
Abstract
Bats (Order: Chiroptera) harbor a high diversity of emerging pathogens presumably because their ability to fly and social behavior favor the maintenance, evolution, and dissemination of these pathogens. Until 2012, there was only one report of the presence of Hantavirus in bats. Historically, it was thought that these viruses were harbored primarily by rodent and insectivore small mammals. Recently, new species of hantaviruses have been identified in bats from Africa and Asia continents expanding the potential reservoirs and range of these viruses. To assess the potential of Neotropical bats as hosts for hantaviruses and its transmission dynamics in nature, we tested 53 bats for active hantaviral infection from specimens collected in Southeastern Brazil. Part of the hantaviral S segment was amplified from the frugivorous Carollia perspicillata and the common vampire bat Desmodus rotundus. DNA sequencing showed high similarity with the genome of Araraquara orthohantavirus (ARQV), which belongs to one of the more lethal hantavirus clades (Andes orthohantavirus). ARQV-like infection was detected in the blood, urine, and organs of D. rotundus. Therefore, we describe a systemic infection in Neotropical bats by a human pathogenic Hantavirus. We also propose here a schematic transmission dynamics of hantavirus in the study region. Our results give insights to new, under-appreciated questions that need to be addressed in future studies to clarify hantavirus transmission in nature and avoid hantavirus outbreaks.
Similar content being viewed by others

Introduction
Hantaviruses (family Hantaviridae) are membrane-enveloped viruses. Their genomes are small and consist of three segmented negative single-stranded RNAs designated large (L), medium (M), and small (S). These segments encode the virus polymerase, glycoproteins (Gn and Gc) and the nucleocapsid (N) protein, respectively1,2. Within natural hosts, hantaviruses do not cause apparent pathogenic effects3. However, transmission to humans can lead to severe diseases and death4,5,6; especially in South America, where hantavirus lethality rate reaches more than 40%7.
Hantaviruses are predominantly rodent-borne pathogens, although insectivore-bat-associated hantaviruses have been reported in the last decade6,8. Until 2012, only two human pathogenic hantaviruses have been reported in bats. The first report found viral antigens and isolated Hantaan orthohantavirus (HTNV) from two broadly distributed insectivorous species: Eptesicus serotinus and Rhinolophus ferrumequinum9. Recently, evidence of a lethal genotype of Andes orthohantavirus (ANDV), Araraquara orthohantavirus (ARQV), was documented by de Araujo et al.10 and Sabino-Santos Jr et al.11 among several Neotropical bats in Brazil. All the remaining reports of Hantavirus infection in bats involved genotypes not known to be pathogenic to humans.
Bats (order: Chiroptera) have highly social behavior that favors the maintenance, evolution, and dissemination of pathogenic viruses12,13. Bats are ranked second when considering top reservoirs of zoonotic viruses, and they host more zoonotic viruses per species than the first place order, rodents14,15. Therefore, the understanding of how these pathogens are transmitted and cause disease in humans is important in public health. Here, we show a systemic infection of hantavirus in Neotropical bats for the first time.
Results
Bats sampling and hantaviral detection
We caught 270 bats from February 2012 to April 2014 with a sampling effort of 56,160 m2h, in southeastern Brazil (Fig. 1). In accordance with legal and ethical statements, whole blood from 53 of the 270 captured bats were screened for antibodies by indirect ELISA using a recombinant N protein of ARQV as antigen11,16,17. Nine phyllostomids bats representing seven species had antibodies to hantaviral N protein antigen and were selected for RT-PCR: Artibeus lituratus (n = 1), A. obscurus (n = 1), A. planirostris (n = 1), Carollia perspicillata (n = 1), Chiroderma villosum (n = 1), Chrotopterus auritus (n = 1), and Desmodus rotundus (n = 3) (for more details see Sabino-Santos Jr et al.)11. Blood, excreta (feces and urine), saliva and organs (heart, liver, kidneys, lungs, and spleen) of all seropositive animals had RNA extracted and screened. Hantaviral RNA was detected only in the blood of two phyllostomids species: the frugivorous Carollia perspicillata and the sanguivorous common vampire bat Desmodus rotundus (Figure S2 and Table S1). In the vampire bat, we amplified a partial S-fragment from the urine, heart, liver, lungs, spleen, and kidneys (Table S1 and Figure S1). Both bats were captured in a riparian forest surrounded by sugarcane monoculture in Batatais County, at sampling point number 3 (Fig. 1A).

Study area, sampling points in Southeastern Brazil; Desmodus rotundus range and one tested bat. (A) The map highlights the five ecologically distinct trap sites in the Northeast of São Paulo State and North region of Minas Gerais State. In green, areas with native vegetation. (B) Distribution area of the common vampire bat D. rotundus34. (C) Photo of D. rotundus captured and tested in the study labeled as GSJ174. Bat picture was taken by Maia FGM. This map was created by Muylaert RL and Sabino-Santos Jr G, and generated using ArcGIS 9 version 9.1 (Environmental Systems Research Institute, USA).
Evidence of active hantaviral infection
We assayed all organ tissues of the Neotropical bats C. perspcillata and D. rotundus by an immuno-histochemistry assay (IHC) detecting the viral nucleocapsid protein. Active infection was suggested in the organs of only D. rotundus. The monoclonal antibody to N protein of HTNV showed an intense cytoplasmic staining in myocardial cells and hepatocytes (Fig. 2B,C,E and F). Organ fragments of Desmodus rotundus were lysed and submitted to western blot (as described in Material and Methods section). Hantavirus nucleocapsid protein, the most synthesized viral protein, was found in all tested organs (except kidneys and liver) from the infected vampire bat, which indicates viral replication (Figs 3, S3).

Immuno-histochemistry (IHC) hantavirus-infected tissues of Desmodus rotundus. (A) Fragment of heart tissue, used as negative control, from a non-infected D. rotundus immune-staining for the virus nucleocapsid in striated cardiac muscle cells. (B) Fragment of heart tissue of D. rotundus showing positive brown immune-staining for the virus nucleocapsid in striated cardiac muscle cells. (C) Represent inset of the same heart section in a higher magnification of a definite area from Fig. 2B. (D) Fragment of liver tissue, used as negative control, from a non-infected D. rotundus immune-staining for the virus nucleocapsid in hepatocyte cells. (E) Liver tissue of D. rotundus showing positive brown immune-staining for the hantavirus nucleocapsid in hepatocyte cells. (F) Represent inset of the same liver tissue section in a higher magnification of a positive area in Fig. 2E. IHC was counterstained with hematoxylin.

Organs tissues lysate from Desmodus rotundus.(A) Negative control and (B) infected organs were lysed as described in Material and Methods and submitted to western blot analyses. Tissue samples were equalized according to the total amount of protein shown by Ponceau (lower pannels) and viral protein was detected using a polyclonal anti-hantavirus hyperimmune murine ascitic fluid against N protein of ARQV (upper panels). *is showing detection of N protein in infected sample.
Phylogenetic relationship of hantaviruses detected
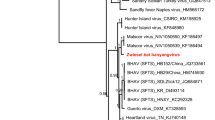
To determine phylogenetic relationships of the hantaviruses detected in D. rotundus and C. perspicillata, a phylogenetic tree based on partial S segment sequences was inferred using Maximum Likelihood (ML) (Fig. 4). In the tree, eight well-supported clades corresponded to already established hantavirus groups: Sigmodontinae, Neotominae, Arvicolinae, Murinae, Soricomorpha, Talpinae, Soricidae and Chiroptera18. Surprisingly, the phylogenetic analysis revealed that the strain detected in C. perspicillata and D. rotundus formed a monophyletic clade closely related to the ARQV variant of ANDV instead of grouping with the “Chiroptera clade,” as shown in Fig. 4. Additionally, the sequence from D. rotundus and C. perspicillata showed over 94% and 99% identity with ARQV respectively, compared to 43.4% averaged identity with hantavirus in the “Chiroptera clade” (Table 1).

Maximum likelihood tree based on partial S segment nucleotides (~264 bp) showing evolutionary relationships of hantaviruses. The scale bar indicates evolutionary distance in numbers of substitutions per nucleotide substitutions per site, and the prime bootstrap support levels were designated. Phylogenies are midpoint rooted for clarity of presentation. Colors on hantaviruses branches indicate viruses described as human pathogen or unknown, as shown in the legend. Hantavirus sequenced in this study is highlighted with green color.
Discussion
Viruses are the most abundant biological entities on the planet Earth, and those originating in animal world cause most of emerging diseases of humans14,15,19. Viruses from bats can become zoonoses that are transmitted to domestic animals and humans presumably due to their ability to fly vast distances disseminating these viruses15. In our study, we found hantaviral RNA closely related to ARQV in urine of the common vampire bat Desmodus rotundus. This vampire bat could be transmitting ARQV-like through contaminated aerosol of its urine that could be inhaled thus infecting other animals20. However, this transmission mechanism should be confirmed by isolation of the hantavirus from urine to investigate if the pathogen shedding by the vampire bat is viable and infectious21.
Lungs are a primary target of infection in rodents and humans since the first detection of HTNV in lung tissue of the striped field mouse (Apodemus agrarius) and bats9,22; although, hantaviruses can also be found in other organs12,23,24. In the present study, we detected the N protein in almost all organs of D. rotundus, particularly in heart and liver, indicating virus replication (Figs 2 and 3). Similarly, other studies have shown liver as a target of infection12,25,26. ARQV-like RNA was present in the blood of the frugivorous bat Carollia perspicillata, and in almost all organs and secretions of the common vampire bat Desmodus rotundus (Figures S1 and S2). This suggests that the vampire bat could be chronically infected, as observed in rodents and soricomorphs6,20.
Although compelling evidence is provided that the vampire bat had viral RNA and antigen in many tissues, this could merely be a spillover infection from ARQV’s natural host, Necromys lasiurus (hairy-tailed bolo mouse). Guo et al.18 suggested that hantaviruses originated in bats and then dispersed among other mammalian reservoirs. However, due to the apparently common evolutionary history of bats with insectivores (shrews and moles), it is more likely that insectivores were the original reservoirs, and from these the virus has spread to bats and rodents6,27,28. There are reports of Sigmondontinae rodents infected in this same trap site region as the bats in the current study7,29,30. It is thought that small rodents may feed in the monocultures, but not necessarily nest in these monocultures. These rodents are more likely to live in the fragments where the vegetation is most preserved, thus sharing the same environment as bats, which depend on natural landscapes to survive environmental degradation31,32,33. D. rotundus feeds on blood of large and medium-sized mammals, including livestock, feral pigs, tapirs and red brocket deer. The bats potentially “take a ride” with the animals and disperse over long distances34,35. D. rotundus is broadly distributed over Latin America and in Brazil in particular, and considered commensal with humans and animals (Fig. 1B). Therefore, gathering our results with those already published, we propose a scheme illustrating the dynamics of transmission of hantavirus in nature in the study region (Fig. 5). In this way, as soricomorphs (shrews and moles) are not present in South America, infected bats could transmit hantavirus to rodents through their excreta. However, viremia of viruses in bats is believed to be low36. On the other hand, it is possible that hantaviruses are still in the stage of adaptation in rodents, as they still suffer a fitness cost3 in which case they would have become infected from bats as suggested by Guo et al.18. In our scenario, rodents would be acting as amplifying hosts where viruses would replicate, increase their load, and then be transmitted to humans via aerosols from their excreta (Fig. 5). Nonetheless, despite low viremia, direct transmission from vampire bats cannot be ruled out since they are in constant contact with humans (Fig. 5). Thus, there are still some questions that need to be addressed. More studies such as a large scale ecological and evolutionary survey of bats for ARQV may clarify how the virus managed to get into a vampire bat, how these bats transmit it, and ultimately if bats can serve as potential reservoirs of ARQV.

A schematic figure illustrating the transmission dynamics of hantavirus, in nature, in the study region, southeastern Brazil.
In short, we show herein that Neotropical bats Carollia perspicillata and Desmodus rotundus can be naturally infected with hantavirus in southeastern Brazil. In our survey, we found the circulating strain to be closely related to ARQV, one of the most virulent and lethal among all hantaviruses in humans. The N protein and/or the partial genome of an ARQV-like were detected in different organs and in the urine of the common vampire bat suggesting that this animal may play a role in hantavirus transmission and dissemination in the study region. These results show that bats are probably playing an under-appreciated part on the maintenance, circulation, and transmission of hantavirus in nature. This report expands the information available to public health authorities for establishing mitigation and surveillance strategies and understanding Hantavirus epidemiology and outbreak risks.
Material and Methods
Legal and Ethical Statements
Captured bats were handled following the guidelines of the American Society of Mammalogists for the use of wild animals in research and biosafety recommendations for working with animals potentially infected with hantavirus37,38. All procedures and protocols performed in the field as part of this study are in accordance with Brazilian environmental and wildlife protection laws and regulations. The study has been approved by the Brazilian Ministry of Environment (protocols 19838-5 and 41709-3). It has also been approved by the Ethics Committee for Animal Research of the University of São Paulo (020/2011) and the Federal University of Minas Gerais (333/2013). The research was approved by state laws (licenses IF-SP/COTEC 260108-007.043 and IEF-MG 012/2014). All laboratory procedures, until viral inactivation, were conducted in our BSL-3 + facility.
Study areas and bat sampling
Bats were captured at five ecologically distinct sites in the northeast region of Sao Paulo State and northern region of Minas Gerais State, in southeastern Brazil (Fig. 1A). Site 1: Jatai Ecological Station, 21°37′ 16″S and 47°49′ 12″W, located at Luis Antonio county, encompasses an area of 9,000 ha and currently is the largest protected area in Sao Paulo State, with continuous cerrado vegetation (a Brazilian savanna-like vegetation) in addition to small enclaves of semideciduous forest patches. Site 2, located in Cajuru county, 21°16′ 31″S and 47°18′ 15″W, with most of the original vegetation (cerrado) converted into mono-specific cultivars. Site 3, located in Batatais county, 20°53′ 28″S and 47°35′ 06″W, is characterized by large sugar cane plantations with small patches of other types of vegetation. Site 4 was located at Sapucaia Ecological Park, which has an area of 37.66 ha and karst topography. Site 5 was located at Lapa Grande Ecological Station with an area of 9,600 ha, where a high concentration of caves and shelters can be found. Both sites 4 and 5 are located in Montes Claros county, 16°43′ 41″S and 43°51′ 54″W, and have a predominant vegetation of cerrado with patches of deciduous seasonal forest (dry forest), and stretches of transition to caatinga (a Brazilian desert-like vegetation). Field trips were conducted from February 2012 to April 2014. Places were sampled twice: in dry season (April to September) and in rainy season (October to March). We used 12 mist-nets in sites 1–3 and 6 mist-nets in sites 4 and 5 (model 716/12 P, 12 × 2,5 m; denier 75/2, 16 × 16 mm net; Ecotone, Gdynia, Poland). Bats were identified following standard ecological taxonomic keys34,39,40 and supplemented when needed by molecular data (e.g., based on a fragment of cyt-b gene)41,42. Vouchers specimens positive for hantaviral RNA, D. rotundus and C. perspicillata, were deposited in the Collection of Mammals from the Center for Taxonomic Collections from the Federal University of Minas Gerais, under numbers: 6948 and 6949, respectively. Saliva and excreta (feces and urine) were collected from all captured bats. According to ethical statements, only one specimen per species per trap-night could be anesthetized and euthanized. Thus, using Halothane TM (Sigma-Aldrich, USA) selected bats were anesthetized, and had blood collected by cardiac puncture. Next, bats were euthanized by deepening anesthesia and had organs collected: spleen, liver, kidneys, lungs, and heart. Organs were collected using sterilized tweezers, one for each specific organ to avoid cross-tissue contamination, washed with sterilized nuclease-free water. One-half of each sample was stored in cryovials with cell culture medium (DEMEM supplemented with 10% fetal calf serum, 1% antibiotic and antimycotic, and 15% nuclease free glycerol) for viral molecular screening, RT-PCR (Reverse Transcription-Polymerase Chain Reaction). The other half sample was placed into cryovials containing paraformaldehyde 4% (Merck, Germany), for immuno-histochemistry assays. All samples, except those in 4% paraformaldehyde, were flash-frozen in liquid nitrogen.
RNA extraction, purification, and sequencing
The RNA was extracted from plasma, feces, urine, and tissues of bats with the QIAamp Viral RNA Mini Kit (QIAGEN, Germany), according to the manufacturer’s instructions. Tissues were homogenized in TissueLyser II (QIAGEN, Germany) as recommended by manufacturer. RNA extracts were quantified in NanoDrop ND1000 (USA) spectrophotometer. For cDNA synthesis, we used the High-Capacity cDNA Reverse Transcription Kit (Life Technologies, USA) following the manufacturer’s instructions. PCRs were performed according to Moreli and others43, for further purification and sequencing. PCR products were purified with ExoSap-TI (Affymetrix, USA) and used for nucleotide sequencing in an Applied Biosystems® - 3500 Genetic Analyzer (Thermo Fisher, USA).
Control plasmids
Rio Mamore orthohantavirus (strain HTN-0007-TPV 4645) were amplified as previously described43. These PCR products (~ 324 bp and ~ 264 bp for Gn and S, respectively) were purified and cloned into pGEM-T Easy Vector (Promega Corporation, WI, USA), according to manufacturers’ guidelines, and used to transform DH10β E. coli cells. Colonies of bacteria were selected in LB medium supplemented with ampicillin 100 ng/ul and confirmed by PCR using same primers of product amplification. Plasmid preparation, from positive clone colonies, was isolated with Plasmid Mini Kits (QIAGEN, Germany) and used as positive controls for PCR reactions.
Immuno-histochemistry assay
To determine the presence of the structural nucleocapsid protein of ARQV in tissue samples from PCR positive bats a protocol previously standardized was performed44. Tissues from bat organs fixed in 4% paraformaldehyde (Benchekroun et al., 2004) were dehydrated in increasing concentration ethanol solutions (50%, 70%, 80%, 90%, and 100%). Then, followed by xylene (1:1 xylene: ethanol and 100% xylene), embedded in paraffin, and 4 μm sections of tissue were obtained in a Leica microtome (Leica RM2125RTS, Heidelberger, BW, Germany). Slides were mounted with Entellan (Merck, Germany). Antigen retrieval in tissue slides was conducted by immuno-histochemistry. First, we used a monoclonal antibody (1:300 dilution in PBS/BSA 1%) against the N protein of HTNV (ab34757, Abcam plc, Cambridge, UK). The tissue in the slide had endogenous peroxidase blocked by 10% hydrogen peroxide and was incubated in humidified chamber at room temperature, for 60 min, washed with PBS, and incubated for 30 minutes with a biotinylated secondary horse antibody (1:300 dilution in PBS/BSA 1%) (BA-2000) (Vectastin® ABC Kit, Vector Laboratories, Burlingame, CA, USA) at room temperature (RT). The signal amplification was obtained by incubation with a polymer conjugated with streptavidin-peroxidase (s2438, Sigma-Aldrich, USA), at RT. Samples were counterstained with Harris hematoxylin (Vector). Positive staining was visualized by Nova Red peroxidase system with Vector Nova Red Substrate kit (SK-4800, Vector Laboratories, USA). Based on comparison with infected HeLa cells as positive control and uninfected heart tissues of Desmodus rotundus used as negative control, tissues showing red-brown cytoplasmic staining were considered as positive for hantaviral antigen.
Western blot
Tissues were incubated in a lysis buffer [10 nM Tri-HCl (pH 8.0), 50 mM NaCl, 50 mM NaF, 1% Triton X-100, 0,1% SDS and 6 mM sodium deoxycholate] supplemented with a protease inhibitor mixture (Sigma-Aldrich, USA). Simultaneous disruption and homogenization were achieved through a high speed shaking of samples in micro-centrifuge tubes with stainless steel beads using TissueLyser II equipment (QIAGEN, Germany). Each of ten cycles of high-speed shaking, samples were followed by ice incubation for 1 min. The supernatant was collected after the lysates were centrifuged at 16.000 g for 20 min at 4 °C to remove tissue debris. Samples were mixed with loading buffer [4% SDS, 160 mM Tris-HCl (pH 6.8), 20% glycerol and 0,1% bromophenol blue] and boiled at 95 °C for 5 min. Proteins were resolved by SDS-PAGE and stained with Coomassie blue; and densitometry analyses of protein bands were calculated to be equalized to same amounts using Image Lab™ Software (Bio-Rad Laboratories, USA). Proteins were resolved by SDS-PAGE and transferred onto a nitrocellulose membrane (Millipore, USA), and blocked with PBS-T (PBS and 0,1% Tween 20) and non-fat dry milk, for one h. Primary antibody (1:500 dilution in PBS/BSA 1%), a hyperimmune murine anti-hantavirus ascitic fluid against the recombinant N protein of ARQV, was incubated overnight at 4 °C. After three washes with PBS-T, membranes were incubated with HRP-conjugated antibody anti-mouse (1:2000 dilution in PBS/BSA 1%) for one h at RT. After washing out the antibodies, proteins were detected by enhanced chemiluminescence ECL (GE Healthcare, USA).
Phylogenetic analysis
Maximum likelihood (ML) phylogenetic tree was constructed using partial S sequences obtained from Desmodus rotundus (urine, blood, and heart) and Carollia perspicillata (blood) and an additional 47 nucleotides of S segment from representative hantaviruses. Multiple sequence alignment (MSA) was conducted by using combinations of Mafft v.745 and Muscle v.3.746. Phylogenies were inferred with IQ-TREE version 1.4.3 software that automatically obtained the best-fit model based on Bayesian Information Criterion. All segments were analyzed under the GTR + I + G4 model substitution model with 1,000 replicates47. Statistical supports for individual nodes were estimated using bootstrap value. The phylogenetic tree was visualized using the FigTree software v.1.4.2. The identity distance for the partial S segment was calculated using Genious 9.1.6 (Biomatters, Auckland, New Zealand). All sequences generated in this study can be retrieved from GenBank under accession numbers MF094267, for D. rotundus and MF094269 for C. perspicillata.
References
Jonsson, C. B., Figueiredo, L. T. M. & Vapalahti, O. A global perspective on hantavirus ecology, epidemiology, and disease. Clin. Microbiol. Rev. 23, 412–441 (2010).
Adams, M. J. et al. Changes to taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses. rch. Virol. 162, (2017).
Kallio, E. R. et al. Endemic hantavirus impairs the winter survival of its rodents host. Ecology 88, 1911–1916 (2007).
Rasmuson, J. et al. Time to revise the paradigm of hantavirus syndromes? Hantavirus pulmonary syndrome caused by European hantavirus. Eur. J. Clin. Microbiol. Infect. Dis. 30, 685–690 (2011).
Gizzi, M. et al. Another case of ‘european hantavirus pulmonary syndrome’ with severe lung, prior to kidney, involvement, and diagnosed by viral inclusions in lung macrophages. Eur. J. Clin. Microbiol. Infect. Dis. 32, 1341–1345 (2013).
Yanagihara, R., Gu, S. H., Arai, S., Kang, H. J. & Song, J. W. Hantaviruses: Rediscovery and new beginnings. Virus Res. 187, 6–14 (2014).
de Figueiredo, G. G. et al. Diagnosis of hantavirus infection in humans and rodents in Ribeirão Preto, State of São Paulo, Brazil. Rev. Soc. Bras. Med. Trop. 43, 348–354 (2010).
Razzauti, M., Plyusnina, A., Sironen, T., Henttonen, H. & Pyusnin, A. Analysis of Puumala hantavirus in a bank vole population in northern Finland: Evidence for co-circulation of two genetic lineages and frequent reassortment between strains. J. Gen. Virol. 90, 1923–1931 (2009).
Kim, G. R., Lee, Y. T. & Park, C. H. A new natural reservoir of hantaviruses: isolation of hantaviruses from lung tissue of bats. Arch. Virol. 134, 85–95 (1994).
de Araujo, J. et al. Detection of hantavirus in bats from remaining rain forest in São Paulo, Brazil. BMC Res. Notes 5, 690 (2012).
Sabino-Santos, G. et al. Evidence of hantavirus infection among bats in Brazil. Am. J. Trop. Med. Hyg. 93, (2015).
Sumibcay, L. et al. Divergent lineage of a novel hantavirus in the banana pipistrelle (Neoromicia nanus) in Côte d’Ivoire. Virol. J. 9, 34 (2012).
Weiss, S. et al. Hantavirus in Bat, Sierra Leone. Emerg. Infect. Dis. 18, 159–161 (2012).
Luis, A. D. et al. A comparison of bats and rodents as reservoirs of zoonotic viruses: are bats special? Proc. R. Soc. B Biol. Sci. 280, 20122753 (2013).
Brook, C. E. & Dobson, A. P. Bats as ‘special’ reservoirs for emerging zoonotic pathogens. Trends Microbiol. 23, 172–180 (2015).
Figueiredo, L. T. M. et al. Expression of a hantavirus N protein and its efficacy as antigen in immune assays. Brazilian J. Med. Biol. Res. 41, 596–599 (2008).
Figueiredo, L. T. M. et al. Evaluation of an enzyme-linked immunosorbent assay based on Araraquara virus recombinant nucleocapsid protein. Am. J. Trop. Med. Hyg. 81, 273–276 (2009).
Guo, W. P. et al. Phylogeny and Origins of Hantaviruses Harbored by Bats, Insectivores, and Rodents. PLoS Pathog. 9, (2013).
Luis, A. D. et al. Network analysis of host-virus communities in bats and rodents reveals determinants of cross-species transmission. Ecol. Lett. 18, (2015).
Easterbrook, J. D. & Klein, S. L. Immunological mechanisms mediating hantavirus persistence in rodent reservoirs. PLoS Pathog. 4, (2008).
Kallio, E. R. et al. Prolonged survival of Puumala hantavirus outside the host: Evidence for indirect transmission via the environment. J. Gen. Virol. 87, 2127–2134 (2006).
Lee, H. W., Lee, P. W. & Johnson, K. M. Isolation of the etiologic agent of Korean Hemorrhagic fever. J. Infect. Dis. 137, 298–308 (1978).
Brummer-Korvenkontio, M. et al. Nephropathia epidemica: detection of antigen in bank voles and serologic diagnosis of human infection. J. Infect. Dis. 141, 131–134 (1980).
Nichol, S. T. et al. Genetic identification of a hantavirus associated with an outbreak of acute respiratory illness. Science (80-.). 262, 914–918 (1993).
Arai, S. et al. Molecular phylogeny of a genetically divergent hantavirus harbored by the Geoffroy’s rousette (Rousettus amplexicaudatus), a frugivorous bat species in the Philippines. Infect. Genet. Evol. 45, 26–32 (2016).
Kang, H. J., Bennett, S. N., Hope, A. G., Cook, Ja & Yanagihara, R. Shared ancestry between a newfound mole-borne hantavirus and hantaviruses harbored by cricetid rodents. J. Virol. 85, 7496–7503 (2011).
Bennett, S. N., Gu, S. H., Kang, H. J., Arai, S. & Yanagihara, R. Reconstructing the evolutionary origins and phylogeography of hantaviruses. Trends Microbiol. 22, 473–482 (2014).
Plyusnin, A. & Sironen, T. Evolution of hantaviruses: Co-speciation with reservoir hosts for more than 100MYR. Virus Res. 187, 22–26 (2014).
Sabino-Santos, G. Jr. Detection of hantavirus in wild rodents and study on its populations dynamics in the Northeastern region of São Paulo State, Brazil. (2010).
Sobreira, M. et al. A serosurvey for hantavirus infection in wild rodents from the states of Rio de Janeiro and Pernambuco, Brazil. Acta Trop. 107, 150–152 (2008).
Gheler-Costa, C., Vettorazzi, C. A., Pardini, R. & Verdade, L. M. The distribution and abundance of small mammals in agroecosystems of southeastern Brazil. Mammalia 76, 185–191 (2012).
Gheler-Costa, C. et al. The effect of pre-harvest fire on the small mammal assemblage in sugarcane fields. Agric. Ecosyst. Environ. 171, 85–89 (2013).
Ripperger, S. P., Kalko, E. K. V., Rodríguez-Herrera, B., Mayer, F. & Tschapka, M. Frugivorous bats maintain functional habitat connectivity in agricultural landscapes but rely strongly on natural forest fragments. PLoS One 10, 1–15 (2015).
Greenhall, A. M., Joermann, G. & Schmidt, U. Desmodus rotundus. Mammalian Species 1–6 (1983).
Galetti, M., Pedrosa, F., Keuroghlian, A. & Sazima, I. Liquid lunch??? vampire bats feed on invasive feral pigs and other ungulates. Front. Ecol. Environ. 14, 505–506 (2016).
Moratelli, R. & Calisher, C. H. Bats and zoonotic viruses: Can we confidently link bats with emerging deadly viruses? Mem. Inst. Oswaldo Cruz 110, 1–22 (2015).
Mills, J. N. et al. Guidelines for Working With Rodents Potentially Infected With Hantavirus. J. Mammal. 76, 716–722 (1995).
Sikes, R. S. & Gannon, W. L. Guidelines of the American Society of Mammalogists for the use of wild mammals in research. J. Mammal. 92, 235–253 (2011).
Reis, N. R. dos, Peracchi, A. L., Pedro, W. A. & Lima, I. P. De. Morcegos do Brasil. Biblioteca Central da Universidade Estadual de Londrina. 1017/CBO9781107415324.004. (2007).
Miranda, J. M. D., Bernardi, I. P. & Passos, F. C. Chave ilustrada para a determinação dos morcegos da Região Sul do Brasil. Biblioteca Central da Universidade Estadual de Londrina (2011).
Larsen, P. A. et al. Phylogenetics and Phylogeography of the Artibeus jamaicensis Complex Based on Cytochrome- b DNA Sequences. J. Mammal. 88, 712–727 (2007).
Larsen, R. J. et al. Evolutionary history of Caribbean species of Myotis, with evidence of a third Lesser Antillean endemic. Mamm. Biol. 77, 124–134 (2012).
Moreli, M. L., Moro De Sousa, R. L. & Figueiredo, L. T. M. Detection of Brazilian hantavirus by reverse transcription polymerase chain reaction amplification of N gene in patients with hantavirus cardiopulmonary syndrome. Mem. Inst. Oswaldo Cruz 99, 633–638 (2004).
Santos, R. I. et al. Experimental infection of suckling mice by subcutaneous inoculation with oropouche virus. Virus Res. 170, 25–33 (2012).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Edgar, R. C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A. & Minh, B. Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Acknowledgements
We appreciate the help and attention of Thiago Neves for all the support in Montes Claros County and field guidance at Sapucaia and Lapa Grande Ecological Parks. We are also grateful to Vinicius Kavagutti, Márcio Schafer, Milene Eigenher, Ariane and Gustavo Crepaldi de Morais for their help in field campaigns. We appreciate all support from the Jatai Ecological Station Manager Edison Montilha; Armando Nascimento who supported us on his farm Santa Gabriela (Batatais County), and José Teotônio (Zezinho) for all support on his farm in Cajuru County and the Secretary of Health from Cajuru city through the official Toninho. We are grateful to Emily Rae Hannon, a native English speaker, for proofread the manuscript. São Paulo State Foundation for Supporting Research (FAPESP) grant nos.: 11/06810-9 and 16/02568-2 to GSSJ; 11/22663-6 to FGMM; 13/06380-0 to RBMJ; 12/03700-0 to TBG; 12/24150-9 to WMS; 15/06142-7 to NBS; 12/04096-0 to RLM; 15/05354-0 to MCP; 11/19897-5 to LKSL; 14/02438-6 to LLPS, EA, and LTMF. Minas Gerais State Foundation for Supporting Research (FAPEMIG) grant no.: APQ-00606-14 to TMV.
Author information
Authors and Affiliations
Contributions
Conception and design of the study: G.S.S.J. and F.G.M.M. Acquisition of data: G.S.S.J., F.G.M.M., R.B.M.J., T.B.G., R.L.M., L.K.S.L., R.S.C., D.M.M., T.M.V., M.C.P., N.B.S., and P.R.M.Z. Analysis and interpretation of data: G.S.S.J., F.G.M.M., R.B.M.J., T.B.G., R.L.M., W.M.S., L.K.S.L., R.S.C., D.M.M., N.B.S., L.L.P.S., E.A., and L.T.M.F. Drafting the manuscript: G.S.S.J., F.G.M.M., R.B.M.J., T.B.G., W.M.S., L.K.S.L., D.M.M., N.B.S., M.C.P., C.B.J., D.G., J.S.B., and L.T.M.F. Contributed with reagents and equipments: G.S.S.J., C.B.J., D.G., J.S.B., N.M.V., L.L.P.S., E.A., and L.T.M.F. All authors revised and agreed with the final version of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sabino-Santos Jr, G., Maia, F.G.M., Martins, R.B. et al. Natural infection of Neotropical bats with hantavirus in Brazil. Sci Rep 8, 9018 (2018). https://doi.org/10.1038/s41598-018-27442-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-27442-w
This article is cited by
-
A novel hantavirus identified in bats (Carollia perspicillata) in Brazil
Scientific Reports (2024)
-
Modeling potential risk areas of Orthohantavirus transmission in Northwestern Argentina using an ecological niche approach
BMC Public Health (2023)
-
A review of the diet of the common vampire bat (Desmodus rotundus) in the context of anthropogenic change
Mammalian Biology (2023)
-
Lack of molecular evidence of fecal-borne viruses in capybaras from São Paulo state, Brazil, 2018–2020: a minor public health issue
Brazilian Journal of Microbiology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
