
Abstract
Long terminal repeat (LTR) retrotransposon is the most abundant DNA component and is largely responsible for plant genome size variation. Although it has been studied in plant species, very limited data is available for cotton, the most important fiber and texture crop. In this study, we performed a comprehensive analysis of LTR retrotransposon families across four cotton species. In tetraploid Gossypium species, LTR retrotransposon families from the progenitor D genome had more copies in D-subgenome, and families from the progenitor A genome had more copies in A-subgenome. Some LTR retrotransposon families that insert after polyploid formation may still distribute the majority of its copies in one of the subgenomes. The data also shows that families of 10~200 copies are abundant and they have a great influence on the Gossypium genome size; on the contrary, a small number of high copy LTR retrotransposon families have less contribution to the genome size. Kimura distance distribution indicates that high copy number family is not a recent outbreak, and there is no obvious relationship between family copy number and the period of evolution. Further analysis reveals that each LTR retrotransposon family may have their own distribution characteristics in cotton.
Similar content being viewed by others
Introduction
Transposons are mobile genetic elements that can be multiplied in the genome using a variety of mechanisms. Transposons play crucial roles in gene expansion, diversification and evolution1,2. Additionally, they are major determinants of genome size in eukaryotes. Indeed, there is a linear correlation between genome size and transposable element content in all eukaryotes3,4,5. The most numerous group of transposons in plants are long terminal repeat (LTR) retrotransposon, which share a unique structural feature, two long terminal repeats, and they typically contain ORFs for a structural protein for virus-like particles, aspartic proteinase, reverse transcriptase, RNase H, and integrase. Occasionally, there is an additional ORF of unknown function6. Two main super families of LTR retrotransposon are Gypsy and Copia, differing in the order of reverse transcriptase and integrase2. An LTR retrotransposon family is a group of elements that have high DNA sequence similarity in their coding region or internal domain, or in their terminal repeat region. According to Wicker2, the sequence similarity was higher than 80%. The lowest taxon of LTR retrotransposon is a particular individual copy, corresponding to a specific transposition and insertion event, and is of particular relevance to genome annotation2.
Cotton (Gossypium spp.) is a remarkably diverse genus, with over 50 recognized species divided into 8 diploid genome groups and a single, monophyletic allopolyploid lineage7. It has been proposed that all diploid cotton species might evolve from a common ancestor that subsequently diversified to 8 groups, including groups A–G and K8,9. DNA sequence data place the origin of Gossypium at about 5 to 10 million years ago, which rapidly diversified into these major genome groups shortly thereafter. Allopolyploid species appeared within the last 1 to 2 million years, which were involved a D-genome species as the pollen-providing parent and an A-genome species as the maternal parent8,9. Gossypium has undergone a threefold increase in genome size due to the accumulation of LTR retrotransposons since its origin10. Further studies show that retrotransposable-mediated gene regulation may modulate the unbalanced expression of homologous gene pairs that results in that great differences in fiber properties among Gossypium species11,12.
Recent whole genome sequencing of two cultivated tetraploid species, island cotton (Gossypium barbadense)13 and upland cotton (G. hirsutum)14, and their two diploid ancestors G. arboretum15, and G. raimondii16,17 make it possible for detailed discovery and cross-species comparison of LTR retrotransposons elements at the whole genome level13,15,16,17,18,19,20. In this study, we annotated the LTR retrotransposon of four Gossypium species (G. barbadense, G. hirsutum, G. arboreum and G. raimondii) by using multiple de novo repeat prediction pipelines with a combination of known repeat elements from the RepBase database21. For the first time, We classified the LTR retrotransposon sequences into the family level, and we also compared the LTR retrotransposon families between G. barbadense13, G. hirsutum14, G. arboreum15 and G. raimondii16. Our results led to the identification of several previously unknown features of Gossypium LTR retrotransposon families associating with copy number, genomic distribution, average element size, and the correlation with the genes. Our results highlight the importance of distinguishing LTR retrotransposon families instead of super families when assessing their impact on gene and genome evolution.
Methods and Materials
Dataset
Genome sequences and annotations of two cultivated tetraploid species, island cotton (G. barbadense)13 and upland cotton (G. hirsutum)14, and their two diploid ancestors G. arboretum15, and G. raimondii16,17 were downloaded from the cotton public available database COTTONGEN22. The genomes of Theobroma cacao, Carica papaya, Arabidopsis thaliana, Populus trichocarpa, Ricinus communis, Durio zibethinus and Glycine max were download from NCBI (ftp.ncbi.nlm.nih.gov/).
LTR retrotransposon identification
Self-comparison approaches were performed by PILER23. Because the genome is too large to align the entire sequence to itself, the genome was split into chunks small enough for PALS23. Each chunk was aligned to itself, then each different pair of chunks was aligned to each other. Signature-based identification of LTR retrotransposon elements were performed using LTR_STRUC24 and LTRharvest25 with the default parameters. Then, all putative LTR retrotransposon elements were classified into super families (such as Copia and Gypsy) by REPCLASS26. Within each super family, CD-HIT27 was used to cluster similar sequences into groups if they shared at least 80% of sequence identity and consensus sequences of every group were created. In addition, the de-novo repeat identification suite RepeatModeler28 was also used to produce the consensus sequences. A library was established by integrating these consensus sequences with Repbase21 and the redundant sequences were removed from the library if they shared 80% sequence identity with any sequence in Repbase. Then, genomes of G. barbadense, G. hirsutum, G. arboretum, and G. raimondii were annotated by RepeatMasker29 with this custom library. In order to analyze the origin of LTR retrotransposon, genomes of T. cacao, C. papaya, A. thaliana, P. trichocarpa, R. communis, D. zibethinus and G. max were also annotated by RepeatMasker with the same library. A set of sequences annotated by the same consensus sequence of the library were defined as a LTR retrotransposon family. A family should consist of at least 10 elements. In this way, more families will be retained so as to understand the characteristics of the low copy family.
Structural analysis
LTR_STRUC was employed to generate a variety of structural features of LTR retrotransposon. By using blastn30, family elements that were generated by other methods were aligned against LTR_STRUC results. For analyzing the structures of LTR retrotransposon, a set of perl script was developed.
Copy number and genome coverage estimation
Copy number and genome coverage of LTR retrotransposon were calculated on RepeatMasker outfiles (.out) using custom perl scripts. The calculation only included sequences longer than 100 nucleotides and short sequences were eliminated. Additionally, if multiply significant hits were detected for a same genome location, only the longer sequence was considered. The distributions and densities of genes were obtained from the GFF3 annotation.
Gossypium genomes were split into contiguous 2-Mb windows. An LTR retrotransposon sequence or gene was assigned to a particular window based on its start point. Circos30 was employed for constructing the diagram. To understand the function of the genes around LTR retrotransposon family, we collected genes locating within both upstream and downstream 20 kb of the LTR retrotransposon family according to the position of transposon and GFF3 annotation files. GO annotation results were plotted by WEGO31.
Kimura distance distribution of LTR retrotransposon elements
Kimura distances32 between LTR retrotransposon elements and its consensus sequence from the library were determined using the RepeatMasker alignments result file (.align).
Phylogenetic analysis
Sequence alignments were performed by Clustal Omega program33 with default options. PHYLIP 3.695 program34 was employed for the neighbor-joining trees.
Results
LTR retrotransposon families in cotton genome
Using a combination of the previously-known sequences from RepBase and newly identified by multiple de novo methods, a Gossypium species-specific LTR retrotransposon library was constructed. The library was composed of 78468 consensus sequences, 38839 of which were classified as Gypsy super family, 11031 were classified as Copia super family, 7250 were classified as other super family, and 21348 could not be classified. Of these consensus sequences, 54649 were identified de novo and 23819 were from Repbase.
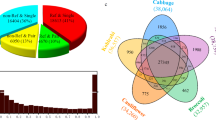
We statistically analyzed the family number of LTR retrotransposons in Gossypium genomes on condition that the family must contain at least 10 copies and the elements must be longer than 100 bp. After such filtering, a total of 24855 families have been identified; G. barabdense, G. hirsutum, G. arboreum, and G.raimondii contain 16634, 15597, 11595, 7208 families, respectively (Supplementary Table S1). The number of LTR retrotransposon families shared between the Gossypium species was shown in Venn35 diagram (Fig. 1). Among them, 2493 (10%) families were shared by all the four Gossypium species, and 10702 (43.1%) families were specific to one specific species; for example, G. barabdense contains 5721 (23%) specific families. In general, most high copy number LTR retrotransposon families are highly copied in all of the four Gossypium species, and most low copy number families have few elements present in all of four Gossypium species (Fig. 2).

Comparison of LTR retrotransposon families between Gossypium genome.

Copy number differences of the same family in different species.
LTR retrotransposon elements were highly unstable in plant genomes, and the instability was associated with illegitimate recombination and unequal homologous recombination. In our study, there are 102 families that are present in G. hirsutum, G. arboreum, and G.raimondii but not in G. barbadense. Structural analysis of these 102 families shows that 79.0%, 75.4% and 82.3% of these elements in G. hirsutum, G. arboreum, and G. raimondii are remnants fragments that were derived from illegitimate recombination. Similar results were obtained from families that only absent in G. hirsutum, G. arboreum and G. raimondii (Fig. 1), that about three-quarter of these elements are remnants fragments. We also found families specific to one of the four Gossypium species (Fig. 1). These families might be originated from duplication events after the Gossypium species separated or might be caused by the loss of the other three species during Gossypium evolution. Comparative analysis shows that only a small part of these families had members in T. cacao, C. papaya, A. thaliana, P. trichocarpa, R. communis, D. zibethinus and G. max genomes, that is to say most families originate after the separated of Gossypium species.
Evolution of LTR retrotransposon family during cotton polyploidization
Polyploidy is a pervasive force in plant evolution. The AD-genome tetraploid G. barbadense and G. hirsutum were derived from progenitor A-genome and D-genome diploids involved in ancestral allopolyploidization. In this study, to understand the evolution of tetraploid Gossypium, we calculated the distribution preferences of LTR retrotransposon families that origin in different evolution stage. There are 4320 LTR retrotransposon families shared by tetraploid G.barbadense, G. hirsutum and A-genome diploid G. arboreum (Fig. 1). However, none of these families originated from D-genome. For these families, there are 111123 copies in A-subgenome and only 5843 copies in D-subgenome in G. barbadense (Fig. 3A). In addition, there are 2613 LTR retrotransposon families shared by G. barbadense, G. hirsutum and D-genome diploid G. raimondii (Fig. 1). The LTR retrotransposon family copies also show unequal distribution in A- and D-subgenome (Fig. 3C,D). In order to make a comparative analysis, we calculated the distribution of 2493 LTR retrotransposon families that shared by the two tetraploid genomes and their A- and D-genome diploid ancestors. The results show that these transposons were approximately equal distributed in the A- and D-subgenomes of the tetraploid genomes (Fig. 3E,F). Therefore, in tetraploid Gossypium species, LTR retrotransposon families from the progenitor D genome may have more copies in D-subgenome, and families from the progenitor A genome may have more copies in A-subgenome.

Copy number distribution in A- and D-subgenomes of the tetraploid genomes.
We also calculated the distribution of tetraploid specific LTR retrotransposon families. First of all, there are 984 families shared by G. barbadense and G. hirsutum (Fig. 1). Secondly there are 1955 families specific to G. hirsutum (Fig. 1). Interestingly, the majority of these transposon families had a tendency to be distributed either in A-subgenome (Left of the figure) or in D-subgenome (Right of the figure, Fig. 3G,H and J). Third, there are 5721 families specific to G. barbadense (Fig. 1), whereas no great preference was detected and there are 302404 copies in A-subgenome and 203721 copies in D-subgenome (Fig. 3I). Therefore, LTR retrotransposon families that insert after polyploid formation may still distribute most of its copies in one of the subgenomes or both subgenomes.
Copy number characterization of LTR retrotransposon family
LTR retrotransposon families with 10–200 copies are most abundant, and families of more than 800 copies are very rare (Fig. 4A). For example, in the G. hirsutum genome, 15143 LTR retrotransposon families have the copy number between 10 and 200, but only 23 LTR retrotransposon families show the copy number with 800 or more. Interestingly, there is a family whose copy number is significantly higher than others in G. hirsutum, G. arboreum and G. raimondii genomes. In both G. hirsutum and G. arboreum genome, the highest copy numbers family RLGy_42738 has 3229, 10055 copies, respectively, followed by the family of 1913, 3830 copies. In G. raimondii genome, the highest copy numbers family RLGy_31317 has 1995 copies, followed by family of the 1076 copies. However, there is not so much difference between the high copy number families in the G. barbadense genome (Supplementary Datasets 1–5). Furthermore, there are five LTR retrotransposon families whose copy number is more than 400 in all of the four Gossypium genomes (Table 1).

Family number, total length and average length of different copy number LTR retrotransposon families.
Large amount of LTR retrotransposon families with 10~200 copies in the Gossypium genome resulting in the total length of these families are the most abundant. In the G. hirsutum genome, LTR retrotransposon families of this copy level is 614.1 Mb, whereas families with 800 or more copies is only 24.1 Mb. These findings are similar in the other three Gossypium genomes (Fig. 4B). Therefore, LTR retrotransposon families with 10~200 copies have a great influence on the genome size, while a small number of high copy LTR retrotransposon families have less contribution to the Gossypium genome size.
The average length of the LTR retrotransposon family can reflect the degree of fragmentation, which may reflect some evolutionary characteristics. In G. arboreum genome, average length of LTR retrotransposon families with 600~1000 copies show little longer. In these families, RLGy_40347 family has 659 copies, with an average length of 9271 bp; RLGy_40817 family has 908 copies, with an average length of 8123 bp. In addition, the average length of RLGy_40479 is 9496 bp, which is the longest among all studied families, with a copy number of 1040. The above three families are all G. arboreum specific. Altogether, there is not much difference in the average length of different copy number families (Fig. 4C).
Distribution of LTR retroelements on chromosomal
LTR retroelement elements are ubiquitous in Gossypium genomes. To be more precise, they are less densely distributed at the end of the A-genome chromosomes, more densely at the head of the D-genome chromosomes. In addition, uniform distribution on the chromosomes 7, 8 10, 12 and 13 of G. arboretum and chromosome A4 of G. hirsutum (Fig. 5).

LTR retrotransposon family elements and gene density of Gossypium chromosomes in 2 Mb unit. The outermost ring is the density of all LTR retrotransposon families elements, after the chromosome ring, followed by RLGy_42738 family, RLCo_3154 family, RLCo_5655 family, RLGy_42774 family and RLCo_258 family.
In order to understand the distribution pattern of each LTR retroelement family in Gossypium genomes, we analyzed 5 families that were shared by all of the four Gossypium species and the copy number is more than 400 (Table 1). In G. barbadense, G. hirsutum and G. raimondii, RLGy_42738 and RLGy_42774 show approximate negative correlation with gene density; however, RLCo_3154 and RLCo_5655 show approximate positive correlations with gene number at the head of the chromosomes. Whereas the distribution of these families in G. arboreum genome show inconsistent correlations with the other three Gossypium genomes. In addition, the distribution of RLCo_258 in all of the four Gossypium genomes had no obvious relationship with gene density, but it had its own unique characteristics and was obviously not random (Fig. 5).
The GO clustering analysis show that the same family in different species and different families in the same species differed. However, in total, the majority of the genes around RLGy_42738, RLCo_3154, RLCo_5655, RLGy_42774 and RLCo_258 under cellular component were involved in cell and cell part, molecular function categories were mostly clustered in binding and catalytic, the genes clustered under biological process mainly involved in cellular process and metabolic process (Supplementary Fig. S1).
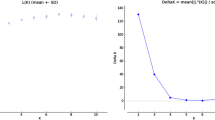
Copy number of LTR retroelement family and evolution
Kimura distances were calculated for all copies of LTR retroelement family by RepeatMasker. Sequence divergence (K-values) is correlated with the transposition history: low K-values copies indicate recent activity, while high K-values elements were generated by more ancient transposition events32. In this study, we generally separated LTR retroelement families into high copy number families (>800 copies), middle copy number families (201~800 copies) and low copy number families (10~200 copies). Profiles were relatively similar not only between species but also between different copy number groups (Fig. 6). First, Despite the dramatic copy number differences across species, all of the four Gossypium genomes had the most LTR retroelement copies at about k = 28; Second, Gossypium genomes generally contain much more recent copies (k < 28) than ancient copies (k > 28). We speculate that LTR retrotransposon elements were eliminated from the genome before the evolution stage of k > 28. This is in agreement with the dynamics of LTR retrotransposons in the rice genome36. Furthermore, a young (k < 3) and small amplification of LTR retrotransposon elements was occurred in Gossypium genomes, such as the low copy number families of G. barbadense, middle and high copy number families of G. arboreum. In addition, G. raimondii high copy number families have a regular amplification over time (k < 26). These young bursts vary among species, revealing that each genome has undergone a particular amplification within very recent evolution history, which may be related to their growth environment.

Kimura distance distribution of LTR retrotransposon. The graphs represent element number (y axis) for high copy number families (>800 copies), middle copy number families (201~800) and low copy number families (10~200) in Gossypium genomes (x axis, K-value from 0 to 60). Due to the large difference, high copy number families was separated from middle copy number families and low copy number families.
The results also show that the high copy number families were not a recent outbreak, but like other families that there are more elements distribute near k = 28. Therefore, there was no significant relationship between the copy number and the evolutionary period.
Phylogenetic analysis
The phylogenetic relationships of the five shared LTR retroelement families (Table 1) among the four Gossypium species further reflected by the tree individually. The results show that all clades are mixed of elements from the four Gossypium genomes, certain elements are more closely related to copies present in other Gossypium species than to the sequences of self-genome (Supplementary Fig. S2).
Discussion
Comparative analysis of tetraploid Gossypium and its diploid progenitors
Allopolyploidization plays a profound impact on genome architecture37. Some of these changes have been linked to novel phenotypic variations in polyploids38. Relative to diploid progenitor cottons, tetraploid cottons (G. hirsutum and G. barbadense) developed superior fiber phenotypes, representing the development of new agronomic traits13. In G. hirsutum genome14, overall gene order and colinearity are largely conserved between the A and D subgenomes and the extant D-progenitor genome (G. raimondii). However, this colinearity was not obvious with the A-progenitor genome14. Furthermore, SNP analysis indicated that the genomic divergence between the two tetraploids was less than the divergence between the tetraploid and its diploid progenitors13. Our analysis shows that the shared families (10410) between tetraploids (G. barbadense and G.hirsutum) were more than the shared families between the tetraploid and its diploid progenitors (Fig. 1). G. hirsutum genome shares 6289 families with G. raimondii and shares 8964 families with G. arboretum; G.barbadense genome shares 5264 families with G. raimondii and shares 7181 families with G. arboretum. This study is an important step in the analysis of the tetraploid cotton genome, which will provide invaluable insights into the biology and evolution of allopolyploidization in cotton and other plants.
Conserved features of LTR retrotransposon
Copy numbers of shared LTR retrotransposon families differ among the four Gossypium genomes (Supplementary Table S1). In addition, the total amount of LTR retrotransposon elements is also quite different. However, copy number and average length characteristics of the LTR retrotransposon family are similar in different Gossypium species (Figs 2 and 4). Furthermore, many researches show that larger genomes tend to have more LTR retrotransposons than smaller genomes3, and we also observed a similar result in Gossypium genomes, except for G. arboreum (Table 2). These features of LTR retrotransposon indicate their importance to the genome evolution.
Distribution characteristics
A large number of transposable elements have been identified from genomes of different species. However, the transposable element distribution shows great or even the opposite characteristics. In maize and sorghum, transposable elements had a strong positive correlation with gene number and have a bias toward insertion near genes, but with a preference for avoiding coding regions39. On the contrary, in Arabidopsis, transposable element distribution analysis suggests a negative correlation between gene and TE density40.
In this study, we annotated the LTR retrotransposon of four Gossypium species and classified these elements into the family level for the first time. One of the most interesting findings is the observation that the distribution of LTR retrotransposon elements had no insertion bias as a whole, however, each family had its own unique distribution characteristics (Fig. 5). Furthermore, if we exclude the special G. arboreum, the same family has similar distribution characteristics in G. barbadense, G. hirsutum and G.raimondii. If so, the distribution characteristic is another conserved feature of LTR retrotransposon families.
Conclusions
The current understanding of LTR retrotransposon increases with an increasing number of sequenced genomes from a broad taxa range. In addition to Arabidopsis41, great progress has been made in LTR retrotransposon studies in corn37, soybean42 and tomato43. In the present study, we identified and classified LTR retrotransposon from genomes of G. barbadense, G. hirsutum, G. arboreum and G. raimondii. Our results suggest that most LTR retrotransposon families have 10~200 copies in the genome. Additionally, there is no obvious relationship between family copy number and the period of evolution. By analyzing 5 of the 2493 shared families, we found that different families have different distribution characteristics; more importantly, the same family has similar distribution character across G. barbadense, G. hirsutum and G. raimondii44,45.
References
Oliver, K. R., McComb, J. A. & Greene, W. K. Transposable elements: powerful contributors to angiosperm evolution and diversity. Genome Biol Evol. 5, 1886–1901 (2013).
Wicker, T. et al. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 8, 973–982 (2007).
Elliott, T. A. & Gregory, T. R. What’s in a genome? The C-value enigma and the evolution of eukaryotic genome content. Philos Trans R Soc Lond B Biol Sci. 370, 20140331 (2015).
Vitte, C. & Panaud, O. LTR retrotransposons and flowering plant genome size: emergence of the increase/decrease model. Cytogenet Genome Res. 110, 91–107 (2005).
Schnable, P. S. et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 326, 1112–1115 (2009).
Havecker, E. R., Gao, X. & Voytas, D. F. The diversity of LTR retrotransposons. Genome Biol. 5, 225 (2004).
Grover, C. E. et al. Re-evaluating the phylogeny of allopolyploid Gossypium L. Mol Phylogenet Evol. 92, 45–52 (2015).
Grover, C. E., Grupp, K. K., Wanzek, R. J. & Wendel, J. F. Assessing the monophyly of polyploid Gossypium species. Plant Systematics and Evolution. 298, 1177–1183 (2012).
Wendel, J. F. & Grover, C. E. Taxonomy and evolution of the cotton genus, Gossypium. American Society of Agronomy (2015).
Hawkins, J. S., Kim, H., Nason, J. D., Wing, R. A. & Wendel, J. F. Differential lineage-specific amplification of transposable elements is responsible for genome size variation in Gossypium. Genome Res. 16, 1252–1261 (2006).
Wang, K., Huang, G. & Zhu, Y. Transposable elements play an important role during cotton genome evolution and fiber cell development. Sci China Life Sci. 59, 112–121 (2016).
Cao, Y. et al. Molecular characterization of a transcriptionally active Ty1/copia-like retrotransposon in Gossypium. Plant Cell Rep. 34, 1037–1047 (2015).
Wang, K. et al. The draft genome of a diploid cotton Gossypium raimondii. Nat Genet. 44, 1098–1103 (2012).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob DNA. 6, 11 (2015).
Yuan, D. et al. The genome sequence of Sea-Island cotton (Gossypium barbadense) provides insights into the allopolyploidization and development of superior spinnable fibres. Sci Rep. 5, 17662 (2015).
Zhang, T. et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat Biotechnol. 33, 531–537 (2015).
Paterson, A. H. et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature. 492, 423–427 (2012).
Li, F. et al. Genome sequence of the cultivated cotton Gossypium arboreum. Nat Genet. 46, 567–572 (2014).
Lin, L. et al. A draft physical map of a D-genome cotton species (Gossypium raimondii). BMC Genomics. 11, 395 (2010).
Liu, X. et al. Gossypium barbadense genome sequence provides insight into the evolution of extra-long staple fiber and specialized metabolites. Sci Rep. 5, 14139 (2015).
Li, F. et al. Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat Biotechnol. 33, 524–530 (2015).
Edgar, R. C. & Myers, E. W. PILER: identification and classification of genomic repeats. Bioinformatics. 21(Suppl 1), i152–158 (2005).
McCarthy, E. M. & McDonald, J. F. LTR_STRUC: a novel search and identification program for LTR retrotransposons. Bioinformatics. 19, 362–367 (2003).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics. 9, 18 (2008).
Feschotte, C., Keswani, U., Ranganathan, N., Guibotsy, M. L. & Levine, D. Exploring repetitive DNA landscapes using REPCLASS, a tool that automates the classification of transposable elements in eukaryotic genomes. Genome Biol Evol. 1, 205–220 (2009).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 28, 3150–3152 (2012).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics. Chapter 4, Unit 4 10 (2009).
Tempel, S. Using and understanding RepeatMasker. Methods Mol Biol. 859, 29–51 (2012).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics. 10, 421 (2009).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Ye, J. et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 34, W293–297 (2006).
Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 16, 111–120 (1980).
Sievers, F. & Higgins, D. G. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol Biol. 1079, 105–116 (2014).
Retief, J. D. Phylogenetic analysis using PHYLIP. Methods Mol Biol. 132, 243–258 (2000).
Shamansky, S. L. & Graham, K. Y. The Venn diagram: a metaphor for life. Public Health Nurs. 16, 1–2 (1999).
Devos, K. M., Brown, J. K. & Bennetzen, J. L. Genome size reduction through illegitimate recombination counteracts genome expansion in Arabidopsis. Genome Res. 12, 1075–1079 (2002).
Sharma, A., Schneider, K. L. & Presting, G. G. Sustained retrotransposition is mediated by nucleotide deletions and interelement recombinations. Proc Natl Acad Sci USA 105, 15470–15474 (2008).
Ma, J., Devos, K. M. & Bennetzen, J. L. Analyses of LTR-retrotransposon structures reveal recent and rapid genomic DNA loss in rice. Genome Res. 14, 860–869 (2004).
Zhang, H. et al. Evolution of the BBAA component of bread wheat during its history at the allohexaploid level. Plant Cell. 26, 2761–2776 (2014).
Chen, Z. J. Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annu Rev Plant Biol. 58, 377–406 (2007).
Sanchez, D. H., Gaubert, H., Drost, H. G., Zabet, N. R. & Paszkowski, J. High-frequency recombination between members of an LTR retrotransposon family during transposition bursts. Nat Commun. 8, 1283 (2017).
Du, J. et al. Bifurcation and enhancement of autonomous-nonautonomous retrotransposon partnership through LTR Swapping in soybean. Plant Cell. 22, 48–61 (2010).
Xu, Y. & Du, J. Young but not relatively old retrotransposons are preferentially located in gene-rich euchromatic regions in tomato (Solanum lycopersicum) plants. Plant J. 80, 582–591 (2014).
Wei, B. et al. Genome-wide characterization of non-reference transposons in crops suggests non-random insertion. BMC Genomics. 17 (2016).
Wright, S. I., Agrawal, N. & Bureau, T. E. Effects of recombination rate and gene density on transposable element distributions in Arabidopsis thaliana. Genome Res. 13, 1897–1903 (2003).
Acknowledgements
This study was sponsored by the National Natural Science Foundation of China (No. 31471548), Innovation Scientists and Technicians Troop Construction Projects of Henan Province (184200510009), State Key Laboratory of Cotton Biology Open Fund (CB2015A21), Key Scientific Research Projects of Henan Higher Education Institution (18A180010), and National Key Research and Development Program (2016YFD0100203).
Author information
Authors and Affiliations
Contributions
Z.L., Y.L., S.Z. and Q.L. performed all the bioinformatics assays, data analyses, and manuscript writing. F.L. and X.W. helped with the Figures. K.W., B.Z. and R.P. conceived and directed the study and manuscript editing. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, Z., Liu, Y., Liu, F. et al. Genome-Wide Survey and Comparative Analysis of Long Terminal Repeat (LTR) Retrotransposon Families in Four Gossypium Species. Sci Rep 8, 9399 (2018). https://doi.org/10.1038/s41598-018-27589-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-27589-6
This article is cited by
-
Molecular insights into the VIRESCENS amino acid sequence and its implication in anthocyanin production in red- and yellow-fruited cultivars of date palm
Scientific Reports (2023)
-
Development of molecular markers based on LTR retrotransposon in the Cleistogenes songorica genome
Journal of Applied Genetics (2022)
-
Preferential insertion of a Ty1 LTR-retrotransposon into the A sub-genome’s HD1 gene significantly correlated with the reduction in stem trichomes of tetraploid cotton
Molecular Genetics and Genomics (2020)
-
In silico mining and FISH mapping of a chromosome-specific satellite DNA in Capsicum annuum L.
Genes & Genomics (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
