbiome being more heritable than others17. Moreover, genetic variation in immune-related pathways between human hosts is found to be associated with microbiome composition, lending support to the idea that aside from environmental factors, immune traits can influence microbiome composition15,18,19. However, it is not known whether it is the differences in overall host genetic background or the differences in specific immune traits that influences the microbiome composition in a given host taxa. Teasing apart these factors would allow to understand the role that the microbiome plays on immune traits and pathogen resistance. This would be especially interesting among invertebrates that lack adaptive immunity.
We used the Daphnia-Pasteuria host-parasite system to address these questions. Genetic variation is observed among natural genotypes of D. magna, and quantitative trait locus (QTL) approaches have been developed to address questions on the genetic basis of parasite resistance20,21. Many microbial parasites infect Daphnia, and one of them, P. ramosa, a gram-positive bacterium of the class Bacilli, was shown to display a co-evolutionary arms race with its hosts22,23. Furthermore, the role of microbiome on Daphnia fitness has been established and the experimental system can be manipulated such that the production of gnotobiotic Daphnia is possible24. These characteristics make Daphnia a good model to disentangle the contribution of host genetic background and microbiome composition, and to determine if the microbiome plays a role in Daphnia-P. ramosa interactions.
The interaction of P. ramosa with D. magna reveals a very high genetic specificity, visible as strong host genotype by parasite genotype interaction with an almost perfect binary infection outcome25,26. When a potential host encounters the parasite spores, the spores are activated followed by attachment to the esophagus wall. After successful attachment, the spores penetrate the foregut epithelium into the host body cavity and start proliferating in the haemolymph. Eventually, the host dies and releases Pasteuria spores in the environment27. Infections are highly virulent with severe reduction in host fecundity and lifespan. Most variation in infection success is explained by the first line of defense, i.e., the ability of the host to prevent attachment to the esophagus, the entry to Daphnia’s foregut25. Susceptibility to the host’s first line of defense can be assessed with an esophagus attachment test, during which fluorescently labeled P. ramosa spores are observed while attaching to the esophagus wall of the host. No esophageal attachment indicates resistance to the parasite25.
The main goal of this study is to determine if the microbiome plays a role in the resistance of D. magna to P. ramosa. We evaluated this by determining the infection phenotype of 8 Daphnia genotypes (=clones) to four P. ramosa strains, followed by comparison of their microbiome structure and composition based on genotype and resistance phenotype (termed resistotype in this study) to four Pasteuria strains. We then performed a reciprocal transplant experiment where we swapped the microbiome of resistant and susceptible D. magna genotypes and tested for resistance to one P. ramosa strain. Furthermore, we compared the resistotype of these D. magna genotypes when they were bacteria-free. We found that D. magna genotypes vary in microbiome composition even after years of culturing them under the same environmental conditions, and that statistical analysis indicated that their microbiome composition can potentially be structured by their resistotype. However, replacing the microbiome of the hosts or making the hosts bacteria-free have not altered their resistance or susceptibility to a P. ramosa pathogen.
Results
Resistotype of D. magna genotypes
Four Pasteuria strains were tested for attachment in the esophagus to identify Daphnia resistotypes (n = 31) (Table 1). Most Daphnia genotypes tested were resistant to Pr_C1 and Pr_P20, while half of the genotypes tested were susceptible to Pr_C19 and Pr_P15. Two genotypes were resistant to all four Pasteuria pathogens (clones Belgium_1, BE-OHZ-M5 and Germany, DE-K35-Inb1), while the Daphnia genotype from Hungary (HU-H0-2) was susceptible to all four pathogens. The Israel (IL-M1-1) clone died in culture and was not tested for Pr_P15 and Pr_P20.
Common microbiome among D. magna genotypes
We sequenced the V3 to V5 hypervariable region of the 16 S rDNA of 31 samples belonging to eight D. magna genotypes including one positive control (mock community) and analyzed 432, 318 sequences after denoising (3893–47570 per sample). Out of 28 genera used in the DNA mock community, 21 were represented at relative abundances similar to the DNA proportions used for sequencing. The use of 97% cut-off in picking Operational Taxonomic Units (OTUs) only gave resolution at the genus or family level identification and not at the strain level (see Supplementary Fig. S1). Strains belonging to Actinobacteria were not seen in the DNA mock community sequences but the V3-V5 16 S rDNA primers were able to capture the majority of the bacterial groups. Actinobacteria sequences were however seen in 4 (out of 31) D. magna samples albeit very rare and low in abundances. Qi et al.28 have seen Actinobacteria sequences in their metagenomics survey of Daphnia microbiome and found these to be abundant in D. pulicaria but very rare in D. magna and D. pulex. The DNA mock community sequences were not included in the downstream analysis.
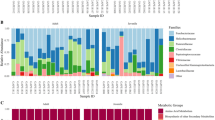
A total of 6548 OTUs (≥97% ID) were represented in the 31 D. magna samples but after removing singletons and chloroplast sequences, only 455 OTUs remained to be used for the analysis. Of these OTUs, 13 were present in 23 (out of 35) of the D. magna samples comprising genera and orders at different relative proportions (Fig. 1). The abundant microbiome populations were dominated by bacteria having homologous 16 S rDNA sequences to Limnohabitans (Beta-proteobacteria, 22–66% of the total counts), Saprospirales (Bacteroidetes, 9%- 54%), and Rhodobacter (Alpha-proteobacteria, 1–22%). Bacterial sequences homologous to Emticicia, Rudanella and Runella (Bacteroidetes, 0.5–3%); Rhizobiales, Ensifer, Sphingopyxis and Blastomonas (Alpha-proteobacteria, 0.2–7%); Ideonella (Beta-proteobacteria, 0.3–5%); Halomonas and Sinobactereacea (Gamma-proteobacteria, ≤ 0.6%) were the other least abundant but common microbiome among D. magna.

Common microbiome found in 75% of D. magna samples (n = 31) originated from different populations. These thirteen genera/families also represent the abundant OTUs in the D. magna microbiome consortium. Bubbles represent OTU counts shown in relative abundance (%).
Bacterial OTUs that were >1% in relative abundance but were only found in 15 out of 31 D. magna samples had homologous 16 S rDNA sequences to the Flavobacteriales of the genera Chryseobacterium and Flavobacterium and the Sphingomonadales of the genera Sphingomonas and Sphingobium. Some bacteria such as Hydrogenophaga, Caulobacter, Myxococcales, Pseudomonas, Pedobacter and Bacillus were abundant in some Daphnia genotypes (Supplementary Fig. S2).
Microbiome similarity by genotype and resistotype
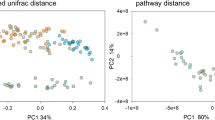
We plotted the principal coordinates of the Weighted and Unweighted UniFrac distances (Fig. 2a,b) to assess the similarity of the microbiome composition between eight Daphnia genotypes. A number of distinct clusters between genotypes were seen, suggesting variation in the microbiome composition between host genotypes. ANOSIM analysis supports statistically significant differences in bacterial community composition between host genotypes (Unweighted UniFrac, ANOSIM R = 0.50, p = 0.001). Similar results were obtained using ADONIS both from Unweighted (R2 = 0.39, p = 0.001) and Weighted UniFrac Distances (ANOSIM R = 0.22, p = 0.016; ADONIS: R2 = 0.42, p = 0.007) and Bray-Curtis Distance (ANOSIM R = 0.22, p = 0.004; ADONIS: R2 = 0.39, p = 0.006). Dissimilarity in the microbiome composition corresponded with the geographic origin of D. magna genotypes. For instance, the Iran clone showed a more distinct microbiome composition than the Finland clone (Unweighted UniFrac distance = 0.67, p < 0.01), even though both were hatched from resting eggs in the lab. A similar observation was seen between the Germany and the Hungary clones (Unweighted UniFrac distance = 0.58, p = 0.052, black and red polygons, respectively, in Fig. 2a,b). Notably, these two Daphnia clones have opposite resistotypes to four P. ramosa strains. The F1 hybrid clone (F1_lab breed) had a microbial composition similar to its parents, the Finland and Germany clones (Unweighted UniFrac distance = 0.50 and 0.53 respectively, p > 0.05), although this sexual offspring has been hatched from ephippia.

Differences in microbial community structure within and between different Daphnia genotypes. Principal Coordinate Analysis based on (a) Weighted and (b) Unweighted UniFrac Distances that were rarified and jacknifed at equal sequence depth of 3893. Similar symbol represents samples belonging to the same Daphnia genotype. Microbial composition differs by Daphnia genotypes as shown by ANOSIM (Weighted UniFrac: R = 0.22, p = 0.016; Unweighted UniFrac: R = 0.50, p = 0.001) and ADONIS (Weighted UniFrac: R2 = 0.42, p = 0.007; Unweighted UniFrac: ADONIS, R2 = 0.39, p = 0.001) statistical analyses with 999 permutations. Polygons show the distinct microbiome between Daphnia genotypes with opposite resistotypes to four Pasteuria pathogen. Red polygon represents the microbiome of Hungary clone (SSSS) and black polygon represents the microbiome of Germany clone (RRRR).
To further determine if the resistotype of the D. magna clones against four P. ramosa strains can explain the variation in the microbiome composition besides the genotypic effect, we assessed the variation of the microbiome composition between Daphnia using their resistotype to individual Pasteuria strains as the constraining variable employing three distance matrices. Distance-based Redundancy Analyses (db-RDA) based on Unweighted UniFrac distance matrix showed significant clustering of Daphnia microbiome by resistotypes to Pr_C1, Pr_P15 and Pr_P20 (p values < 0.05, Fig. 3a,c and d) but not with Pr_C19 (p > 0.05, Fig. 3b). However, db-RDA performed on Weighted UniFrac distance matrix did not show significant clustering in the microbiome composition between Daphnia based on resistotypes to Pr_C1, Pr_C19, Pr_P15 and Pr_P20 (Weighted Unifrac: p = 0.07, 0.63, 0.11 and 0.07, respectively; Supplementary Fig. S3A–D). Similarly, db-RDA analysis performed using Bray-Curtis distance matrix also did not show significant clustering in the microbiome composition between Daphnia resistotypes to Pr_C1, Pr_C19 and Pr_P15 but showed significant microbiome clustering based on Pr_20 (Bray-Curtis: p = 0.08, 0.56, 0.07 and 0.05, respectively; Supplementary Fig. S4A–D). The analyses suggest that the microbiome composition between D. magna could potentially be influenced by their resistotype to specific P. ramosa strains.

Biplot of db-RDA analysis using Unweighted UniFrac Distance, showing the clustering of microbiome composition between D. magna samples, with the analysis constrained by their resistotype to each P. ramosa strain (a) Pr_C1, (b) Pr_C19, (c) Pr_P15, and (d) Pr_P20. R (resistant) and S (susceptible) act as centroids to the clusters. Red colored samples belong to the susceptible D. magna resistotype and black colored samples belong to the resistant D. magna resistotype for the respective P. ramosa strains. Pseudo-F and p values < 0.05 denotes statistically significant variation between the microbiome of the Daphnia resistotypes.
Microbiome composition of highly-resistant and highly-susceptible Daphnia
We compared the alpha diversity and bacterial composition of the highly-resistant (Germany, RRRR) and highly-susceptible Daphnia (Hungary, SSSS) to gain an insight on the differences of the microbiome composition between the two extreme Daphnia resistotypes. These clones were used in the reverse microbiome transplant experiment described below. Daphnia genotype from Germany is significantly less species rich than the Hungary genotype based on Chao1 and number of observed OTUs metrics (Students T-test; p = 0.03 and 0.03, respectively). In terms of bacterial abundance, the highly-resistant German genotype contains a much higher proportion of bacteria from the family Saprospirales (p = 0.05), Sphingomonadaceae, Sphingobacteriaceae (p = 0.05) and Rhizobiaceae while it has a significantly lower proportion of bacteria from Comamonadaceae (p = 0.01), Rhodobacteraceae (p = 0.008) and Cytophagaceae (p = 0.06) (Fig. 4) than the highly-susceptible Hungarian genotype.

Differences in microbiome composition between highly-resistant (RRRR) D. magna genotype from Germany (DE-K35-Inb1) and highly-susceptible (SSSS) D. magna genotype from Hungary (HU-HO-2) shown as average relative abundance at the family level. Statistical comparison was performed using Student’s T-test with levels of significance denoted by *for p < 0.05 and ** for p < 0.01.
Microbiota swapping between D. magna of opposing resistotypes
We used Hungarian (SSSS) and German (RRRR) genotypes for the reverse microbiota transplant experiment to determine if microbiota swapping will change the original resistotype of the respective Daphnia clone. Pasteuria attachment tests showed that Pr_C1 attached to the esophagus wall of all Hungarian genotype replicates, regardless if the animal is bacteria-free, if it contains its native microbiota or if the Pasteuria-susceptible Daphnia contains the microbiota of a Pasteuria-resistant German genotype (Table 2). Similarly, Pasteuria Pr_C1 failed to attach on the esophagus wall of all Germany genotype replicates in all treatments. This suggests that the respective Daphnia genotypes retained their infection phenotype regardless of their microbiota or even in the absence of microbiota.
Discussion
Genetics and microbiota can have important functions in health and performance of animals. Here we report that genotypes of the model system D. magna from various geographic backgrounds still differed in their microbiome composition even after years of culturing the animals under the same dietary and environmental growth conditions. This indicates that the effects of the host genotype and of the host microbiota on the expression of phenotypic traits can be confounded. Both effects can be very important in the ecology and evolution of the system, but in different ways. For example, genes are only transmitted vertically, while microbiota can be transmitted vertically and horizontally. During migration, microbiota may be lost and in new habitats, new members may be acquired. To understand the expression and evolution of phenotypic traits, it is therefore necessary to disentangle host genetics from microbiota effects. Here we tested whether the first line of defense (i.e., the attachment of parasite in the esophagus or foregut entry) characterized by resistotypes of the water flea D. magna, are influenced by host genetic differences, or by the host’s microbiota as has been suggested in other systems.
We found that genotypic variation and parasite susceptibility and resistance of D. magna to specific P. ramosa strains, can correlate with differences in the microbial composition of the daphnid hosts (see Figs 2–3). In humans and mice, host genetics is reported to be a factor in shaping the composition and diversity of the microbiome17,29 and the abundance of some members of microbiota are associated with immune-related genes18,30,31. However, despite significant variation in the microbiome composition and structure between daphnid hosts (especially between two highly-opposite resistotypes), we have not seen a microbiota influence on the resistance trait of D. magna against the parasite P. ramosa (at least with Pr_C1 strain) after performing a reciprocal microbiome exchange. Remarkably, the resistotypes of the two Daphnia genotypes to the parasite were also not altered even when the animals were in a bacteria-free state, which strongly supports our conclusion, that the microbiome may not play a role in the innate immunity (first line of defense) of D. magna against a P. ramosa pathogen. These results are in direct contrast from what is reported in other taxa such as in bumblebees where it has been shown that gut microbiome plays a protective role against trypanosomatid parasites5, with the host genotype playing a secondary role in their immunity8, and in wild Anopheles gambiae where a member of the midgut microbiome renders the insect resistant to the malaria-causing Plasmodium32. In a marine crustacean Palaemon macrodactylus, a bacterium constantly isolated from the eggs can protect the shrimp embryo from a fungal pathogen by releasing an anti-fungal chemical33. In human disease-related pathogenesis, a fecal microbiota probiotic could combat infection of the pathogen Clostridium difficile to alleviate suffering from chronic intestinal bowel disorder34,35. Furthermore, the human gut microbiota member, Bacteroides fragilis, has been proposed as a potential probiotic to improve autistic behavioral symptoms36. The disparity between our study and the emerging paradigm that the microbiome plays a major role in the resistance against parasite infection or disease pathogenesis emphasizes the idea that the microbiome’s role in immunity could be organism-dependent (or at least parasite-dependent) and should not be generalized across all taxa. Notably, the Daphnia-Pasteuria system exhibits strong host-parasite species interaction, with host infection rate dependent on the presence of diverse parasite strains that would specifically infect matching host genotypes37. The co-evolutionary arms race between Daphnia and Pasteuria may be a stronger driving factor on the evolution of innate immunity in D. magna against the pathogen and could mask the influence of microbiota (if there is any). Moreover, the difference of the Daphnia-Pasteuria system compared to other host-parasite systems may lie with how the Pasteuria infection process takes place, the decisive step being the attachment of Pasteuria to the esophagus. P. ramosa can only infect Daphnia if it manages to attach to the esophagus of the daphnid host25. It is evident that this inhibition of attachment is accomplished by the epithelial immunity or barrier defenses present in the esophagus (the esophagus being the extension of the foregut) of resistant Daphnia, and appears to be entirely independent from the microbiome influence. Interestingly, the absence of microbiome in resistant Daphnia did not alter the barrier defenses. Bacteria-free organisms are known to have abnormal and immature gut development38,39,40 which impairs mucosal immunity in the gut leading to disease susceptibility. In tse-tse flies, microbiota-free organisms are susceptible to trypanosome infection because of a compromised peritoneal matrix that act as a physical barrier between lumen and epithelial cells in their midgut6.
In this study, we only evaluated the influence of the microbiome on D. magna’s first line of defense against pathogen infection, which is the prevention of infection via resistance against gut entry. Microbiota may possibly play a role on Daphnia’s second line of defense, which is the prevention of pathogen growth inside the body or infection tolerance by reducing the damage of the infection through fitness compensation. Indeed, Pasteuria susceptible Daphnia can mount an immune response by increasing the number of phagocytes in its haemolymph when infected41, or by maintaining similar levels of fecundity before total sterilization42. To investigate the possible role of the microbiome on Daphnia’s second line of defense, a Pasteuria susceptible Daphnia transplanted with various microbiome members could be evaluated against pathogen growth followed by fitness measurement in the presence of infection.
Because Daphnia is being used as a model system for environmental genomics43, exploring the possible contribution of the microbiome on the adaptive response of Daphnia to changing environments could be interesting. We have previously shown that the microbiome has a significant effect on Daphnia fitness24 and there is a significant variation in the microbiome composition between Daphnia genotypes (in this study). Host adaptation to changing environmental conditions could shape their microbiome composition and could confer differences in fitness between Daphnia genotypes. The genetic basis of the differing tolerance of natural populations of Daphnia to high copper concentrations44,45 and temperature46 has been investigated previously. The interaction of host genetics and the microbiome in this context could be an interesting area for further investigation.
Conclusion
This is the first study to show that despite considerable differences in the microbiome composition between Daphnia genotypes, particularly that of P. ramosa-susceptible and P. ramosa-resistant Daphnia, reciprocal swapping of their microbiota did not alter the susceptibility or resistance of the respective daphnid hosts against a Pasteuria pathogen, as demonstrated by attachment test of the parasite in the host’s esophagus. More importantly, despite the known contribution of the microbiome to host immunity, a Pasteuria-resistant Daphnia that was raised bacteria-free was able to maintain its barrier defenses against parasite entry, suggesting that the microbiome does not influence the first line of defense in Daphnia. Further investigation of the microbiome’s role on host immunity could be focused on the second line of defense.
Methods
Animals
Eight D. magna genotypes were collected from different geographic regions, either as planktonic individuals or as resting eggs which were subsequently hatched and kept as clonal lines for several years (Table 1). Two clones included are the parents of a standing QTL panel47,20, one of which (FI-Xinb3) was selfed three times, while the other (DE-K35-Iinb1) was selfed once. The F1_lab_breed (QTL-IXF-1) genotype is the sexual offspring of these two parent clones and was hatched from a resting egg in the lab47. Animals were maintained in artificial Daphnia medium (ADaM48, for at least 20 generations under standardized environmental conditions; 20 °C, 16:8 light dark photoperiod, the green algae Scenedesmus sp. as only food and animals cultured.
Only female adults were sampled for the 16 S rDNA profiling. Five jars with 80 ml ADaM containing two Daphnia of each genotype were grown for 4 weeks under standard laboratory conditions fed with 10 million cells algae per jar every other day. ADaM was replaced every week and only 2 adult individuals were kept in the jar every ADaM change. Animals were washed twice in sterile ADaM to remove unassociated bacteria before storage in −20 °C prior to DNA extraction.
Resistotype determination via esophagus-attachment test
Different D. magna genotypes were attachment-tested against four Pasteuria clones/isolates as described by Duneau et al.25 Daphnia show a binary infection outcome to Pasteuria pathogens; susceptible animals allow P. ramosa attachment to the esophagus or to the hindgut while resistant animals display no sign of attachment. P. ramosa Pr_C1 and Pr_C19 clones were isolated from infected D. magna from Moscow (Russia) and Gaarzerfeld (Germany), respectively, and previously characterized in Luijckx et al.26. Pr_C1 and Pr_C19 mode of attachment is through the esophagus. P. ramosa Pr_P15 and Pr_P20 are isolates that have been propagated in the same D. magna host clones several times, but where not cloned. Pr_P20 was isolated from an infected D. magna female that was collected in 2011 from Aegelsee, Switzerland. Pr_P20 attaches to the host in the esophagus. Pr_P15 is derived from an infected D. magna collected in Belgium and is unique in that it attaches usually to the hindgut of the host.
P. ramosa spores were fluorescently labeled and attachment tests performed as described by Duneau et al.25. Briefly, spores were labeled with fluorescein-5(6)-isothiocyanate (FITC) dye at a concentration of 1 mg in 0.1 M NaCO3 (pH 9.1). The solution was kept in the dark for 2 hours and washed 3–5 times via centrifugation. Daphnia were exposed to resuspended spores (5000 spores per individual) for 1 hour in the dark. Animals were then examined for spore attachment with an inverted epifluorescent microscope (Leica DM 2500) at 400× magnification. Resistotypes were defined as 4 letter codes (e.g. RSSR) composed of R (for resistance, no attachment) and S (susceptibility, attachment) for P. ramosa strains Pr_C1, Pr_C19, Pr_P15, Pr_P20, respectively.
DNA extraction
Frozen animal tissues were bead-beat with two pieces of acid-washed and autoclaved 1.0 mm beads and a 100 µl of 0.1-mm silica zirconian beads in PCR water in a FastPrep F120 cell disruptor (Qbiogene, USA) for 30 s at 4 m·s−1. DNA extraction was continued following the steps described in the DNeasy Blood and Tissue Kit (Qiagen, Netherlands). PCR water was used as a control for the extraction procedure and went through the same extraction and PCR amplification steps as the Daphnia samples.
454 Barcode Tagging
Two PCR reactions steps were carried out in amplifying the V3-V5 hypervariable region of the 16 S rDNA with Roche 454 barcodes. The first PCR amplification step involved using the primer pairs F (5′-ACACGGYCCARACTCCTAC-3′) and R (5′-TTGCWTCGAATTAAWCCAC-3′) that targets the 327–969 bp of the 16 S rDNA using Phusion High Fidelity Taq (New England Biolab, USA) with this PCR program: 98 °C for 1 min, 15 cycles of 98 °C for 10 s, 50 °C for 20 s and 72 °C for 30 s with a final extension time of 72 °C for 5 min. PCR reactions were treated with ExoSap (Fermentas, USA), to remove unincorporated nucleotides and primers. A second PCR reaction step was prepared using the first PCR purified product as DNA template and re-amplified with F/R primer pairs containing the Libl fusion sequence and barcodes (PFx: 5′-CCATCTCATCCCTGCGTGTCTCCGACTCAGbarcodeF primer-3′.
PRx: 5′-CCTATCCCCTGTGTGCCTTGGCAGTCTCAGR primer-3′) using the same PCR program and cycles. Amplified products were purified using Agencourt Ampure XP beads (Beckman and Coulter, USA) and eluted in sterile TE buffer. DNA concentrations were measured using Qubit 2.0 Fluorometer (Invitrogen, USA) and a pool of equimolar amounts (30 ng/µL) of barcoded products were sequenced on 454 GS-FLX instrument with LibL Titanium chemistry (Roche, USA) at Microsynth (Balgach, Switzerland). A DNA mock community consisting of 76 bacterial strains belonging to 28 genera and families of various phyla was also tagged with a barcode and was sequenced as a positive control (see Supplementary Table S1). The mock community consisted of bacterial isolates previously isolated from Daphnia animals either freshly collected from the field or maintained in the lab, the algal food cultures (Scenedesmus sp.) or ADaM (Daphnia culture medium). Isolates were chosen to represent different bacterial phylogenies and based on the availability of bacterial cultures.
Sequence Data Processing and Analysis
Demultiplexed samples were denoised and pre-clustered at 99% level similarity using the method described by Reeder and Knight49. Samples were checked for chimeras, low quality sequence and short sequence reads (<150 bp) prior to post-analysis with the Quantitative Insights Into Microbial Ecology (QIIME 1.9.150). OTUs were clustered at 97% sequence similarity with UClust51. Representative sequences were picked for taxonomic identification based on Blast 2.2.2252. OTU representative sequences were aligned with Pynast53, while gaps in the alignment were filtered using the default 16 S rDNA Greengenes alignment lane mask before making the phylogenetic tree using FastTree 2.1.354. Singletons and chloroplast sequences were filtered out of the OTU table before downstream analyses.
Microbial community composition analysis
To control for sequencing effort, samples were rarified at a minimum of 10 sequences and a maximum depth of 3893 sequences in steps of 50, and performing 20 iterations for each step. The microbial diversity for each D. magna sample was assessed using 4 alpha diversity indices (Observed_OTUs, Phylogenetic Diversity, Chao1 and Shannon index) at 3893 sequence depth and statistically compared using Student’s T-tests in JMP 12.0 (Cary, NC, USA). Since our main objective is to compare the microbial community between genotypes with contrasting resistotypes for the reciprocal transplant experiment, only alpha diversity of RRRR (Germany, n = 5) and SSSS (Hungary, n = 3) host genotypes were reported in this study.
For microbial composition assessment of Daphnia genotypes and resistotypes, the OTU table was rarified at an equal depth of 3893 sequences to generate pairwise distance matrices. Weighted and Unweighted UniFrac and Bray-Curtis distance matrices derived from this rarified beta diversities were used to statistically test for differences in microbial composition using ANOSIM and ADONIS methods with 999 permutations. Distance-based redundancy analysis (db-RDA) was also applied to determine the clustering of D. magna microbiome composition when constrained by their resistotype to each P. ramosa strain.
Reciprocal transplant of microbiota between susceptible and resistant Daphnia
To determine if microbiota influences the resistotype of D. magna to Pasteuria infection, we removed the bacteria, or swapped the microbiota of two Daphnia genotypes that were previously tested to be highly-susceptible (SSSS, Hungary clone) and highly-resistant (RRRR, Germany clone) to four P. ramosa clones and isolates (see Table 1). The detailed methods for synchronizing the animals, the production of bacteria-free Daphnia from asexual eggs, the preparation of bacterial suspension, preparation of axenic Scenedesmus obliquus algae and subsequent PCR screening for bacteria-free Daphnia and algae were previously described in Sison-Mangus et al.24. Briefly, eggs from 100 individuals of synchronized Hungarian and German Daphnia mothers were extracted from the mother’s brood pouch and washed with sterile ADaM three times. Eggs were treated with freshly made Ampicillin (1 mg/mL) and Kanamycin (100 μg/mL) for 48 hours until development of hatchlings. Sterile ADaM and antibiotics were replaced each day. The bacteria-free state of the hatchlings after antibiotics treatment was verified by 16 S rDNA PCR amplification.
Two independent reciprocal transplant experiments were performed using the Hungarian (SSSS) and the German (RRRR) clones as respective experimental animals. After antibiotics removal, bacteria-free Daphnia hatchlings were divided into groups representing 3 treatments: 1) Bacteria-free Daphnia, 2) Daphnia inoculated with microbiota from the same clone, and 3) Daphnia inoculated with microbiota coming from clone with opposite resistotype (e.g., German hatchlings received the microbiota from Hungarian clone while the Hungarian hatchlings received the microbiota from German clone). Bacterial suspensions were harvested from the respective Daphnia clone sources following the method of Sison-Mangus et al.24 with 1 ml bacterial suspension given to respective hatchlings from each group. The optical density (OD600) of the bacterial suspension ranged from 0.72–0.73. Hatchlings were exposed to bacteria for 24 hours and subsequently washed once before placing in individual experimental jars with 25 million axenic algal food cells.
Hatchlings were grown under the same environmental conditions as above and maintained daily as in Sison-Mangus et al.24. P. ramosa Pr_C1 stick testing was done at Day 6 post-hatching using the method described above, but with antibiotic treated spores. For this, the FITC-dyed P. ramosa spores were treated with triple antibiotic solution (Ampicillin (2 mg/mL), Kanamycin (1 mg/mL) and Tetracyclin (100 μg/mL) for 36 hours in the dark prior to stick testing. This is to prevent unwanted bacteria that maybe present from Pasteuria spore solution from contaminating the experimental treatments during the attachment test. Preliminary test showed that P. ramosa C1 spores remain viable (able to attach to the esophagus of Daphnia) after being treated with antibiotics.
Data availability
The raw sequencing reads have been deposited at the NCBI Short Read Archive under BioProject ID PRJNA407858 with the following BioSample accession numbers: SAMN07665251 to SAMN07665254 for IR-GG1–1 clone, SAMN07665255 to SAMN076652588 for IL-M1-1 clone, SAMN07665259 to SAMN07665261 for HU-HO-2 clone, SAMN07665262 to SAMN07665264 for BE-OHZ-M5 clone, SAMN07665265 to SAMN07665267 for BE-OHZ-M10 clone, SAMN07665268 to SAMN07665272 for FI-Xinb3 clone, SAMN07665273 to SAMN07665277 for DE-K35-Iinb1 clone, SAMN07665278 to SAMN07665281 for QTL-IXF-1 clone and SAMN07695326 for Positive Control.
References
Browning, J. A. & Frey, K. J. Multiline Cultivars as a Means of Disease Control. Annu. Rev. Phytopathol. 7, 355-+ (1969).
Sherman, P. W., Seeley, T. D. & Reeve, H. K. Parasites, Pathogens, and Polyandry in Social Hymenoptera. Am. Nat. 131, 602–610 (1988).
Altermatt, F. & Ebert, D. Genetic diversity of Daphnia magna populations enhances resistance to parasites. Ecol Lett 11, 918–928, https://doi.org/10.1111/j.1461-0248.2008.01203.x (2008).
Jaenike, J., Unckless, R., Cockburn, S. N., Boelio, L. M. & Perlman, S. J. Adaptation via symbiosis: recent spread of a Drosophila defensive symbiont. Science 329, 212–215, https://doi.org/10.1126/science.1188235 (2010).
Koch, H. & Schmid-Hempel, P. Socially transmitted gut microbiota protect bumble bees against an intestinal parasite. Proceedings of the National Academy of Sciences 108, 19288–19292, https://doi.org/10.1073/pnas.1110474108 (2011).
Weiss, B. L., Wang, J., Maltz, M. A., Wu, Y. & Aksoy, S. Trypanosome infection establishment in the tsetse fly gut is influenced by microbiome-regulated host immune barriers. PLoS Pathog 9, e1003318, https://doi.org/10.1371/journal.ppat.1003318 (2013).
Koehler, S., Doubsky, J. & Kaltenpoth, M. Dynamics of symbiont-mediated antibiotic production reveal efficient long-term protection for beewolf offspring. Frontiers in Zoology 10, https://doi.org/10.1186/1742-9994-10-3 (2013).
Koch, H. & Schmid-Hempel, P. Gut microbiota instead of host genotype drive the specificity in the interaction of a natural host-parasite system. Ecol. Lett. 15, 1095–1103, https://doi.org/10.1111/j.1461-0248.2012.01831.x (2012).
Garrett, W. S. et al. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell 131, 33–45, https://doi.org/10.1016/j.cell.2007.08.017 (2007).
Turnbaugh, P. J., Baeckhed, F., Fulton, L. & Gordon, J. I. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host & Microbe 3, 213–223, https://doi.org/10.1016/J.Chom.2008.02.015 (2008).
Vijay-Kumar, M. et al. Metabolic Syndrome and Altered Gut Microbiota in Mice Lacking Toll-Like Receptor 5. Science 328, 228–231, https://doi.org/10.1126/science.1179721 (2010).
Muegge, B. D. et al. Diet Drives Convergence in Gut Microbiome Functions Across Mammalian Phylogeny and Within Humans. Science 332, 970–974, https://doi.org/10.1126/science.1198719 (2011).
David, L. A. et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559-+, https://doi.org/10.1038/nature12820 (2014).
Ley, R. E. et al. Evolution of mammals and their gut microbes. Science 320, 1647–1651, https://doi.org/10.1126/science.1155725 (2008).
Jones, R. T., Sanchez, L. G. & Fierer, N. A Cross-Taxon Analysis of Insect-Associated Bacterial Diversity. Plos One 8, https://doi.org/10.1371/journal.pone.0061218 (2013).
Leamy, L. J. et al. Host genetics and diet, but not immunoglobulin A expression, converge to shape compositional features of the gut microbiome in an advanced intercross population of mice. Genome Biology 15, https://doi.org/10.1186/s13059-014-0552-6 (2014).
Goodrich, J. K. et al. Human Genetics Shape the Gut Microbiome. Cell 159, 789–799, https://doi.org/10.1016/j.cell.2014.09.053 (2014).
Blekhman, R. et al. Host genetic variation impacts microbiome composition across human body sites. Genome Biology 16, https://doi.org/10.1186/s13059-015-0759-1 (2015).
Bonder, M. J. et al. The effect of host genetics on the gut microbiome. Nat. Genet. 48, 1407–1412, https://doi.org/10.1038/ng.3663 (2016).
Routtu, J. & Ebert, D. Genetic architecture of resistance in Daphnia hosts against two species of host-specific parasites. Heredity 114, 241–248, https://doi.org/10.1038/hdy.2014.97 (2015).
Luijckx, P., Fienberg, H., Duneau, D. & Ebert, D. A Matching-Allele Model Explains Host Resistance to Parasites. Curr. Biol. 23, 1085–1088, https://doi.org/10.1016/j.cub.2013.04.064 (2013).
Decaestecker, E. et al. Host-parasite ‘Red Queen’ dynamics archived in pond sediment. Nature 450, 870–873, https://doi.org/10.1038/nature06291 (2007).
Ebert, D. Host-parasite coevolution: Insights from the Daphnia-parasite model system. Curr Opin Microbiol 11, 290–301, https://doi.org/10.1016/j.mib.2008.05.012 (2008).
Sison-Mangus, M. P., Mushegian, A. A. & Ebert, D. Water fleas require microbiota for survival, growth and reproduction. Isme Journal 9, 59–67, https://doi.org/10.1038/ismej.2014.116 (2015).
Duneau, D., Luijckx, P., Ben-Ami, F., Laforsch, C. & Ebert, D. Resolving the infection process reveals striking differences in the contribution of environment, genetics and phylogeny to host-parasite interactions. BMC Biol 9, 11, https://doi.org/10.1186/1741-7007-9-11 (2011).
Luijckx, P., Ben-Ami, F., Mouton, L., Du Pasquier, L. & Ebert, D. Cloning of the unculturable parasite Pasteuria ramosa and its Daphnia host reveals extreme genotype-genotype interactions. Ecol Lett 14, 125–131, https://doi.org/10.1111/j.1461-0248.2010.01561.x (2011).
Ebert, D. In Evolution and Modern Society: An Introduction to how Evolution Shapes our Lives (eds R. E. Lenski & J. Losos) (Princeton University Press 2016).
Qi, W., Nong, G., Preston, J. F., Ben-Ami, F. & Ebert, D. Comparative metagenomics of Daphnia symbionts. BMC Genomics 10, 172, https://doi.org/10.1186/1471-2164-10-172 (2009).
Benson, A. K. et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proceedings of the National Academy of Sciences of the United States of America 107, 18933–18938, https://doi.org/10.1073/Pnas.1007028107 (2010).
Knights, D. et al. Complex host genetics influence the microbiome in inflammatory bowel disease. Genome Medicine 6, https://doi.org/10.1186/s13073-014-0107-1 (2014).
Khachatryan, Z. A. et al. Predominant Role of Host Genetics in Controlling the Composition of Gut Microbiota. Plos One 3, https://doi.org/10.1371/journal.pone.0003064 (2008).
Cirimotich, C. M., Ramirez, J. L. & Dimopoulos, G. Native Microbiota Shape Insect Vector Competence for Human Pathogens. Cell Host & Microbe 10, 307–310, https://doi.org/10.1016/J.Chom.2011.09.006 (2011).
Gilturnes, M. S., Hay, M. E. & Fenical, W. Symbiotic Marine-Bacteria Chemically Defend Crustacean Embryos from a Pathogenic Fungus. Science 246, 116–118, https://doi.org/10.1126/Science.2781297 (1989).
Bakken, J. S. et al. Treating Clostridium difficile Infection with Fecal Microbiota Transplantation. Clin Gastroenterol Hepatol, https://doi.org/10.1016/j.cgh.2011.08.014 (2011).
Cui, B. T. et al. Fecal microbiota transplantation through mid-gut for refractory Crohn’s disease: Safety, feasibility, and efficacy trial results. Journal of Gastroenterology and Hepatology 30, 51–58, https://doi.org/10.1111/jgh.12727 (2015).
Hsiao, E. Y. et al. Microbiota Modulate Behavioral and Physiological Abnormalities Associated with Neurodevelopmental Disorders. Cell 155, 1451–1463, https://doi.org/10.1016/j.cell.2013.11.024 (2013).
Ganz, H. H. & Ebert, D. Benefits of host genetic diversity for resistance to infection depend on parasite diversity. Ecology 91, 1263–1268 (2010).
Hooper, L. V. Bacterial contributions to mammalian gut development. Trends Microbiol. 12, 129–134, https://doi.org/10.1016/J.Tim.2004.01.001 (2004).
Rawls, J. F., Samuel, B. S. & Gordon, J. I. Gnotobiotic zebrafish reveal evolutionarily conserved responses to the gut microbiota. Proc Natl Acad Sci USA 101, 4596–4601, https://doi.org/10.1073/pnas.0400706101 (2004).
Bates, J. M. et al. Distinct signals from the microbiota promote different aspects of zebrafish gut differentiation. Dev Biol 297, 374–386, https://doi.org/10.1016/j.ydbio.2006.05.006 (2006).
Auld, S. K. J. R., Edel, K. H. & Little, T. J. The Cellular Immune Response of Daphnia Magna under Host-Parasite Genetic Variation and Variation in Initial Dose. Evolution 66, 3287–3293, https://doi.org/10.1111/j.1558-5646.2012.01671.x (2012).
Vale, P. F. & Little, T. J. Fecundity compensation and tolerance to a sterilizing pathogen in Daphnia. J. Evol. Biol. 25, 1888–1896, https://doi.org/10.1111/j.1420-9101.2012.02579.x (2012).
Colbourne, J. K. et al. The ecoresponsive genome of Daphnia pulex. Science 331, 555–561, https://doi.org/10.1126/science.1197761 (2011).
Lopes, I., Baird, D. J. & Ribeiro, R. Genetic determination of tolerance to lethal and sublethal copper concentrations in field populations of Daphnia longispina. Arch. Environ. Contam. Toxicol. 46, 43–51 (2004).
Hochmuth, J. D., De Meester, L., Pereira, C. M., Janssen, C. R. & De Schamphelaere, K. A. Rapid Adaptation of a Daphnia magna Population to Metal Stress Is Associated with Heterozygote Excess. Environ Sci Technol 49, 9298–9307, https://doi.org/10.1021/acs.est.5b00724 (2015).
Yampolsky, L. Y., Schaer, T. M. & Ebert, D. Adaptive phenotypic plasticity and local adaptation for temperature tolerance in freshwater zooplankton. Proc Biol Sci 281, 20132744, https://doi.org/10.1098/rspb.2013.2744 (2014).
Routtu, J., Jansen, B., Colson, I., De Meester, L. & Ebert, D. The first-generation Daphnia magna linkage map. BMC Genomics 11, https://doi.org/10.1186/1471-2164-11-508 (2010).
Klüttgen, B., Dülmer, U., Engels, M. & Ratte, H. T. ADaM, an artificial freshwater for the culture of zooplankton. Water Res. 28, 743–746, https://doi.org/10.1016/0043-1354(94)90157-0 (1994).
Reeder, J. & Knight, R. Rapidly denoising pyrosequencing amplicon reads by exploiting rank-abundance distributions. Nat. Methods 7, 668–669, https://doi.org/10.1038/nmeth0910-668b (2010).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat Meth 7, 335–336 (2010).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461, https://doi.org/10.1093/bioinformatics/btq461 (2010).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic Local Alignment Search Tool. J. Mol. Biol. 215, 403–410, https://doi.org/10.1006/jmbi.1990.9999 (1990).
Caporaso, J. G. et al. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26, 266–267, https://doi.org/10.1093/bioinformatics/btp636 (2010).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2-Approximately Maximum-Likelihood Trees for Large Alignments. Plos One 5, https://doi.org/10.1371/journal.pone.0009490 (2010).
Acknowledgements
We acknowledge the help of Elodie Burcklen for the preparation of samples for sequencing, Jürgen Hottinger for help with the Daphnia cultures, Kristina Mueller for the help with attachment testing and Mahendra Mariadassou for denoising the 16 S rDNA amplicon sequences in this study. This study was supported by an ERC Advanced Investigator Grant and by the Swiss National Science Foundation.
Author information
Authors and Affiliations
Contributions
M.P.S.M. and D.E. conceived the study. M.P.S.M., D.E. and C.M. designed the study. C.M. conducted the esophagus-attachment test. M.P.S.M. conducted the Daphnia experiments and performed the microbiome data analysis. All authors contributed to the final version of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sison-Mangus, M.P., Metzger, C.M.J.A. & Ebert, D. Host genotype-specific microbiota do not influence the susceptibility of D. magna to a bacterial pathogen. Sci Rep 8, 9407 (2018). https://doi.org/10.1038/s41598-018-27681-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-27681-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
