
Abstract
Similar to the onset phase of inflammation, its resolution is a process that unfolds in a manner that is coordinated and regulated by a panel of mediators. Lipoxin A4 (LXA4) has been implicated as an anti-inflammatory, pro-resolving mediator. We hypothesized that LXA4 attenuates or prevents an inflammatory response via the immunosuppressive activity of Stem Cells of the Apical Papilla (SCAP). Here, we report for the first time in vitro that in a SCAP population, lipoxin receptor ALX/FPR2 was constitutively expressed and upregulated after stimulation with lipopolysaccharide and/or TNF-α. Moreover, LXA4 significantly enhanced proliferation, migration, and wound healing capacity of SCAP through the activation of its receptor, ALX/FPR2. Cytokine, chemokine and growth factor secretion by SCAP was inhibited in a dose dependent manner by LXA4. Finally, LXA4 enhanced immunomodulatory properties of SCAP towards Peripheral Blood Mononuclear Cells. These findings provide the first evidence that the LXA4-ALX/FPR2 axis in SCAP regulates inflammatory mediators and enhances immunomodulatory properties. Such features of SCAP may also support the role of these cells in the resolution phase of inflammation and suggest a novel molecular target for ALX/FPR2 receptor to enhance a stem cell-mediated pro-resolving pathway.
Similar content being viewed by others


Introduction
The role of inflammation in tissue regeneration is multi-faceted. According to current thinking, early pro-inflammatory signaling is detrimental while anti-inflammatory signaling may be beneficial for stem cell activity1. In the presence of an inflammatory environment (e.g., high levels of tumor necrosis factor α (TNF-α) and interferon-γ (IFN-γ)), mesenchymal stem cells (MSC) may be activated and assume immunosuppressive functions by secreting high levels of anti-inflammatory soluble factors (e.g., indoleamine-pyrrole 2,3-dioxygenase, prostaglandin E2, nitric oxide, transforming growth factor β, heme-oxygenase2). In the absence of an inflammatory environment (i.e., low levels of TNF-α and IFN-γ), MSC adopt a pro-inflammatory phenotype, thus enhancing the immune response by secreting chemokines that recruit leukocytes to a site of tissue injury (e.g., chemokine ligands such as CCL3, 4, 5, CXCL9, and CXCL10)3,4.
Similar to the onset phase of inflammation, its resolution is coordinated and regulated by a panel of mediators including specialized pro-resolving lipid mediators. These mediators are derived from polyunsaturated fatty acids and include lipoxins, maresins, resolvins and protectins5.
Lipoxin A4 (LXA4), for example, is a pro-resolving mediator secreted by immune cells such as neutrophils and macrophages. Lipoxins regulate functions of the innate immune system and also modulate the adaptive immune system by decreasing memory B-cell responses6. These actions are mediated by the activation of ALX/FPR2 receptor, a specific G-protein-coupled receptor that binds LXA4 with high affinity7.
Stem Cells from the Apical Papilla (SCAP) are dental mesenchymal stem cells characterized by their pluripotency and ability to differentiate into several cell-restricted lineages8. Under specific conditions in vitro, SCAP are reported to differentiate in functional dentinogenic cells, which are capable of producing typical dentin-like structures9,10; these findings suggest that SCAP could be a potential source of odontoblast progenitors. Similar to bone marrow mesenchymal stem cells, SCAP appear to possess immunomodulatory properties. For example, SCAP are able to inhibit T-cell proliferation in a mixed lymphocyte reaction11. Cell-cell contact and/or paracrine mechanisms are putatively involved, however, the exact mechanism of this interaction is currently unknown. Under clinical conditions, reparative efforts of odontoblast-like cells occur in inflammatory conditions. Therefore, we hypothesized that LXA4 may attenuate or prevent the inflammatory response via the immunosuppressive activity of SCAP. The aim of our study, therefore, was to investigate interactions of LXA4 with SCAP, while focusing on the role of LXA4 binding to ALX/FPR2 receptor.
Results
SCAP maintain stem cell properties and share similar surface markers with periodontal ligament cells (PDLC)
SCAP and PDLC were isolated from different donors (Fig. 1A). Both cell populations presented typical homogeneous fibroblast-like morphology and were able to generate fibroblast-like colonies from single cells after 8 to 12 days of culture (Fig. 1B). Morphological features of fresh and post-thaw cells were similar. To characterize the SCAP population in vitro, we performed flow-cytometric analysis of mesenchymal stem cell surface markers. Both SCAP and PDLC expressed specific MSC antigens (CD90, CD105, CD146 and STRO-1) and were negative for a hematopoietic marker, CD45 (Fig. 1C). Next, we sought to determine whether SCAP would undergo osteogenic and chondrogenic differentiation. Under osteo-inductive conditions for 3 weeks, SCAP produced mineralized extracellular matrix that was stained with alizarin red. After 21 days in chondrogenic conditions, deposition of chondrogenic-like matrix was revealed by alcian blue staining. Results were essentially the same at different passages (3 and 9) (see Fig. 1D). To demonstrate any impact of passaging on SCAP and PDLC viability and on expansion capacity, a trypan blue exclusion test was done and population doubling times were calculated. Cell viability in SCAP and PDLC was comparable and high from passages 1 to 8 (Fig. 1E). Cell doubling time was stable (around 48 h) and similar between SCAP and PDLC from passage 1 to 8 (Fig. 1F). In order to obtain more purified populations, SCAP sorted with immunomagnetic separation using STRO-1, CD73, CD90 and CD105 were characterized and compared with non-purified (mixed) SCAP and human gingival fibroblasts (Supplementary File S1). There was no effect of SCAP passages on in vitro differentiation and cell surface markers (Supplementary File S1A,C. ALX/FPR2 has been recently identified in PDLC12. Thus, in our study, we used PDLC as a positive control in the experiments.

Characterization of stem cells of the apical papilla (SCAP) in comparison with periodontal ligament fibroblast (PDLC). (A) Freshly extracted human third molar. The arrows indicate apical papilla tissue of immature tooth. (B) Generation of fibroblast colonies from single cells after 8 to 12 days of culture. Representative phase contrast microscopic photographs of generation and expansion of SCAP and PDLC. Cells have elongated shapes and grow attached to substrata. Scale bar, 25 μm. (C) Flow cytometry analysis of representative histograms at passage 3 (P3) showed that SCAP and PDLC expressed cell surface human mesenchymal stem markers (CD90, CD105, CD146 and STRO-1) and lacked the expression for leukocyte common antigen (CD45) (in red) compared with their appropriate isotype controls (dash line). (D) Differentiation of SCAP to odontoblast-like and chondrocyte-like phenotype. Unsorted SCAP at passage 3 and 9 were subjected to differentiation media for 2 weeks, which resulted in deposits positive for alizarin red and alcian blue stain, respectively. (E) The cell viability (trypan blue exclusion assay) of SCAP and PDLC was stable and similar from P1 to P8 for both populations. (F) Cell doubling times were stable and similar from P1 to P8 for both populations.
SCAP normally express ALX/FPR2 and this receptor is overexpressed when inflammatory stimuli are applied
In order to explore the roles of the LXA4-ALX/FPR2 axis in SCAP, we investigated the expression of ALX/FPR2 under resting and simulated inflammatory conditions. To demonstrate the surface and intracellular expression of ALX/FPR2, we used flow cytometry of intact and permeabilized cells. Intracellular expression of ALX/FPR2 was higher than surface expression. PDLC and Human peripheral blood mononuclear cells (PBMC) were used as positive controls (Fig. 2A,B). Confocal microscopy confirmed expression at the protein level of ALX/FPR2 in SCAP (Fig. 2C). Then, we investigated the effect of various doses of two inflammatory factors (TNF-α and lipopolysaccharide (LPS)) on ALX/FPR2 expression using flow cytometry. We found that 1 μg/mL of LPS had a maximal inductive effect in SCAP at 24 hours, as shown by flow cytometric analysis. Only the highest dose (10 and 100 ng/mL) of TNF-α upregulated the expression of ALX/FPR2 in SCAP at 24 hours. We showed that TNF-α (10 ng/mL) in combination with LPS (1 μg/mL) also upregulated expression of ALX/FPR2 in SCAP at 24 h, but to a lesser degree than LPS alone (1 μg/mL) (Fig. 2D).

Expression of formyl peptide receptor 2 (ALX/FPR2) in SCAP is upregulated under inflammatory condition. (A) Flow cytometry analysis of representative histograms at passage 3 (P3) showed that SCAP and PDLC expressed surface (S) and intracellular (IC) ALX/FPR2. ALX/FPR2 antibody (red) and secondary antibody staining with appropriate isotype controls (dash line) (n=6). (B) Quantification of ALX/FPR2 expression by flow cytometry analysis shown as MFI (Mean Fluorescence Intensity) in SCAP and Peripheral Blood Mononuclear Cells (PBMC). **p < 0.01. (C) Representative confocal images of ALX/FPR2 distribution in permeabilized SCAP and PDLC. No immunostaining was observed in control conditions with an isotype control. Anti-ALX/FPR2 (green), nuclei (blue). Original magnification 43x (n=4). (D) 1 μg/mLof LPS and highest dose of TNF-α upregulated expression of ALX/FPR2 in SCAP at 24h. The expression of ALX/FPR2 was analyzed by flow cytometry using RFI (Relative Fluorescence Intensity). RFI presented here as a ratio between median fluorescence intensity (MFI) from experimental groups (grey histograms) and MFI from control group (black histogram). ***p < 0.001 versus control. (E) Lipopolysaccharide (LPS) stimulation of SCAP led to a significant increase of ALX/FPR2 mRNA expression compared to control after 24h. ALX/FPR2 mRNA expression was quantified by RT-PCR. *p < 0.05 versus control.
Finally, we confirmed by RT-PCR that SCAP express ALX/FPR2. Moreover, SCAPs significantly boosted ALX/FPR2 mRNA expression after they were incubated in LPS (1 μg/mL) for 24 h (Fig. 2E).
LXA4 enhances proliferation and migration of SCAP via ALX/FPR2 receptor
Potential cytotoxic effects of LXA4 in SCAP were evaluated in the absence or presence of LPS using 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assays. LXA4 (1, 10 and 100 nM) and a vehicle did not affect cell viability. A viability decrease was observed after application of 1 μg/mL of LPS only (Fig. 3A). Cells pretreated with LXA4 for 30 min showed no significant increase in viability compared with cells that were treated with LPS only (Fig. 3A). Because SCAP may play a role in resolution of inflammation, their self-renewal and migration capabilities are fundamental to achieve a pathophysiological impact. Therefore, we determined whether these functions could be modulated by LXA4. Proliferation was evaluated by cell count and trypan blue exclusion. At 48 h and 72 h, LXA4 in different doses increased proliferation, and this effect was suppressed by the formyl peptide receptor antagonist, WRW4 (Trp-Arg-Trp-Trp-Trp-Trp-CONH2) (Fig. 3B). Then, in an in vitro wound healing assay to assess migration of SCAP, we found that 1 nM and 100 nM LXA4 significantly augmented migration capacities, accelerating wound closure of SCAP at 24 h and 48 h (Fig. 3C,D). W50 (defined as the time to reduce wound width by 50%) was shortened from 24 to 18 hours when 100 nM of LXA4 was used (Fig. 3E). This effect was abrogated by WRW4 (Fig. 3C–E), which, by itself, did not affect W50 of cells exposed to human serum (Fig. 3F).

ALX/FPR2 dependent stimulation of SCAP cell viability, proliferation, migration by lipoxin A4 (LXA4). (A) MTT method was used to analyze the cell viability. Cell viability was reduced by 11% after cells were incubated in LPS (1 μg/mL) for 24 h, and with no effect on the pretreatment of LXA4 (1, 10 and 100 nM). ***p < 0.001 versus control. (B) Proliferation was evaluated by MTT proliferation test. SCAP exposed to increasing concentrations of LXA4 (0.01–100 nM) for 24, 48, and 72 hours displayed a concentration and time-dependent increment in proliferation, with a maximum at 48 and 72 hours.This effect was suppressed by ALX/FPR2 selective antagonist peptide WRW4 (Trp-Arg-Trp-Trp-Trp-Trp-CONH2). Data are expressed as proliferation index (cell number with LXA4 per cell number with vehicle). *p < 0.05 WRW4 + 0.01 nM LXA4versus 0.01 nM LXA4; **p < 0.01 WRW4 + 1 nM LXA4 versus 1nM LXA4; ***p < 0.001 WRW4 + 100 nM LXA4 versus 100 nM LXA4. (C,D) Analysis of SCAP migration after exposure to LXA4 (1 nM and 100 nM) or vehicle. Human Serum 10%, and human serum 10% +WRW4 were uses as positive controls. Migration was evaluated by a wound scratch healing test. Wound closure was quantified at 3, 6, 24, and 48 hours post-wounding, using ImageJ software. *p < 0.05 **p < 0.01 ***p < 0.001 versus vehicle. (D) Representative images of wound closure at 3, 6, 24, and 48 hours (magnification: x4). (E) Kinetic to reduce wound closure of SCAP treated with LXA4 (100 nM) in comparison with vehicle and with SCAP treated with WRW4 before LXA4 (100 nM). *p < 0.05 LXA4 vs. vehicle; #p < 0.001 LXA4 + WRW4 vs. LXA4. (F) Effects of WRW4 treatment on Human serum (HS)-induced migration.
LXA4 inhibits LPS-induced production of inflammatory cytokines, chemokines and vascular endothelial growth factor (VEGF) by SCAP
To investigate whether the production of inflammatory cytokine IL-6, chemokines (IL-8, CCL2, CXCL10, CCL11) and growth factor VEGF was inhibited by LXA4 in SCAPs, multiplex analyses of cytokines and chemokines were performed. Here, SCAP were pretreated with 1 μg/mL LPS for 24 hours and compared with a control group (vehicle only). Stimulation of SCAP with LPS significantly upregulated the secretion of inflammatory cytokine IL-6, chemokines (IL-8, CCL2, CXCL10, CCL11) and of growth factor VEGF in comparison with control conditions. LXA4 significantly inhibited the LPS-induced increase in a concentration-dependent manner. To evaluate the role of the ALX/FPR2 in the anti-inflammatory effects of LXA4, SCAPs were treated with WRW4 (10 μM, 30 min) prior to treatment with LXA4 (100 nM). Pretreatment with WRW4 tended to inhibit these effects in response to LXA4, however the difference was not statically significant except for VEGF (Fig. 4).

Result of multiplex bead-based assay showing inhibition with dose effect of LXA4 on IL-6, IL-8, CXCL10, CCL2, CCL11 and VEGF secretion by SCAP. SCAP were pretreated with vehicle (0.035% ethanol) or various concentrations of LXA4 (1, 10 and 100 nM) for 30 min in the absence or presence of WRW4 (30 min before LXA4 treatment) followed by stimulation with LPS (1 μg/ml) for 24 h. The concentration IL-6, IL-8, CXCL10, CCL2, CCL11 and VEGF in culture media was measured using a multiplex bead-based assay. **p < 0.01 compared with LPS in the absence of LXA4; ##p < 0.01 compared with control cells (vehicle).
LXA4 enhances the immunomodulatory potential of SCAPs
We sought to investigate the effect of LXA4 on immunomodulatory potential of LPS-stimulated SCAP. SCAP possess low immunogenicity and can also suppress the one-way mixed lymphocyte reaction (MLR) in a dose-dependent manner13. First, we sought to investigate the effect of different concentrations of LXA4 (0, 1, 10, and 100 nM) on SCAP in MLR. SCAP were co-cultured with phytohemagglutinin (PHA)-stimulated PBMC in different ratios. Pretreatment of SCAP with 100 nM LXA4 significantly amplified the suppressive effect on PBMC when 1:5 and 1:10 dilution ratios were used (Fig. 5A).

LXA4 enhanced immunomodulatory properties of SCAP on PBMC stimulation and proliferation. (A) SCAP were first stimulated with LXA4 100 nM or LXA4 100 nM + LPS (1 μg/ml) or WRW4 + LXA4 100 nM + LPS (1 μg/ml) during 24 h. PBMC were stimulated with PHA and co-cultured with SCAP at different ratio (1:1, 1:5, 1:10). After 5 days, Stimulation index was calculated with MTT assay. *p < 0.05. Columns containing the same letter or letters are not significantly different (p> 0.05). (B) SCAP (red dots) were first stimulated with LXA4 only or LXA4 + LPS (1 μg/ml) for 24 h. CFSE-labeled PBMC (solid round cells) were stimulated with PHA in the lower chamber of a Transwell. SCAP were co-cultured with the stimulated PBMC (ratio SCAP/PBMC 1:10) either in direct contact or separated by a Transwell membrane. After 5 days of co-culture, proliferating PBMC were assessed by CFSE staining and flow cytometric analysis. *p < 0.05. (C) SCAP were first stimulated with LXA4 (0, 1, 10 or 100 nM) during 24 h and co-cultured with (PHA)-stimulated PBMC at different ratio (1:1, 1:5, 1:10). After 5 days, the effect of the different dose of LXA4 on PBMC proliferation was assessed by MTT. ***p < 0.001 compared with control cells (vehicle). (D) SCAP were first stimulated with LXA4 100 nM or LXA4 100 nM + LPS (1 μg/ml) during 24 h. PBMC were stimulated with PHA and co-cultured with SCAP at different ratio (1:1, 1:5, 1:10). After 5 days, dead cells assessment was calculated by trypan blue uptake. PHA phytohaemagglutinin A, CFSE carboxyfluorescein succinimidyl ester.
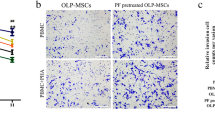
According to the pro-inflammatory effect of LPS on SCAP population (Fig. 4), LPS may significantly influence the immunomodulatory properties of SCAP on the proliferation of PBMC. Therefore, we then examined the effect of LXA4 on SCAP in a mixed leukocyte reaction where SCAP were pre-stimulated with LPS. Proliferation of PHA-stimulated PBMC served as a positive control. The proliferation of PBMC was analyzed using the MTT method. SCAP suppressed proliferation of PHA-stimulated PBMC in a dose dependent manner (Fig. 5B). Pretreatment with LPS (1 μg/mL) did not inhibit the suppressive effect of SCAP on PBMC proliferation. However, pretreatment of SCAP with 100 nM of LXA4 significantly increased the suppressive effect on PBMC when 1:5 and 1:10 dilution ratios were used (Fig. 5B). This effect was abrogated by WRW4 (Fig. 5B).
To elucidate whether the immunosuppressive action of LXA4 on SCAPs relies on secretion of soluble factors or requires cell-to cell contact, we performed other MLR using transwell inserts with a pore diameter of 0.4 μm. Percentages of proliferating cells were calculated by the carboxyfluorescein diacetate succinimidyl ester dilution method using flow cytometry. We confirmed results shown in Fig. 5B with PBMC diluted in 1:10 ratio. SCAP suppressed PHA-induced PBMC proliferation. LPS (1 μg/mL) did not inhibit the suppressive effect of SCAP on PBMC proliferation. However, we demonstrated that the suppressive action of 100 nM LXA4 was efficient only when cells were in direct contact (Fig. 5C).
Finally, to measure viability of PBMC after PHA stimulation, the trypan blue uptake method was used. There was no difference in viability of PBMC (trypan blue uptake) between groups with LPS and LPS + LXA4 (Fig. 5D).
Discussion
The apical papilla of developing teeth represents an enriched source of stem cells. These so-called Stem Cells of the Apical Papilla (SCAP) are considered as a potential source for dental pulp tissue regeneration and seem to be involved in the interplay between the processes of inflammation and regeneration14.
In this study, we identified for the first time that SCAP express the lipoxin receptor ALX/FPR2. The expression of ALX/FPR2 is upregulated by a variety of inflammatory stimulants such as TNF-α, interferon-γ as well as ligands for TLR-2, 3, 4, 7 and 9 in different cell types15,16. Although the effect was apparently weaker in comparison with microglial cells or microvascular endothelial cells, we demonstrated that important inflammatory mediators of pulpal and periapical diseases such as TNF-α and LPS upregulated ALX/FPR2 expression in SCAP. Further, we demonstrated that the LXA4-ALX/FPR2 axis increased proliferation and migration of SCAP. Pretreatment with LXA4 abrogated the paracrine activity of SCAP stimulated with LPS. Interestingly, we also showed that LXA4 enhanced immunomodulatory properties of SCAP.
Mesenchymal Stem Cells (MSC), SCAP and fibroblasts share common features (for instance their set of cell surface markers is negative for CD45 and positive for CD90, CD105) and they can also be induced to differentiate into adipocytes, chondrocytes and osteoblasts. Moreover, SCAP have immunomodulatory properties similar to MSCs17. SCAP were first characterized based on the expression of the surface marker STRO-118. In other experiments STRO-1 sorted SCAP were also compared with CD73, CD90 and CD105-sorted SCAP, with non-purified SCAP and with human gingival fibroblasts for phenotyping and in vitro osteogenic differentiation (Supplementary File S11A,C). However, STRO-1 + SCAP represent a small subset of SCAP population and are not representative of SCAP population present in the apical papilla19. Therefore we used a classical outgrowth technique for isolation of SCAP8,20. As expected, the effect of SCAP passages did not alter in vitro differentiation and cell surface markers of the cells (Supplementary File S2), demonstrating the stemness of the unsorted SCAP population.
It has been shown that not all cells from apical papilla are stem cells, e.g., fibroblasts have been noted18. However, in our study, we directly utilized the whole heterogeneous cell population, since there has been no compelling evidence that a purified stem cell subpopulation would be more homogeneous than original population21.
LXA4 was the first identified endogenous ligand for ALX/FPR222. This receptor has been shown in several cell types, including leukocytes, microglia, endothelial and epithelial cells23. Of note, other pro-resolving mediators (resolvins) are also ALX/FPR2 ligands, and both, lipoxins and resolvins, have been used with promising results in periodontal disease and pulpitis models24,25.
Only recently, the expression of ALX/FPR2 was described in human Mesenchymal Stem Cells and periodontal ligament cells. In acute lung injury, human MSC promoted the resolution of inflammation and prolonged survival of mice in part through exogenous LXA426. We found that SCAP expressed a relatively low level of ALX/FPR2 in comparison with PBMC, however, this expression was upregulated by pro-inflammatory factors (TNF-α and LPS). These results are in line with other studies using microglial or endothelial cells16,27. Although no mechanistic studies have been performed at this point, we may hypothesize that ALX/FPR2 is upregulated through c-Jun N-Terminal protein kinase and transcription factor NF-κB signaling pathways as discussed in other studies16.
Cell proliferation and migration are critical processes in regeneration of connective tissue and, in particular, of dental pulp and dentin. In our study, we demonstrated that the LXA4-ALX/FPR2 axis was involved in SCAP proliferation and migration. This result is consistent with earlier studies showing the role of the formyl peptide receptor in cell migration and proliferation of stem cells28,29. The effect of chemotactic receptors/ligands to facilitate SCAP recruitment has already been explored. For instance, SCAP can be chemo-attracted by the Stromal Cell-derived Factor-1α/ chemokine receptor 4 axis30. We may speculate that the recruitment and expansion of SCAP could be further modulated by LXA4.
To date, there is only limited data regarding the behavior of SCAP in an inflammatory context. Because of the presence of Gram-negative bacteria in the pulp space of infected teeth, LPS from Gram-negative bacteria has been used to stimulate toll-like receptor 431. Interestingly, in dental follicle cells, only LPS from Escherichia coli, but not from Porphyromonas gingivalis, induced expression of cytokines and chemokines32. In our study, we used LPS from E. coli. LPS markedly increased production of inflammatory cytokines, chemokines and vascular endothelial growth factor (VEGF) by SCAP. In another study, LPS from E. coli induced production of pro-inflammatory cytokines and chemokines including IL-6, IL-8, and TNF-α in SCAP33. We have shown here that LXA4 reduced LPS-induced production of cytokines, chemokines and VEGF in a dose dependent manner with the maximum effect of 100 nM LXA4. In microglial cells or corneal fibroblasts, comparable concentrations of LXA4 were used and inhibited pro-inflammatory cytokine such as IL-1β, ΤΝF-α as well as nitric oxide production in a concentration-dependent manner27. This inhibitory effect of LXA4 on LPS-induced inflammatory mediators was ALX/FPR2 mediated, since it was attenuated when SCAP were pretreated with the formyl peptide receptor antagonist WRW4 (Trp-Arg-Trp-Trp-Trp-Trp-CONH2). These results are in agreement with data from ALX/FPR2−/− mice, where LPS treatment showed an increase in pro-inflammatory cytokines28.
However, WRW4 did not completely block the action of LXA4, in particular in response to IL-6 and IL-8. WRW4’s action is believed to be through inhibition of agonist binding to ALX/FPR2, and inhibition of intracellular calcium release34. Moreover, WRW4 can block the activation of ERK1/2 and p38 MAPK signaling35. Although lipoxins and epi-lipoxins exert their anti-inflammatory effects through signals generated by binding to ALX/FPR2, lipoxins have also been found to interact with other receptors like G protein-coupled receptor 32 (GPR 32)7, aryl hydrocarbon receptor36, and high affinity cysteinyl leukotriene receptor37. Interestingly, human mesenchymal stem cells express both the cysteinyl leukotriene type 1 receptor and the aryl hydrocarbon receptor. These receptors, upon positive activation, may potentially regulate the MSC-associated immunomodulatory function in particular secretion of cytokines such as IL-6 and IL-838.
Only limited data exists regarding the effect of lipoxins on adaptive immune cells; one study described that LXA4 decreased IgM and IgG production by activated human B cells through ALX/FPR2-dependent signaling6. Recent reports suggested that dental mesenchymal stem cells, in particular SCAP, could suppress mitogen- or allogenic-stimulated proliferation of PBMC or T-cells in vitro at a ratio SCAP/PBMC higher than 1/1039,40,41. A lower mesenchymal stem cell number in culture was non-suppressive42,43. Therefore, aiming to see the immunomodulatory effect of SCAP in vitro with conditions that could mimic in vivo situation, we decided to study the following SCAP:PBMC ratio 1:10; 1:5 and 1:1. In our study, SCAP suppressed phytohemagglutinin-stimulated PBMC proliferation in a dose-dependent manner. Moreover, this inhibitory effect was increased by LXA4 pretreatment and was also observed when SCAP and PBMC were in direct contact. Such a modulation of the pulpal inflammatory response has been suggested as a way for optimizing endodontic therapeutics44.
Taken together, our data demonstrate that LXA4, a potent endogenous signal involved in the resolution phase of inflammation, can modulate the behavior of stem cells in inflammatory conditions in vitro. Our findings may help to elucidate mechanisms, by which stem cells can influence the outcome of inflammatory processes and disorders. We demonstrated that ALX/FPR2 receptor is essential for recruitment, proliferation and immunomodulatory functions of SCAP. Thus, ALX/FPR2 receptor may represent a novel molecular target for development of new drugs for therapies that require enhancement of stem cell-mediated pro-resolving functions.
Materials and Methods
Isolation, Culture, and Characterization of SCAP and PDLC
Apical papilla tissue was collected from third molars extracted from medically healthy patients (16 to 25 years old); informed consent had been obtained in accordance with a protocol approved by the Institutional Review Board at the Arthur A. Dugoni School of Dentistry at the University of the Pacific (IRB protocol #16-128). SCAP culture was established as previously described45. Similarly, periodontal ligament cells (PDLC) were obtained from scraped PDL tissue. Cells were cultured in alpha minimum essential medium (α-MEM) supplemented with 1% L-glutamine, 1% penicillin/streptomycin/amphotericin B (all ThermoFischer Scientific, Pittsburgh, PA, USA) and 10% human serum (HS) (Sigma-Aldrich, St Louis, MO, USA). SCAP and PDLC at passage 4 were analyzed for cell surface antigen expression by flow cytometry using Guava easyCyte 8HT flow cytometer (EMD Millipore, Billerica, MA, USA). Fluorochrome-conjugated monoclonal mouse anti- human against CD45-APC/Cy7, CD90-PE-CY7, CD105-APC, CD146-PerCP/Cy5.5 and STRO-1- FITC or isotype controls (all from BioLegend, San Diego, USA) were used. Data were analyzed using InCyte 2.5 software (EMD Millipore). Cells counts were performed at each passage, and the population doubling times (PDTs) were calculated.
In vitro differentiation assays
To show a multipotentiality, osteogenic and chondrogenic pathways of differentiation were evaluated using alizarin red S and alcian blue, respectively (Sigma-Aldrich). Briefly, cells were plated in 12-well plates at 30,000 cells/well and cultured for 21 days with the appropriate media being replaced every 2–3 days.
LPS, TNF-α application
When the cells reach 80%–90% confluence, they were incubated in starvation medium (1% HS) for 12 hours. Then, cells were treated with various concentrations of Escherichia coli O111:B4 Lipopolysaccharide (LPS) (Sigma-Aldrich) and/or various concentrations of Recombinant Human TNF-α (R&D Systems, Minneapolis, USA).
Analysis of ALX/FPR2 Expression
Flow cytometry
Surface and intracellular expression of the lipoxin receptor, ALX/FPR2, was evaluated in PDLC and SCAP from different donors using a monoclonal anti-ALX/FPR2 primary antibody (Thermo Fischer Scientific) and incubated with an anti-mouse PE-conjugated secondary antibody (BioLegend). For intracellular staining, cells were permeabilized with Tween-20 (Thermo Fischer Scientific). Secondary antibody-matched controls were used to assess unspecific fluorescence.
Immunofluorescence and Confocal Microscopy
SCAP and PDLC grown on glass coverslips were fixed with 4% paraformaldehyde, permeabilized with 0.2% Tween-20 for 20 minutes, and neutralized with 1% bovine serum albumin (BSA) (Affymetrix, Santa Clara, CA, USA) for 30 minutes. Incubation with primary mouse anti-ALX/FPR2 antibody (1:500) was followed by exposure to secondary FITC conjugated goat anti-Mouse (BioLegend) (1:1000). After washing in PBS, samples were stained with ProLong® Gold Antifade Mountant with DAPI (Thermo Fischer Scientific). Samples were examined with a Leica TCS-SPE II confocal laser-scanning microscope (Leica, Mannheim, Germany) at 10X and 43X original magnification. Images were acquired with LAS AF V.3 software (Leica).
Cells-to-CT 1-Step quantitative RT-PCR
SCAP stimulated with LPS (1 μg/mL) for 24 h were lysed using the Cells-to CT™ 1-Step TaqMan® Kit (Thermo Fischer Scientific). The resulting lysate was then used for one-step real-time RT-PCR with a TaqMan® gene expression assay for ALX/FPR2 (assay Hs00265954_m1), and with glyceraldehyde 3-phosphate dehydrogenase as an endogenous reference (assay Hs02786624_g1), using a StepOnePlus™ apparatus (Applied Biosystems). The relative ALX/FPR2 gene expression was determined using a comparative delta-delta cycle threshold method (DDCt) with a control group as a calibrator46.
MTT Analysis for Cell Viability
SCAPs were seeded in 96-well plates at a density of 7 × 10³ cells, pretreated with different concentrations of LXA4 (Cayman Chemical, Ann Arbor, MI, USA) for 30 min, and incubated with or without LPS (1 μg/mL) for 24 h in the continued presence of LXA4. Wells containing only culture medium only served as controls for non-specific dye reduction. Then, the medium was removed and cells were incubated with 0.5 mg/mL of 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) (ThermoFischer Scientific) in full medium at 37 °C, 5% CO2. After 4 hours of incubation at 37 °C, the medium was removed and the formazan crystals dissolved in DMSO (Sigma-Aldrich). Absorbance was measured at 540 nm using a VersaMax™ Microplate Reader. The results were expressed as the percentage of surviving cells compared to control cells.
Proliferation Assay
SCAP (2 × 103 cells per well) were seeded in 96-well microplates with complete medium overnight. The following day, cells were exposed to LXA4 (0.01 to 100 nM), the formyl peptide receptor antagonist WRW4 (Trp-Arg-Trp-Trp-Trp-Trp-CONH2) (R&D Systems) (10 µM, for 30 minutes) before LXA4 or vehicle (0.035% ethanol) diluted with 1% HS α-MEM. Vehicle controls were defined as 1 × PBS with 0.035% ethanol by volume, equivalent to the highest concentration of lipoxins used in the experiment (100 nM) LXA4. Cells were enumerated after 24, 48, and 72 hours using trypan blue exclusion test. Proliferation was calculated as ratio between LXA4-and vehicle-treated SCAPs. LXA4-affected cellular proliferation index was calculated as ratio between LXA4 and WRW4 + LXA4-treated cells.
In Vitro Wound Healing Assay
SCAP (5 × 104 cells per well) were grown in 24-well plates in complete medium until 80% to 90% confluency was reached. A disposable plastic pipette tip (200 μl) was used to make a scratch across the monolayer of cells. Cell debris was rinsed away by medium and increasing concentrations of LXA4, with or without WRW4 were added to each well. SCAPs maintained in complete medium served as a control of the maximal healing capacity. Cell wounds were monitored at 0, 3, 6, 24, and 48 hours using a phase contrast microscope equipped with a digital camera. Each well was photographed at four locations along the original wound boundary and wound width was quantified using ImageJ software.
Multiplex analysis of cytokines and chemokines
SCAPs were seeded in 24-well plates at a density of 5 × 104 cells, pretreated with different concentrations of LXA4 and/or WRW4 for 30 min, and incubated with or without LPS (1 μg/mL) for 24 h, then, culture media were harvested. Cytokines and chemokines were analyzed with the HCYTMAG-60K-PX41 Milliplex kit (EMD Millipore). The procedure described in the manufacturer’s manual was followed. Plates were analyzed using a Magpix platform (Luminex) equipped with the xPONENT 3.1 software and the concentration of each cytokine was calculated from raw data. Analyte concentrations were determined by a five-parameter logistic curve.
Mixed leukocyte reactions (MLR)
SCAP 5 × 104 were seeded in wells of a 24-well plate or into a transwell inserts for 24-well plates (0.4 μm) (Corning, NY, USA) in RPMI medium +10% HS (ThermoFischer Scientific). SCAP were stimulated with LPS (1 μg/mL) for 24 h, before application of LXA4 (1 to 100 nM), and/or WRW4 (10 μM), while control SCAPs were left untreated. SCAP were washed twice in PBS with 10% Human Serum, treated with mitomycin C (Sigma-Aldrich) at the final concentration of 25 μg/mL for 30 min, and then washed 4 times with PBS + 10% Human Serum. Human peripheral blood mononuclear cells (PBMC) (AllCells, Alameda, CA, USA) in dilution ratios 1:1, 1:5 and 1:10 were stimulated with 5 μg/mL phytohemagglutinin (PHA) (Sigma-Aldrich) and immediately added to the wells containing SCAP. Negative controls (PBMC only) and positive controls (PBMC + PHA) were used. Results of MLR were evaluated after five days of cultivation.
MTT assay for stimulation index of PBMC
The proliferation of PBMCs was analyzed using the MTT method. Stimulation index (SI) values were calculated by the following formula: SI = proliferation of stimulated PBMC with or without SCAP/proliferation of unstimulated PBMC alone47.
CFSE staining to estimate percentage of proliferating PBMC
PBMCs were labeled with 2 µM carboxyfluorescein diacetate succinimidyl ester (CFSE) (ThermoFischer Scientific) before being added to the mixed cell culture. Percentage of proliferating cells was calculated by the CFSE dilution method using flow cytometry.
Trypan blue uptake for measurement of PBMC viability
After 5 days of MLR, PBMCs were harvested and suspended in 0.4% trypan blue. Dead cells (colored by trypan blue), were counted and the percentage of positive cells to total cells was calculated.
Statistical Analysis
All results were expressed as the means ± standard error of the mean (SEM) of triplicate measurements; all experiments were repeated at least 3 times, except for multiplex analysis of cytokines which were done in duplicates. Data were analyzed and visualized using GraphPad Prism 7.0. Statistical analyses were performed using Student’s t tests or 1-way analysis of variance (ANOVA) followed by Dunnett post-tests. P < 0.05 was regarded as statistically significant.
References
Kizil, C., Kyritsis, N. & Brand, M. Effects of inflammation on stem cells: together they strive? EMBO Rep. 16, 416–26 (2015).
Waterman, R. S., Tomchuck, S. L., Henkle, S. L. & Betancourt, A. M. A new mesenchymal stem cell (MSC) paradigm: polarization into a pro-inflammatory MSC1 or an immunosuppressive MSC2 phenotype. PLoS One 5, e10088 (2010).
Ren, G. et al. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell 2, 141–150 (2008).
Shi, Y. et al. How mesenchymal stem cells interact with tissue immune responses. Trends Immunol. 33, 136–143 (2012).
Serhan, C. N., Chiang, N. & Dalli, J. The resolution code of acute inflammation: Novel pro-resolving lipid mediators in resolution. Semin. Immunol. 27, 200–215 (2015).
Ramon, S., Bancos, S., Serhan, C. N. & Phipps, R. P. Lipoxin A4 modulates adaptive immunity by decreasing memory B-cell responses via an ALX/FPR2-dependent mechanism. Eur. J. Immunol. 44, 357–369 (2014).
Bäck, M. et al. Update on leukotriene, lipoxin and oxoeicosanoid receptors: IUPHAR Review 7. Br. J. Pharmacol. 171, 3551–3574 (2014).
Potdar, P. D. & Jethmalani, Y. D. Human dental pulp stem cells: applications in future regenerative medicine. World J. Stem Cells 7, 839–851 (2015).
Sonoyama, W. et al. Mesenchymal stem cell-mediated functional tooth regeneration in swine. PLoS One 1, e79 (2006).
Huang, G. T. J. et al. The hidden treasure in apical papilla: the potential role in pulp/dentin regeneration and bioroot engineering. J. Endod. 34, 645–651 (2008).
Ding, G. et al. Suppression of T cell proliferation by root apical papilla stem cells in vitro. Cells. Tissues. Organs 191, 357–364 (2010).
Cianci, E. et al. Human periodontal stem cells release specialized proresolving mediators and carry immunomodulatory and prohealing properties regulated by lipoxins. Stem Cells Transl. Med. 5, 20–32 (2016).
Liu, J. et al. Concise reviews: Characteristics and potential applications of human dental tissue-derived mesenchymal stem cells. Stem Cells 33, 627–38 (2015).
Sharpe, P. T. Dental mesenchymal stem cells. Development 2273–2280, http://publications.ki.se/xmlui/handle/10616/42291 (2016).
Cui, Y. H. et al. Up-regulation of FPR2, a chemotactic receptor for amyloid β 1-42 (Aβ42), in murine microglial cells by TNFα1. Neurobiol. Dis. 10, 366–377 (2002).
Mou, H. et al. Proinflammatory stimulants promote the expression of a promiscuous G protein-coupled receptor, mFPR2, in microvascular endothelial cells. Inflammation 35, 656–664 (2012).
Denu, R. A. et al. Fibroblasts and Mesenchymal Stromal/Stem Cells Are Phenotypically Indistinguishable. Acta Haematol. 53705, 85–97 (2016).
Sonoyama, W. et al. Characterization of the Apical Papilla and Its Residing Stem Cells from Human Immature Permanent Teeth: A Pilot Study. J. Endod. 34, 166–171 (2008).
Bakopoulou, A., Leyhausen, G., Volk, J., Koidis, P. & Geurtsen, W. Comparative characterization of STRO-1neg/CD146pos and STRO-1pos/CD146pos apical papilla stem cells enriched with flow cytometry. Arch. Oral Biol. 58, 1556–1568 (2013).
Aguilar, P. & Lertchirakarn, V. Comparison of stem cell behaviors between indigenous high and low-CD24 percentage expressing cells of stem cells from apical papilla (SCAPs). Tissue Cell 48, 397–406 (2016).
Gong, T., Heng, B. C., Lo, E. C. M. & Zhang, C. Current Advance and Future Prospects of Tissue Engineering Approach to Dentin/Pulp Regenerative Therapy. Stem Cells Int. 9204574 (2016).
Fiore, S., Maddox, J. F., Perez, H. D. & Serhan, C. N. Identification of a human cDNA encoding a functional high affinity lipoxin A4 receptor. J. Exp. Med. 180, 253–60 (1994).
Maderna, P. & Godson, C. Lipoxins: resolutionary road. Br. J. Pharmacol. 158, 947–959 (2009).
Dondoni, L. et al. Effect of the pro-resolution lipid mediator Resolvin E1 (RvE1) on pulp tissues exposed to the oral environment. Int. Endod. J. 47, 827–834 (2014).
Van Dyke, T. E. et al. Proresolving nanomedicines activate bone regeneration in periodontitis. J. Dent. Res. 94, 148–56 (2015).
Fang, X. et al. Human mesenchymal stem (stromal) cells promote the resolution of acute lung injury in part through lipoxin A4. J. Immunol. 195, 875–881 (2015).
Xu, M. X. et al. Resolvin D1, an endogenous lipid mediator for inactivation of inflammation-related signaling pathways in microglial cells, prevents lipopolysaccharide-induced inflammatory responses. CNS Neurosci. Ther. 19, 235–243 (2013).
Wang, G. et al. Formylpeptide receptors promote the migration and differentiation of rat neural stem cells. Sci. Rep. 6, 25946 (2016).
Viswanathan, A., Painter, R. G., Lanson, N. A. & Wang, G. Functional Expression of N -Formyl Peptide Receptors in Human Bone Marrow-Derived Mesenchymal Stem Cells. Stem Cells 25, 1263–1269 (2007).
Liu, J. Y., Chen, X., Yue, L., Huang, G. T. J. & Zou, X. Y. CXC chemokine receptor 4 is expressed paravascularly in apical papilla and coordinates with stromal cell-derived factor-1 α during transmigration of stem cells from apical papilla. J. Endod. 41, 1430–1436 (2015).
Botero, T. M., Shelburne, C. E., Holland, G. R., Hanks, C. T. & Nör, J. E. TLR4 mediates LPS-induced VEGF expression in odontoblasts. J. Endod. 32, 951–5 (2006).
Morsczeck, C. O., Dress, J. & Gosau, M. Lipopolysaccharide from Escherichia coli but not from Porphyromonas gingivalis induce pro-inflammatory cytokines and alkaline phosphatase in dental follicle cells. Arch. Oral Biol. 57, 1595–1601 (2012).
Zhang, J. et al. Human stem cells from the apical papilla response to bacterial lipopolysaccharide exposure and anti-inflammatory effects of nuclear factor I C. J. Endod. 39, 1416–1422 (2013).
Bae, Y.-S. et al. Identification of Peptides That Antagonize Formyl Peptide Receptor-Like 1-Mediated Signaling. J. Immunol. 173, 607–614 (2004).
He, H. Q. & Ye, R. D. The formyl peptide receptors: Diversity of ligands and mechanism for recognition. Molecules 22, (2017).
Schaldach, C. M., Riby, J. & Bjeldanes, L. F. Lipoxin A4: A new class of ligand for the Ah receptor. Biochemistry 38, 7594–7600 (1999).
Chandrasekharan, J. A. & Sharma-walia, N. Lipoxins: nature’s way to resolve inflammation. J. Inflamm. Res. 8, 181–192 (2015).
de Almeida, D. C., Evangelista, L. S. M. & Câmara, N. O. S. Role of aryl hydrocarbon receptor in mesenchymal stromal cell activation: A minireview. World J. Stem Cells 9, 152–158 (2017).
Dokic, J. et al. Characterization and immunosuppressive properties of mesenchymal stem cells from periapical lesions. J. Clin. Periodontol. 39, 807–816 (2012).
Tomic, S., Djokic, J., Vasilijic, S., Vucevic, D. & Todorovic, V. Immunomodulatory Properties of Mesenchymal Stem Cells Derived from Dental Pulp and Dental Follicle are Susceptible to Activation by Toll-Like Receptor Agonists. Stem Cells Dev. 20, 695–708 (2011).
Ding, G. et al. Effect of cryopreservation on biological and immunological properties of stem cells from apical papilla. J. Cell. Physiol. 223, 415–422 (2010).
Yazid, F. B., Gnanasegaran, N., Kunasekaran, W., Govindasamy, V. & Musa, S. Comparison of immunodulatory properties of dental pulp stem cells derived from healthy and inflamed teeth. Clin. Oral Investig. 18, 2103–2112 (2014).
Fang, L., Lange, C., Engel, M., Zander, A. R. & Fehse, B. Sensitive balance of suppressing and activating effects of mesenchymal stem cells on T-cell proliferation. Transplantation 82, 1370–1373 (2006).
Cotti, E. et al. The Influence of Adalimumab on the Healing of Apical Periodontitis in Ferrets. J. Endod. 43, 1841–1846 (2017).
Peters, O. A. et al. Effects of two calcium silicate cements on cell viability, angiogenic growth factor release, and related gene expression in stem cells from the apical papilla. Int. Endod. J. 49, 1132–1140 (2015).
Livak, K. J. & Schmittgen, T. D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 25, 402–408 (2001).
Liu, J. et al. Suppression of human peripheral blood lymphocyte proliferation by immortalized mesenchymal stem cells derived from bone marrow of Banna Minipig inbred-line. Transplant. Proc. 36, 3272–3275 (2004).
Acknowledgements
This study was supported by Research Pilot Project Award DRES03-Activity 114 from the University of the Pacific, Arthur A. Dugoni School of Dentistry.
Author information
Authors and Affiliations
Contributions
A.G., M.T., O.A.P. contributed to conception, design, data acquisition, analysis, interpretation and drafted the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gaudin, A., Tolar, M. & Peters, O.A. Lipoxin A4 Attenuates the Inflammatory Response in Stem Cells of the Apical Papilla via ALX/FPR2. Sci Rep 8, 8921 (2018). https://doi.org/10.1038/s41598-018-27194-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-27194-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
