
Abstract
DNA elimination is a radical form of gene silencing and occurs both in somatic and germ cells. The programmed DNA elimination occurs during gametogenesis in interspecies hybrids that reproduce by hybridogenesis (stick insects, fishes, and amphibians) and concerns removal of whole genomes of one of the parental species and production of clonal gametes propagating the genome of the other species. The cellular mechanisms differ considerably in hybridogenetic insects and fishes but remains unknown in edible frogs Pelophylax esculentus, natural hybrids between Pelophylax lessonae and Pelophylax ridibundus. Here we report DNA elimination mechanism in early developing gonads of diploid and triploid hybrid frogs, studied by TEM, immunofluorescence, and cytochemistry. In gonocytes of both sexes (primary oogonia and prespermatogonia), micronuclei emerge as detached nuclear buds formed during interphase. We found depletion of nuclear pore complexes in micronuclear membrane and chromatin inactivation via heterochromatinization followed by degradation of micronuclei by autophagy. Micronuclei formation does not lead to apoptotic cell death showing that genome elimination is a physiological process. Chromatin elimination via micronuclei in P. esculentus is unique among hybridogenetic animals and contributes to broadening the knowledge about reproductive modes in animals.
Similar content being viewed by others

Introduction
The physiological programmed DNA elimination, known also as chromatin diminution or chromosome elimination, is a radical form of gene silencing and has been described in more than a hundred animal species from nine taxonomic groups, ranging from Protista (ciliates), invertebrates (nematodes, copepods, insects, arachnids) to vertebrates (lampreys, hagfish, birds, and marsupials)1,2. The amount of the eliminated genomic DNA ranges from about 20% in the hagfish and nematodes to 95% in ciliates. In invertebrates, programmed DNA elimination occurs during early embryonic development and concerns both somatic1,2 and germline cells3,4. In the latter case, DNA is eliminated from early gonocytes (primary oogonia or prespermatogonia) or during the first meiotic division of spermatogenesis and concerns determination of the male sex5,6,7,8. Similar mechanism was also described in a cartilagous fish Hydrolagus colliei3 and in a passerine bird, the zebra finch4,5. Genome elimination from germline cells occurs also during sporogenesis in plant hybrids between Brachiaria ruziziensis and B. brizantha6.
A peculiar kind of programmed DNA elimination occurs in interspecies hybrids that reproduce by hybridogenesis and concerns removal of whole genomes of one of the parental species. The hybrids are restored in each generation by mating with a species whose genome was eliminated from the germline7. Hybridogenesis has emerged independently in a few evolutionary distant animal groups (insects, fishes, amphibians). Although its result is the same, i.e. elimination of one of the parental genomes from the germline cells and clonal propagation of the other one, the cellular mechanisms differ considerably. Elimination of genomes may occur pre-meiotically during the phase of gonocyte proliferation or during meiosis. This specific process was originally described in a female hybrid fish Poeciliopsis monacha-lucida (Cyprinidae)8 but later was also found in other interspecies hybrid fishes from the genus Hypseleotris9 and Hexagrammos10, as well as in the female hybrid stick insects (Phasmodea, Insecta)11,12,13,14. Genome elimination was also found in a variety of triploid hybrid organisms: among fishes Squalius alburnoides15, Phoxinus eos-neoagaeus16 and Misgurnus anguilicaudatus complex (Cobitidae)17,18,19,20; amphibians: the Batura toads of Bufotes viridis group21 and the edible frog Pelophylax esculentus (formerly Rana esculenta)22,23.
Hybridogenesis has also been discovered in the western Palearctic water frogs of the Pelophylax esculentus complex24,25. Pelophylax lessonae (LL) and P. ridibundus (RR) are diploid species, whereas P. esculentus is their interspecific hybrid represented by diploid (RL) or triploid (RRL, LLR), and occasionally non-viable tetraploid (RRLL) and pentaploid (RRLLL) males and females that usually coexist with one of the parental species in the same populations26,27,28,29. The hybrids eliminate one of the parental genomes from their germline cells before meiotic recombination, duplicate the remaining one and undergo meiosis; however, the recombination occurs between the identical copies of the duplicated chromosomes, as was evidenced in diplotene oocytes30,31,32. As the result, the retained genome is transmitted clonally and F1 hybrids are restored de novo in each generation when mating with this parental species, whose genome was eliminated33,34. The retention and duplication of one of the genomes (R; ridibundus) was first detected by Tunner and Heppich35 and Tunner and Heppich-Tunner36 in juvenile hybrid females.
Cytogenetic mechanisms of the programmed DNA elimination remain poorly understood in the majority of cases with exception of the ciliate protozoa37. Chromosome elimination in insects and birds (finches) involves a wide spectrum of epigenetic modifications including heterochromatinization, histone H3/H4 acetylation, H3S10 phosphorylation, and DNA methylation38,39. Ravi and Chan40 and Sanei et al.41 found that the mechanism underlying haploidization in plants is also connected with changes of histone H3 in centromeric regions of chromosomes that compromises the assembly site for the kinetochore complex. Similar processes are involved in unipolar spindle formation and monocentric cell divisions during spermatogenesis in insects Mycophila speyeri42, Sciara coprophila43,44, and fishes Poeciliopsis monacha-lucida45.
A different mechanism involves formation of micronuclei and concerns both the programmed and non-programmed DNA elimination. Micronuclei are specific extranuclear structures morphologically similar to the main cell nucleus, surrounded by the double membrane, and containing the eliminated DNA. They are usually formed during aberrant mitoses displaying chromosome missegregation and contain chromosomes that are not bound to the spindle and lag behind during anaphase and telophase46,47,48,49,50. Micronuclei may also arise at interphase as the result of nuclear budding47,48,49,51. However, the fate of encapsulated chromatin remains unknown in a majority of cases. Micronuclei were observed in somatic cells of plant hybrids49, in male germline cells in the zebra finch4,5, and in somatic cells of the sea lamprey52. Moreover, they are well-known structures that appear as a result of chromosome missegregation and reflect chromosome instability in many kinds of cancer cells47 and a variety of cells exposed to genotoxic agents53.
Micronuclei (described formerly as nucleus-like bodies, NLB54) were also observed in the germline cells of developing gonads in Pelophylax esculentus34,54,55, however the cytological mechanisms of their appearance remained unknown. From the earliest stages of differentiation, tadpole gonads are composed of somatic cells (pre-follicular in the ovaries and pre-Sertoli in the testes) and germline cells. Germ cells that arise from primordial germ cells of an embryo are gonocytes, which proliferate before and after sexual differentiation of gonads, eventually giving rise to the limited pools of primary oogonia in females56,57,58 and prespermatogonia in males58. These events take place only during larval and juvenile periods of individuals’ lives. Prespermatogonia are the only germ cells in a male gonad before sexual maturation and afterwards they transform into spermatogonial stem cells59. The situation is different in females where meiosis starts early, so in developing ovaries we can observe both mitotically dividing gonocytes and meiocytes up to early diplotene that form a limited stockpile for the entire female’s life (for details see Ogielska et al.60). Meiocytes (but not gonocytes) in amphibians form isogenic groups and are encapsulated inside cysts; only in females diplotene oocytes are individual cells (each inside its own ovarian follicle). Because of the large variety of names used in the literature, we decided to apply a general name “gonocytes” for female and male germline cells during the phase of mitotic multiplication before formation of cysts and meiotic entry in larval stages. Nuclei of gonocytes in Pelophylax, as in other ranid frogs, are spherical and smooth in outline in contrary to most amphibian species studied so far, where nuclei are highly lobulated56,61.
In the present paper we provide results of the first complete examination of micronuclei in the gonocytes of diploid and triploid water frog hybrids. We focused on ultrastructure, spatial relationship with gonocyte nuclei, time of appearance, and presumed mechanisms of micronuclei formation and their fate. The aims of the study were to check whether micronuclei in P. esculentus undergo epigenetic chromatin modifications and alterations of nuclear envelope. We consider that micronucleation is a physiological process, which does not lead to gonocyte death, whereas micronuclei, as carriers of the eliminated DNA, are directed to autophagic process while the main nuclei remain intact.
Results
Morphology (TEM and fluorescence microscopy)
The difference between size and morphology of somatic and germ cells was evident in tadpole gonads. Somatic cells had pyramidal nuclei with clearly visible heterochromatin patterns, whereas interphase gonocytes had bigger spherical nuclei, 10–15 µm in diameter, with smooth outline and euchromatin content (Fig. 1a). Most gonocytes were in interphase and all encountered mitoses had regular distribution of segregating chromosomes.

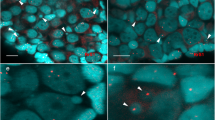
Morphology of the micronuclei in gonocytes of P. esculentus. (a) Immunofluorescent staining of frozen tissue sections of testis from male at 28 Gosner stage: cell membrane (wheat germ agglutinin, WGA, red) and nuclear pore complex proteins, NPC (Nup F/G, green), DNA counterstained with DAPI (blue). Confocal image represents projection of 4 z-sections (0.46 µm thick) to highlight the weak NPC staining at the rim of micronucleus (arrowhead) contained inside the cell boundaries probed with WGA. Note the weaker chromatin signal in nuclei of germ cells (n), corresponding to euchromatin, and stronger signal in nuclei of somatic cells (s), corresponding to heterochromatin. (b–e) TEM images of micronuclei present in prespermatogonia (b,c) and gonocytes (d,e). Micronuclei were found in both diploid (b,d,e) and triploid (c) hybrids during the premetamorphosis stage of somatic development according to Gosner (1960), stage 36 (d,e) or during metamorphosis climax, stage 46 (b,c). Double membrane envelope was surrounding chromatin in micronuclei, similarly as in main nuclei (inset in d). Chromatin condensation level differed from euchromatin (c–e) to heterochromatin (b,d). Cytoplasm of germ cells was filled with membranes (al), mitochondria and nuage material (asterisk) was present in the vicinity of cell nucleus. (e) Note the nuclear pore complexes (arrowhead) abundant in nuclear envelope of the main nucleus and annulate lamellae but rare in micronuclei. al- annulate lamellae, n – nucleus, m – mitochondria, mn – micronucleus, s – somatic cells. Scale bars (a) 5 µm, (d-e) 2 µm.
In cytoplasm of gonocytes of hybrid tadpoles, both diploid (RL) and triploid (RRL and LLR) we observed chromatin-positive bodies, i.e. micronuclei (Fig. 1a). Micronuclei were small spherical structures morphologically resembling the main nucleus (Figs 1b–e, 2a,b) and containing DNA, as confirmed by DAPI staining (Fig. 1a DAPI). They had the double membrane envelope that resembled that of the main nucleus but was almost depleted of the nuclear pore complexes (Fig. 1d,e). The cytoplasm of gonocytes contained intracellular membranes (endoplasmic reticulum, annulate lammelae) lying close to the nuclear envelope (Fig. 1b,c,e). Micronuclei differed in size (0.8–4 μm) and number (1–5) per one cell, and were localized at various distances from the main interphase nucleus (Fig. 1b–e), as confirmed in 3D reconstructions from confocal images (Fig. 2c). The size and number of micronuclei per one cell was analysed in various tadpole developmental stages from sexual differentiation until completion of metamorphosis (Gosner62 stages 28–46) and genomic composition (RL, RRL, RLL). In total number of 907 micronuclei analysed most frequently found were micronuclei of diameter 1.1–2 and 2.1–3 μm (37.82 and 38.26%, respectively), the smallest were 0.8–1 μm (3.42%), and the biggest were 3.1–4 μm (20.5%) (see Supplementary Fig. 1 and Supplementary Table 1). The prevailing group of cells possessed one micronucleus (67.2% of all cells), two micronuclei were found inside smaller group of cells (21.67%), three micronuclei were present in 9.08%, and four and five micronuclei were rarely seen (1.76% and 0.29%, respectively of total 683 cells) (see Supplementary Fig. 2 and Supplementary Table 2). Plasma membrane staining using wheat germ agglutinin (WGA) confirmed the presence of micronuclei inside the cytoplasm of gonocytes in both sexes (Fig. 1a). Frequency of micronucleated gonocytes in diploid and triploid hybrids was recently reported and ranged from 15 to 35% (see55); whereas in gonads of the parental species we have not found any cellular structures similar to micronuclei.

Level of nuclear pore complex proteins is reduced in nuclear envelopes of micronuclei in contrast to the main nucleus. Immunofluorescent staining of frozen tissue sections of ovaries from P. esculentus diploid female at 29 Gosner stage: nuclear pore complex proteins, NPC (Nup F/G, green), DNA counterstained with DAPI (blue). Germ cells in the centre of the images (n) have higher expression level of NPC proteins in the nuclear envelope in contrast to somatic cells, visible in the surrounding tissue. The NPC proteins staining pattern in MN is patchy, dotted (arrowhead) or absent (arrow). (a) Gonocyte with 3 micronuclei of various sizes. Micronucleus in the middle has patchy NPC proteins staining similar to main nucleus. (b) On the left side is gonocyte with MN devoid of NPC proteins (arrow), on the right side gonocyte with dotted NPC proteins signals in two MN of various sizes (arrowheads). Confocal images represent projection of 2 z-sections (a) or single z-section (b) (0.46 µm). (c) 3D model of reconstructed fluorescent signal volumes from cell visible in Fig. 9d, NPC proteins (green) and DNA (blue). Note that NPC signal is discontinuous probably due to differential distribution of nuclear pore complexes in the nuclear envelope and threshold settings excluding cytoplasmic signal. Scale bars 5 µm.
Nuclear envelope antigens in micronuclei
The TEM examination of the nuclear envelope revealed the reduced number of the nuclear pore complexes (NPC) in the micronuclei (Fig. 1e - arrowheads). We examined the density and distribution of NPC using antibody reacting with a conserved domain of FG-repeat nucleoporins. NPC protein signals were detected as dots or patches in the nuclear envelopes of nuclei of somatic cells and gonocytes in ovaries and testes of all examined individuals (Figs 1a and 2). Interestingly, gonocytes revealed much higher expression level of NPC proteins comparing to somatic cells, as well as the presence of cytoplasmic signal, which was absent in somatic cells (Fig. 2). The same staining pattern was observed in P. lessonae (Supplementary Fig. 3) and P. ridibundus (not shown), as well as in hybrid gonads (Figs 1, 2). A portion of gonocytes displayed micronuclei surrounded by NE with similar or lower staining level of FG-repeat nucleoporins as compared to the main nucleus (Fig. 2a - arrowhead, lower micronucleus). Micronuclei with condensed heterochromatic DNA displayed lower expression level of NPC proteins than in nuclear envelope of the main nucleus. Among 106 micronuclei analysed in 80 cells, FG-repeat nucleoporins were detected in 61% as patches (Fig. 1a) or dots on the chromatin rim (Fig. 2b - arrowheads). We found that 15% of micronuclei showed the rim staining of entire envelope and 46% possessed only 1–3 signals, while the signal was absent in 39% of micronuclei (Fig. 2a,b - arrows, Fig. 3a). Significant portion of gonocytes displayed at the same time micronuclei with remnants of nucleoporins together with nucleoporins-negative ones (Fig. 2a). We have supported morphological observations by quantitative densitometric analysis of relative fluorescence signal for NPC proteins present in nuclear envelopes (ratio of mean fluorescence values of micronucleus to main nucleus). The ratios were significantly different in the three groups of micronuclei, classified according to morphological NPC proteins expression pattern. The highest values were found in micronuclei possessing rim signal of entire envelope, lower values were noted in micronuclei having dots and patches, and the lowest values in those without nuclear envelope signal (Fig. 3b, Supplementary Table S3). Additionally our analysis showed that in the majority of MN the mean ratio value was 0.64 or lower, revealing depletion of NPC from micronuclear envelope (Supplementary Table S3). The results of TEM and NPC proteins level analyses indicated that the envelopes of micronuclei were gradually destroyed, which most probably affected its function.

Quantitative analysis of nuclear pore complex proteins in micronuclei. (a) Frequency of micronuclei (MN) with various distribution of nuclear pore complex proteins (Nup) assessed by morphological criteria. (b) Ratios of Nup relative fluorescence of micronuclei (MN) to main nuclei (N) according to three classes of Nup distribution (rim, dots/patches, absent) in gonocytes of hybrid frogs.
Histone modifications of chromatin in micronuclei
Hybrid gonocytes were harbouring micronuclei, in which chromatin was at various stages of condensation (Fig. 1b-e), either more condensed than in the main nucleus (Figs 1a, 4a - arrows, B - arrowhead) or at similar condensation level (Fig. 2b - arrow). Morphological observations were in agreement with densitometry analysis of fluorescence intensity after DAPI staining. Similar condensation level of chromatin was observed in 51% of micronuclei, 43% showed higher condensation, and 6% displayed lower signal (Supplementary Fig. S4), as compared to main nuclei. For more detailed characterization of chromatin state (eu- or heterochromatic) we performed two indirect approaches to show immunofluorescent staining of epigenetic modifications of histones: (1) against acetyl-histone H4 (H4Ac) as a marker of transcriptionally active chromatin (euchromatin), and (2) against histone H3 tri-methylated at lysine 9 (H3K9me3) as a marker of heterochromatin. Chromatin modification pattern in the main nuclei of gonocytes was similar both in the parental species and in the hybrids. Most of the interphase nuclei of gonocytes were euchromatic, as was detected with antibodies against H4Ac (Fig. 4a). On the contrary, we detected a reduced level of the H4Ac staining in all observed micronuclei. As detected by immunofluorescence intensity analysis, 42% (36 micronuclei) exhibited reduction of the intensity of H4Ac signal from 0% to 50% comparatively to main nuclei (Fig. 4a - upper arrow and arrowhead, Supplementary Fig. S5); 53% (46 micronuclei) showed reduction of signal from 50% to 100% comparatively to main nuclei, while 5% (4 micronuclei) exhibited reduction of signal higher than 100% comparatively to main nuclei (Fig. 4a - lower arrow, Supplementary Fig. S5). Heterochromatin detected with antibody against H3K9me3 histone mark was observed in compact chromatin regions of interphase nuclei of somatic and in some germ cells. A portion of micronuclei (50 micronuclei, i.e. 40%) revealed increased fluorescence signal after detection of H3K9me3 modification from 0.1% to 50% in comparison to main nuclei (Fig. 3b - arrow, Supplementary Fig. S6); 22% (28 micronuclei) showed increased signal from 50% to 100%, and 15% (19 micronuclei) showed more than 100% increase of the signal (Fig. 3b - arrowhead, Supplementary Fig. S6). Interestingly, we also found that 23% (19 micronuclei) demonstrated reduction of the H3K9me3 mark (see Supplementary Fig. S6). Thus, the results of epigenetic histone modification approaches are in agreement with both the ultrastructure observations and DAPI-positive compact staining in micronuclei in gonocytes of hybrid tadpoles (Figs 1 and 2). This in turn suggest that DNA present in micronuclei behaved differently than in the main nuclei and most probably became inactivated by heterochromatinization.

Chromatin in the micronuclei in contrast to gonocytes nuclei is inactivated and accumulates heterochromatin modification. 3D imunofluorescence of whole mount gonads of diploid P. esculentus females at 32 Gosner stage: (a) Chromatin in the micronuclei, but not in the gonocytes nuclei, revealed decreased level (arrow, upper micronucleus) or even absence (arrow, lower micronucleus) of acetylated histone Н4, the modification corresponding to active chromatin. Some micronuclei still retain level of H4Ac similar to main nucleus (arrowhead). (b) Chromatin in the micronuclei accumulates H3K9me3 modification indicating its inactivation and heterochromatinization (arrowhead). Some micronuclei still retain level of H3K9me3 similar to main nucleus (arrow). Images represent single z-section of the gonad. Scale bars 10 µm.
Formation and degradation of micronuclei
To answer the question whether micronuclei detached from the main nuclei by budding, we analysed confocal microphotographs of gonocytes labelled for plasma membrane and NPC proteins (WGA and FG-repeat nucleoporins) in search of micronuclei situated in the closest vicinity of the main nuclei. In several interphase gonocytes we found nuclear blebs similar to micronuclei in shape and size protruding from the main nuclei (Fig. 5a,c - arrowheads), which we interpreted as micronuclei budding. We could distinguish an extruded DAPI-positive content of a forming micronucleus surrounded by nuclear envelope displaying a weak NPC protein signal (Fig. 5a,b - arrowhead, Supplementary Movie S1), representing an early stage of micronucleus formation. At the same time we could see an aggregation of Nup signal in nuclear membrane around the base of the budding chromatin (Fig. 5a,b - arrowhead). The next step of budding was illustrated by the finger-shaped chromatin extrusion tightly attached to the main nucleus (Fig. 5c–f - arrowhead, Supplementary Movie S2). Finally, micronuclei were separated from the main nucleus by NPC proteins-positive membrane protruding from the main nucleus toward the micronucleus (Fig. 5a,c,e - arrow). In the already separated micronuclei we observed a lower level (Fig. 5a - double arrowhead) or lack of NPC proteins (Fig. 5c - double arrowhead). This suggests that either during chromatin budding the nuclear membrane surrounding micronucleus was depleted of majority of NPC proteins or micronuclei are extruded from the main nuclei at nuclear envelope regions of low NPC proteins level.

Formation of micronuclei from nuclear extrusions in gonocytes. Immunofluorescent staining of frozen tissue sections of ovaries from P. esculentus diploid females at 41 (a,b) and 44 (c–f) Gosner stages: nuclear pore complex proteins, NPC (Nup F/G, green), DNA counterstained with DAPI (blue). (a–c) Single confocal sections of DNA and NPC proteins (0.46 µm). (b,d) 3D reconstructions of natural confocal sections showing the views of germ cells from the angle to visualise chromatin budding (arrowheads). (e,f) 3D models of reconstructed fluorescent signal volumes from cell visible in c,d, (e) NPC proteins and DNA, note that NPC signal is discontinuous probably due to differential distribution of nuclear pore complexes in the nuclear envelope and threshold settings excluding cytoplasmic signal, (f) DNA. (a,b) Early stage of micronucleus formation: gonocyte with small chromatin extrusion possessing a weak NPC protein signal in nuclear envelope and strong DAPI signal (arrowhead), representing emerging micronucleus containing heterochromatin. Second micronucleus with punctate NPC protein signal (a, arrowhead). (c,d,f) Middle stage of micronucleus formation: extruded chromatin has finger-like shape and is connected with main chromatin mass in the cell nucleus (arrowhead). Note the discontinuous NPC signal in the nuclear envelope at budding site. Late stage: micronuclei (double arrowhead, a–e) are separated from the main nucleus by nuclear envelope with strong NPC protein signal (arrow), either protruding towards the micronucleus (a) or flat (c,e). There is no distinct NPC protein signal at the nuclear envelope in micronucleus (c) as well as in finger-like extrusion (arrow, c,e). Scale bars 5 µm.
To further investigate the fate of micronuclei we examined the gonocyte morphology in TEM images. Along with regular and intact micronuclei we also found micronuclei presenting various signs of degeneration. We observed sequestration of the cytoplasm by lipid double membranes, apparently endoplasmic reticulum and annulate lamellae, surrounding heterochromatinized intact micronuclei (Fig. 6a - arrowheads), which might reflect an initial stage of autophagy. We also found membranes assembling on the surface of micronucleus envelope (Fig. 6b - arrowheads) or encapsulating degenerating micronuclei (Fig. 6c - arrowheads) that we interpreted as autophagosomes (nucleophagosomes according to Mijaljica and Devenish63). The presence of vacuolar spaces, lysosomal vesicles (Fig. 6c), and disruption of micronucleus envelope (Fig. 6b - arrows) suggests the autophagic degradation of chromatin inside micronuclei. In some micronuclei we found membranes inside the chromatin territory (Fig. 6b - note the membranes inside micronucleus, D - arrowheads), which suggested an invasion of endoplasmic reticulum membranes and might represent nuclear envelope collapse and disruption of micronuclei.

Chromatin in micronuclei is degraded by autophagy. Transmission electron microscopy analysis of autophagosome formation around micronuclei and invasion of tubular membranes inside the chromatin territory. Micronuclei were found in diploid hybrid males (a) and females (b–d) during the premetamorphosis stage of somatic development according to Gosner (1960), (a–d) stages 37, 41, 31, 36 respectively. (a) Sequestration of the cytoplasm by lipid double membranes (arrowheads) surrounding large heterochromatinized, intact micronucleus. (b) Micronucleus accompanied by two membranes assembling on the surface (arrowheads): cap-shaped (at the bottom) and linear (upper part), inside the chromatin the invading membranes are visible. Arrows point the disruptions of nuclear envelope in micronucleus (c) Autophagosome membranes (arrowheads) encapsulating micronucleus are partially double- or single layered. Vacuolar spaces are signs of degeneration (white spaces) and vesicles similar to lysosomes are visible. (d) Micronucleus with tubular membranes invading chromatin. n – nucleus, m – mitochondria, mn – micronucleus. Scale bars 2 µm.
To support the ultrastructure investigation, we applied immunofluorescence examination using microtubule-associated protein light chain 3 (LC3) as the marker of phagophore and autophagosome formation. The cytoplasm of gonocytes in hybrid gonads (Fig. 7a,b), as well as in P. lessonae and P. ridibundus, was filled with punctate signals, corresponding to the free form of LC3 proteins residing in that compartment. The prevailing portion of micronuclei (76%) displayed no LC3 signal whereas 14% had only weak punctate signals, similar to the free cytoplasmic form of the protein. Nevertheless, in 10% of all observed micronuclei (9 of 68 cells analysed), chromatin was decorated by LC3 aggregates corresponding to the membrane-bound form of LC3. The aggregates were visible as strong dots situated at the periphery of highly condensed chromatin in micronuclei (Fig. 7a - arrowhead) or as clumps at one of their sides; at the same time the lower density and fragmentation of chromatin inside the micronuclei were observed (Fig. 7b - arrowhead). This may represent stages of phagophore and nucleophagosome formation, respectively, as well as chromatin degradation inside micronuclei. To confirm the presence of intracellular membranes surrounding micronuclei we applied lectin cytochemistry using wheat germ agglutinin (WGA), a useful marker of plasma membranes, as well as post-Golgi membranes and phagophore structures in cytoplasmic compartment of a cell. In some cases we found micronuclei accompanied by a patchy WGA signal at one of their sides; which may represent the Golgi-derived membranes of autophagosome forming around micronuclei (Fig. 7c - arrowheads). We supported morphological observations by quantitative densitometric analysis of relative fluorescence signal of LC3 present inside nuclei, micronuclei, and around micronuclei, i.e. in the area of intensive signal reflecting the nucleophagophore membranes (ratio of mean fluorescence values of micronucleus to nucleus (see Supplementary Table S4). The ratios were significantly different in the three analysed groups of micronuclei, the highest values were found in micronuclei possessing LC3 aggregates, lower values were noted in micronuclei displaying dots and patches, and the lowest values in micronuclei without nuclear envelope signal (see Fig. 8a). Within the group of LC3 aggregates we also measured the signal intensity around the micronuclei, which was about twice as high as inside micronuclei (Fig. 8b). We performed TUNEL assay on paraffin sections of gonads followed by LC3 immunocytochemistry to answer the question whether chromatin in micronuclei is experiencing degradation. However, our results shown in Supplementary Fig. S7 and Supplementary Table S5 did not reveal TUNEL signal in micronuclei, while apoptotic gonocytes showed accumulation of double strand breaks in nuclear DNA. Similarly we could not detect LC3 signals in micronuclei when a staining was applied subsequently after TUNEL assay. However, we cannot exclude that Proteinase K digestion of the tissue during TUNEL protocol affected LC3 molecules, which are small proteins, and potentially may have lost their affinity to antibodies. Taken together, these results show possible nuclear envelope collapse and support our hypothesis of the autophagic removal of micronuclei, since DNA-nicks generated by the lysosomal enzyme deoxyribonuclease II did not allowed for TUNEL detection.

Autophagosome markers are accumulated in the micronuclei. Immunofluorescence staining of paraffin sections from gonads of diploid female at 35 Gosner stage: LC3 autophagosome marker (red), DNA counterstained with DAPI (blue) (a,b). (a) Dotted and linear aggregates of LC3 (arrowhead) on the surface of highly condensed micronuclear chromatin may represent the early stage of autophagosome formation. (b) Large LC3 aggregate (arrowhead) on the micronucleus and between the chromatin clumps. Note that DNA is less condensed and fragmented with an empty interior which may represent the later stage of autophagic process connected with DNA degradation. Second micronucleus (arrow) is highly heterochromatinized and apparently not autophagic. (c) Immunofluorescence of frozen tissue sections of an ovary from diploid female at 29 Gosner stage showing wheat germ agglutinin (WGA, red) staining of cellular membranes, DNA counterstained with DAPI (blue). Note the cap-shaped WGA aggregates (arrowheads) at the surface of micronuclei which we interpret as staining of fusion site of autophagosome membranes. The irregular shape of the left micronucleus could reflect chromatin degradation. Additional strong intra-cytoplasmic agglomerates or dots, visible in upper and lower cells, could be trans-Golgi cisternae and post-Golgi membranes, e.g. lysosomal membranes. Confocal images represent projection of 9 z-sections (a) or single z-section (b,c) (0.46 µm). N – nucleus. Scale bars 5 µm.

Quantitative analysis of LC3 autophagosome marker presence in micronuclei. (a) Ratios of LC3 relative fluorescence of micronuclei (MN) to main nuclei (N) according to 3 classes of LC3 distribution (aggregates, dots/patches, absent) in gonocytes of hybrid frogs. (b) Ratios of LC3 relative fluorescence of micronuclei (MN) to main nuclei (N) in gonocytes of hybrid frogs. In case of LC3 aggregates and dots/patches the signal was measured inside whole area of a micronucleus (black bars) or juxtaposed outside the perimeter (solid grey) of chromatin in MN, related to aggregating membranes of forming autophagosomes.
Micronucleated gonocytes and apoptosis
To find out whether micronuclei formation was a possible cause of gonocyte death, we analysed the indirect immunofluorescence labelling of cells with antibodies to active caspase-3, which is a reliable marker of apoptosis. Cells were assessed as apoptotic when active caspase-3 staining was present in the cytoplasm and chromatin structure in cell nuclei was amorphous and condensed, and thereby lacking heterochromatin/euchromatin structure, as compared to surrounding germ cells. In the parental species gonads, where micronuclei were absent, we did not found apoptotic signals in the gonocytes (Supplementary Fig. S3); the only exceptions were gonocytes situated in the degenerating distal portion of the testes, a normal process of male gonad morphogenesis (P. lessonae, Supplementary Fig. S3; see61,64). On the contrary, we found numerous apoptotic gonocytes in hybrid gonads (Fig. 9a–c, see also the recent publication of our team65). Among this group there were cells possessing FG-repeat nucleoporins at the nuclear membrane of nucleus (Fig. 9b), or lacking the nuclear rim staining (Fig. 9c). We found only two gonocytes displaying strong active caspase-3 signal in the cytoplasm, apoptotic morphology, and the presence of 2 or 3 spherical chromatin blebs, 0.6–1 µm in diameter, which might be considered as micronuclei (Fig. 9c).

Presence of micronuclei does not lead to apoptotic death of gonocytes. Immunofluorescent staining of frozen tissue sections of gonads from P. esculentus diploid female (a–c, e) and male (d) at 34 Gosner stage: active caspase-3 (red), NPC proteins (Nup F/G, green), DNA counterstained with DAPI (blue). Gonocytes with positive expression of active caspase-3 showing various stages of degeneration: (a) the view of ovary section with interphase, mitotic and apoptotic germ cells, (b) germ cells possessing NPC proteins at the NE of nucleus containing flat/amorphous chromatin without heterochromatin/euchromatin structure and similar DNA condensation level as surrounding germ cells, may reflect the early stage of apoptosis (c) germ cell with pycnotic nucleus and three micronuclei completely devoid of NPC proteins (arrows) showing later stage of apoptosis. Apoptotic cells possessing micronuclei were very rare in gonads. (d,e) Majority of germ cells possessing micronuclei of various sizes and NPC proteins level represent the normal cellular morphology and absence of active caspase-3 apoptotic marker, suggesting that DNA elimination in micronuclei does not affect normal cellular processes. (d) Micronucleus with condensed chromatin decorated by NPC proteins dots (arrowhead), second micronucleus is lacking NPC proteins, (e) big micronucleus with highly condensed chromatin and weak NPC proteins signal at the nuclear membrane (arrowhead) and small micronuclei lacking NPC proteins signal (arrows). Confocal images represent single z-section (a–c,e) or projection of 12 z-sections (d) (0.46 µm) to image micronuclei at different levels in the cell. Scale bars (a) 10 µm, (b–e) 5 µm.
We analysed 31 micronucleated cells that did not exhibit active caspase-3 signal. Morphology and nuclear structure was normal and similar to surrounding gonocytes. We could detect strong expression of FG-repeat nucleoporins at the nuclear envelope of the main nucleus. Among this group of cells, the nuclear envelope in micronuclei had reduced level or punctate signals of NPC proteins (Fig. 9d,e - arrowhead), but with similar frequency we could also find micronuclei without FG-repeat nucleoporins (Fig. 9d,e - arrows). Despite of chromatin degeneration and reduced FG-repeat nucleoporin expression in micronuclei, no active caspase-3 signal was present in the cells. These results demonstrate that micronuclei formation as such was not related to apoptosis and thereby should be considered as a unique physiological process characteristic of P. esculentus hybrid gametogenesis.
Discussion
Micronuclei have never been observed in the parental species, and have never been reported in gonocytes of any amphibian species, neither in primary oogonia nor in prespermatogonia. In the ranid frogs, they apparently are not nuclear lobes because the shape of gonocytes’ nuclei is mostly spherical or ovoid, and only slightly irregular in outline, but not lobed as is the case of most amphibian species56,57,58,61. The same was confirmed in the present paper using confocal microscopy of wheat germ agglutinin stained gonads where all encountered micronuclei were situated between the nucleus and the cell membrane. Ultrastructural examination of the hybrid gonocytes revealed that the nuclear membrane, at least the lipid bilayer, was well preserved in the majority of micronuclei, but the nuclear pore complexes were absent or observed occasionally. The reduced density and abnormal distribution of pore complex structures in the micronuclear envelope was further confirmed in immunofluorescence approach by the use of FG-repeat nucleoporins as a nuclear membrane marker66,67. Unfortunately, widely used lamin B1 antibodies were not useful in gonocytes, although they gave strong signal in somatic cells (data not shown). The reduced pore density was found in the radiation-induced micronuclei in mammalian cells68, moreover in the sea lamprey, micronuclei did not possess neither lamin B1 nor nuclear pore O-linked glycoproteins52. These alterations of nuclear envelope were connected with subsequent chromatin degradation in micronuclei as was also shown by other authors for lamin B169. These findings support our point of view that disruption of nuclear pore complex structures may precede chromatin degradation in micronuclei in hybrid gonocytes.
Heterochromatinization of genetic material destined for elimination accompanies almost all known cases of programmed DNA elimination in different organisms52,70,71,72. During elimination of germline-restricted chromosomes in the finches and the Sciara flies, and whole paternal genome elimination in mealy bugs Planococcus citri, strong heterochromatinization and abnormal H3S10 phosphorylation caused improper chromatin compaction before mitosis or meiosis. As a result, chromosomes failed to attach to the spindle microtubules and thereby were lost5,70,73,74,75. During the programmed genome rearrangement in the sea lamprey DNA accumulation of H3K9me3 and 5MeC heterochromatinization marks in micronuclei might play functional roles in maintaining chromatin compaction or positioning the eliminated chromatin52. It seems evident that formation of micronuclei in gonocytes of the hybridogenetic P. esculentus follows the same cellular mechanisms as in other organisms, as well as in cancer cell lines, i.e. during both the programmed and non-programmed DNA elimination. The lower level or absence of H4Ac and accumulation of H3K9me3 histone methylation confirmed transcriptional silencing and heterochromatinization of chromatin encapsulated in micronuclei. Our data from TEM showed both eu- and heterochromatic micronuclei content, which suggests that micronuclei are first detached and then heterochromatinized. However, at the moment we cannot unambiguously state when heterochromatinization of the eliminated DNA starts. Together with different levels of heterochromatinization observed ultrastructurally and by means of immunofluorescence densitometric analyses, we can assume that micronuclei contain chromatin that no longer plays its function and is directed for degradation.
In the present study we confirmed our initial suggestion about formation of micronuclei in gonocytes during interphase via budding from the main nucleus34,54. We found several gonocytes with micronuclei budding from the main nucleus, where chromatin was in tight contact with the nuclear chromatin mass (for details see Fig. 5c–f). The nuclear envelope surrounding chromatin at the budding site was depleted of majority of nuclear pore complex proteins, which might explain the low level of pore complexes observed in the prevailing group of micronuclei. Even more severe disruption of micronuclear membrane lacking lamin B1 and nuclear pore O-linked glycoprotein was observed in the sea lamprey somatic cells where micronuclei were formed from lagging chromatin during mitosis and were encapsulated de novo by nuclear membranes52. At the earliest stages of ovaries and testes differentiation studied herein we did not find lagging chromosomes in the gonocytes of diploid and triploid hybrids; the same observations were provided by Dedukh et al.55. However, lagging chromosomes were observed in some gonocytes at later stages around completion of metamorphosis34,54, so we cannot rule out the possibility of mitotic micronuclei formation. Moreover, they do not stand in contradiction with each other: budding from interphase nuclei is the primary event, whereas the post-mitotic encapsulation of chromosomes lagging behind the mitotic spindle is the secondary event. Coexistence of the two processes of micronuclei formation was previously reported in interspecific hybrids of plants49. Most probably, the elimination of chromatin in hybridogenetic frog gonocytes is a multistep process, which is not always precise and some chromosomes may lack active centromeres; to solve the problem, a more detailed study focused on mitotic spindle organization, centromere distribution, and lagging chromosomes is needed.
Rello-Varona et al.76 reported that discontinuity of the nuclear membrane, reduced density of chromatin, and presence of lysosomal vesicles engulfed in autophagosomes around micronuclei indicated their degradation. We obtained similar results and confirmed that micronuclei in the hybrid gonocytes are directed to autophagy. The TEM images of gonocytes revealed considerable amount of membranes lying around micronuclei that were most probably precursors of autophagosomes. We found structures representing all steps of autophagy63,77,78 beginning from cytoplasm sequestration to phagophore, and finally to autophagosome formation. Autophagic degradation of micronuclei was also confirmed by LC3 immunostaining. This protein is present in the cytoplasm in a free form and is aggregating on the membranes during formation of phagophore and further autophagosome closure53,79. In our study only 10% of all micronuclei displayed LC3-positive aggregates, which may reflect a rapid turnover of autophagic micronuclei, as suggested by Rello-Varona et al.76. Autophagy is also playing an important role in the programmed DNA elimination in a unicellular eukaryote Tetrahymena thermophila, in which several nuclei are selectively degraded during sexual reproduction80,81. The next evidence for autophagic removal of micronuclei in hybrid frogs comes from lectin cytochemistry. WGA-positive vesicles accompanying micronuclei may represent either staining of the terminal portion of the isolation membranes (phagophore) assembling on the surface of micronuclei, or the fusion site at the rims of closing nucleophagosome membranes. These results are consistent with TEM images showing the lipid double membranes surrounding some micronuclei. Similar staining pattern was obtained in cardiomyocytes82, and in Golgi vesicles and autophagosomes of rat hepatocytes83, where autophagic degradation was confirmed. Except structures typical of autophagy, we also noticed membranes invading chromatin inside some micronuclei. Invasion of membranes may indicate abnormal nuclear lamina organization and subsequent chromatin degradation69. Moreover, invagination of nuclear membrane was also reported during micronuclei formation via budding of B chromosome from interphase nuclei in Sciara coprofila51. Chromatin encapsulated in micronuclei from P. esculentus gonocytes does not accumulate double strand breaks (no TUNEL reaction) characteristic of apoptosis and hydrolysis by deoxyribonuclease I (DN-ase I), the enzyme generating 3′-hydroxyl groups during DNA fragmentation. Our data are in contradiction with the majority of reports where micronuclei formed from lagging chromosomes or budding from interphase nuclei display TUNEL signal49,52,84. The study of Hatch et al.69 does not report DN-ase I cleavage of chromatin in micronuclei during catastrophic nuclear envelope collapse. The evident lack of this cleavage in the case of micronuclei in hybrid frogs can indirectly suggest DNA degradation by autophagy, where lysosomal acidic hydrolase, deoxyribonuclease II (DN-ase II), is active, producing 3′-phosphate groups in digested DNA molecules which are not detected in TUNEL assay85,86. This evidence implicates that micronuclei are more likely degraded via autophagy and not by apoptotic-like processes. The final fate of micronucleated chromatin needs further investigation in terms of other autophagy markers (Beclin-1 and 2, Atg proteins family) and presence of lysosomal membrane markers (LAMP2), as well as accumulation of γH2AX as a marker of DNA repair. Summarizing our observations of nuclear pore complex depletion and degradation of micronuclei, we can present the following scenario of micronuclei fate: from budding and detachment of chromatin during interphase to final degradation by nucleophagy (as summarized in Fig. 10).

(a) diagram summarizing the supposed process of micronuclei formation (a–d) and degradation (e–h) in gonocytes of a hybrid P. esculentus. (a) Gonocyte nucleus with NPC proteins in nuclear envelope (green), (b,c) emerging chromatin extrusions (buds), (d,e) fully separated micronucleus with lower expression of NPC proteins, (f) heterochromatinized micronucleus possessing partial NPC signal, (g) strongly heterochromatinized micronucleus with LC3-positive membranes (phagophore, red), (h) nucleophagic micronucleus with abnormal chromatin structure and disrupted membranes.
A recent study on cancer cell lines experimentally induced to form micronuclei by drug treatment reported the coincidence of apoptosis and the presence of micronuclei87. The authors favoured explanation that the malfunctioning of gene expression in the micronuclei might trigger apoptosis, but they could not exclude the harmful effect of severe DNA damage in micronuclei. In the case of hybrid frog early gametogenesis we proved that the prevailing group of micronucleated gonocytes was not directed to apoptosis, so we assume that micronuclei formation during hybridogenetic frog gametogenesis is a physiological process. Degenerating organelles are neutralized and eventually the activation of cell death is prevented due to autophagy that plays a role of the survival factor63,88. Because the majority of gonocytes survives at least during premetamorphosis and early gonad development, we suppose that micronuclei formation is a part of the programmed genome rearrangement. The presence and further disruption of micronuclei do not necessarily lead to cell death and do not prevent cell divisions47,52,69 as also shown in our study. In hybridogenetic frogs, it seems probable that gonocytes in developing gonads would divide several times until the whole DNA of the eliminated genome is removed. Micronuclei were detected at similar time of tadpole development (around metamorphosis) when Tunner and Heppich35, Heppich et al.89, and Tunner and Heppich-Tunner36 reported the gradual loss of the lessonae chromosomes from the gonocytes of P. esculentus. The process of genome elimination in water frogs seems to be long and inaccurate, although the production of micronuclei as such do not cause gonocyte death. Development and differentiation of gonads in both sexes are delayed at least one year, as compared to the parental species, and a great bulk of germ cells degenerate at this time34,65,90. This raises the question, which cellular mechanisms may be compromised during the genome elimination. We can only hypothesize that the loss of some genes maintaining the proper cell cycle progression or required for cell viability, which were entrapped into micronuclei, exposes possible deleterious mutations on the clonally transmitted chromosomes and eventually leads to apoptotic cell death87,91.
Micronuclei, putatively carrying the eliminated genome, have never been observed in somatic cells of hybridogenetic animals although as a rule they have two different chromosome sets: one from the mother and the second from the father. The homeologue chromosomes of both the parental species of P. esculentus are similar, however, in the ridibundus chromosomes heterochromatin is more abundant, DNA content is higher, and they are longer than the lessonae ones23,92,93,94,95. Apparently, the chromosome incompatibility as such is not the only prerequisite for genome elimination from the germline cells. We suspect that there is an unknown mechanism(s) that acts only in cells committed to undergo meiosis that may be triggered during gonocyte proliferation.
Conclusions
Chromatin elimination via micronuclei in P. esculentus is unique among hybridogenetic animals and contributes to broadening the knowledge about reproductive modes in animals. Here we show for the first time that formation of micronuclei in early pre-meiotic germ cells (gonocytes) of a hybrid frog is a physiological process that may be considered as the programmed DNA rearrangement leading to eventual elimination. Micronuclei contain chromatin directed for degradation, which is the essence of genome elimination in hybridogenetic gametogenesis. In the light of our observations presented in this study we suggest the following mechanism of micronuclei formation and degradation in hybrid water frogs: (a) budding of chromatin into micronuclei encapsulated by double membrane with lower density of nuclear pore complexes, (b) subsequent heterochromatinization and loss of transcriptional activity followed by (c) probable degradation of DNA by autophagy (nucleophagy). The process is the same in females and males, both diploid (RL) and triploid (RRL and LLR). Our study on cellular level is a necessary basis for further genetic and epigenetic research on hybridogenesis in Pelophylax water frogs. Comparing the mechanism of micronuclei formation, chromatin silencing and heterochromatinization, as well as degradation via autophagy described in hybridogenetic frogs, other organisms, and cancer cells, we conclude that some aspects of the DNA elimination process may be evolutionarily conserved.
Materials and Methods
Animals
All procedures with both adults and tadpoles were performed in accordance with relevant guidelines and regulations. The capture of adult frogs was approved by the Polish General and Regional Directorates for Environmental Protection (DOPg 4201-02-74/05, DOP-oz.6401.02.2.2013.JRO, DOP-oz.6401.02.2.2013.2014.JRO.as, WPN.6205.28.2014.IW.2, DZP-WG.6401.02.5.2015.JRO, WPN.6401.177.2016.IL). The adult individuals used for in vitro crosses were sampled from different locations in Poland, according to data collected in Supplementary Table S6A. P. lessonae (females N = 8, males N = 16) were obtained from populations of L-E and R-E-L type systems: P. ridibundus (females N = 11, males N = 6) were collected from R-E and R-E-L population systems. P. esculentus (RL females N = 7, RL males N = 3, RLL males N = 1) were taken from E-E population systems in Wysoka Kamieńska from north-western Poland96, and Uciechów from south-western Poland, as well as from other locations (Supplementary Table S6B).
All further experimental procedures were accepted by the Local Commission for Ethics in Experiments on Animals in Wrocław, Poland (103/2007, 7/2013, 27/2016). The collected animals were kept in humid boxes and subsequently used for in vitro crossing experiments according to the standard procedures for water frogs97. Gravid females were injected intraperitoneally with 6,25 mg/kg of body weight salmon luteinizing hormone-releasing hormone (LHRH, H-7525.0001, Bachem) in amphibian PBS (APBS, pH 7.4, 11.2 mM NaCl, 0.22 mM KCl, 0.8 mM Na2HPO4, 0.14 mM KH2PO4) 24 hours before the procedure. Males and females were sacrificed after anesthetizing in 0.5% solution of ethyl 3-aminobenzoate methanesulfonate (MS-222, Sigma Chemical Co.) in APBS. Tissues and organs were dissected for DNA (phalange) and chromosomal (intestine) analyses, as well as for in vitro fertilization of eggs (testes). Tadpoles of various phenotypes (LL, RR, RL, RRL, RLL), resulting from controlled in vitro crosses, were reared in a greenhouse in PPE tanks, in a concentration of c.a. 10 tadpoles per litre of water, and fed with frozen lettuce and fish food. Altogether, 7 tadpoles of P. lessonae (progeny of 4 pairs), 2 of P. ridibundus (progeny of 2 pairs), and 46 diploid and 11 triploid tadpoles of P. esculentus (progeny of 24 pairs) were used (for details see Supplementary Table S7). Staging of somatic development of tadpoles followed Gosner62. Gonads for morphological and immunohistochemical analyses were dissected from tadpoles after anesthetizing by immersion in 0.25% solution of MS-222 (Sigma Chemical Co.) in APBS.
Taxonomic evaluation of animals
Taxonomic evaluation of adult frogs collected for crossing experiments was performed first according to morphological criteria. Ploidy of P. esculentus individuals was estimated before performing crosses using erythrocyte long axis measurements according to Kierzkowski et al.98. The blood smears were obtained after gentle cutting one of the phalanges of a hind limb and air-dried. Measurement of the erythrocytes long axis was performed under Axiostar Plus microscope (Zeiss) using 20× lens and KS400 software (Zeiss). Diploid erythrocytes were 23.4–24.9 µm long and triploid were 29.5–33.3 µm. Species level of parental individuals and tadpoles (used in the examination of nucleoporins, caspase-3 and autophagy) were confirmed by allele size polymorphism in serum albumin introne-1. DNA amplification was done according to Hauswaldt et al.99 with some modification in PCR protocol following Kolenda et al.100 (see Supplementary Table S6C). The PCR reaction was performed in a C1000 Thermal Cycler (Bio-Rad, USA): initial denaturation for 3 min at 95 °C followed by 35 cycles of 30 s denaturation at 94 °C, 30 s annealing at 53 °C, and 60 s elongation at 72 °C, and final extension for 7 min at 72 °C. Three µl of PCR products were separated electrophoretically on a 1.5% agarose gel and compared with a 100 bp size marker.
Further analysis of ploidy and genomic composition was done for adult individuals of P. esculentus and for progeny sourcing from P. esculentus parents, using microsatellites analysis of sequences differentiating ploidy, FISH with telomeric probe or AMD-DAPI (details in Supplementary Tables S6C and S7).
Microsatellites analysis
Six nuclear microsatellite loci were used: Re1Caga10, RICa1b6, RICa1b5, Ga1a19red, Rid059A, Rrid013A (details in Supplementary Table S6C). DNA amplification was performed in one multiplex PCR set with forward primers fluorescently labelled with 6-FAM, NED, PET and VIC. Reaction was carried out in 6 µl, containing 1 µl DNA, 3 µl 2 × Multiplex PCR Master MIX (EURx Ltd., Poland), 0.06 µl (10 µM) of both forward and reverse primers (a total of 0.72 µl of primers mix). The PCR reaction was conducted in a C1000 Thermal Cycler (Bio-Rad, USA) under following conditions: initial denaturation for 15 min at 95 °C followed by 30 cycles of 30 s denaturation at 94 °C, 90 s annealing at 60 °C, and 60 s elongation at 72 °C, and final extension for 30 min at 60 °C. Separation of PCR products was outsourced at a commercial facility (Genomed, Warsaw, Poland). Ploidy level and genomic composition were determined using GENEMAPPER v. 3.5 (Applied Biosystems, USA).
Fluorescence in situ hybridization
For analysis of epigenetic chromatin modification during early gonadal development, we took P. lessonae, as well as diploid and triploid hybrid tadpoles which were identified using FISH with TTAGGG probe. This method is based on difference between parental species in amount and localization of interstitial telomeric sites on NOR-bearing chromosome. Earlier we found that P. ridibundus NOR bearing chromosome has two interstitial telomeric sites while P. lessonae NOR bearing chromosome - only one interstitial telomeric site. Thus distinguishing of P. lessonae and/or P. ridibundus NOR bearing chromosomes allows to infer karyotype composition of parental species as well as diploid and triploid hybrid frogs32,93.
FISH with single-stranded oligonucleotide telomeric probes (TTAGGG)5 conjugated with Cy3 was performed on metaphase chromosomes as described previously32,93. Before hybridization metaphase plates were pre-treated with RNase A (100–200 μg/ml) for 1 h, pepsin (0.01% in 0.01 N HCl) for 10 min and then post-fixed in formaldehyde (1% in PBS, 50 mM MgCl2) for 10 min. Hybridization mixture (40% formamide, 2.4 × SSC, and 12% dextran sulphate, 5 ng/μl labelled probe and 10–50-fold excess of tRNA) was added to metaphase chromosomes. After denaturation at 82 °C for 5 min slides were incubated at room temperature for 12–18 h. Then slides were washed three times in 2 × SSC at 42 °C. After FISH chromosomal preparations were mounted in DABCO antifade solution containing 1 mg/ml DAPI.
AMD-DAPI chromosomal staining
The method is based on difference in centromeric regions staining pattern between parental species, where ridibundus chromosomes display intensive signals, whereas lessonae do not stain or stain weakly. Genome composition was determined on 10–20 metaphase plates. Intestinal epithelial cells were put on a slide in a drop of 70% acetic acid and squashed under a coverslip. The slides were put on dry ice block until frozen, and then coverslips were removed. Chromosomes were stained with Actinomycin D- 4′,6-diamidino-2-phenylindole (DAPI) as described by Heppich et al.89. Slides were examined using Zeiss Axioskop microscope (Zeiss) equipped with a fluorescence lamp and appropriate filters.
Transmission electron microscopy
Gonads samples for TEM examination were fixed in 2.5% glutaraldehyde (Serva Electrophoresis, Heidelberg, Germany). After 24 hours, the specimens were washed in the cacodylate or phosphate buffers (0.1 M, pH 7.4, Serva) and postfixed for 1 h in 1% osmium tetroxide (Serva). Subsequently, the specimens were dehydrated in ethyl alcohol and twice in pure acetone (Chempur, Poland). Afterwards samples were embedded in epoxy resin (Epon 812, Serva). Epon blocks were cut into semithin (600 nm thick) sections and stained with toluidine blue (Serva). Finally, ultrathin (50 nm thick) sections were prepared using an ultramicrotomes Power Tome XL (RMC, Tucson, USA) and Reichert Ultracut E. Ultrathin sections were counterstained with uranyl acetate and lead citrate (Serva), and examined in a TEM JEM-1011 (JEOL,Tokyo, Japan) or Zeiss EM 900. Digital micrographs were collected with the use of TEM imaging platform iTEM1233 equipped with the Morada Camera (Olympus, Münster, Germany).
Immunofluorescence on tissue sections
Immunofluorescence studies were performed on tissue sections of P. esculentus, P. lessonae and P. ridibundus gonads collected at 28–45 Gosner stages. Nuclear envelope staining was carried out using mouse monoclonal antibodies raised against F/G repeat nucleoporins, mAb414 (diluted 1:250, Abcam ab24609, recommended for Xenopus leavis), recognizing proteins of the external cytoplasmic part, central core and inner nuclear basket (Nucleoporins p62, Nup153, Nup214, Nup358), previously used in Xenopus66,67. Apoptosis was detected using rabbit antibodies recognizing active caspase-3 (diluted 1:250, Abcam ab13847, recommended for Xenopus), previously used in P. esculentus – complex65. A positive control for the proper antibody staining pattern in the gonads were degenerating cells in parental species shortening testes64. Autophagy was examined with rabbit antibody anti LC3 (diluted 1:200, Novus Biologicals NB100–2331, predicted to react with Xenopus based on 100% sequence homology), recognizing both LC3A and LC3B proteins. Wheat germ agglutinin conjugated with Texas Red (WGA, 5 µg/ml, Molecular Probes/Life Technologies) was used for counterstaining of plasma membrane. WGA can bind sialic acid and N-acetylglucosamine in the integral membrane glycoproteins present in post-Golgi membranes as well as in plasma membrane and is a marker of autophagosome formation83.
Gonads were dissected under Stemi Zeiss stereomicroscope from 23 individuals: 3 P. lessonae, 2 P. ridibundus and 18 P. esculentus (16 RL, 2 RRL). For nucleoporins and caspase-3 staining tissues were fixed in 2% PFA in APBS for 1, 5-2 hours at RT, washed 3 × 15 min in PBS and incubated for 2–4 hours in 12.5% and overnight in 25% sucrose in PBS at 4 °C, then mounted in tissue freezing medium (Leica Biosystems), frozen at −20 °C and stored at −80 °C until analysis. Cryostat sections, 7 µm thick, were cut on CM1850UV microtome (Leica Biosystems), mounted on Superfrost Plus microscope slides (Thermo Scientific), washed in PBS (pH 7.4, 14 mM NaCl, 0.27 mM KCl, 1 mM Na2HPO4, 0.18 mM KH2PO4) with 0.05% Tween 20 (Sigma) (PBST), incubated with WGA for 10 min at RT, then permeabilized in 0.5% Triton X-100 (Sigma) in PBS for 15 min at RT. For LC3A/B stainings tissues were fixed for 24 hours in 2% PFA in APBS at 4 °C, washed in 10 changes of PBS, then dehydrated through graded alcohol solutions and xylene and embedded in paraffin. Paraffin tissue sections, 7 µm thick, were cut using RM 2255 microtome (Leica Biosystems), deparaffinized in xylen and hydrated in graded alcohol. Heat induced antigen retrieval was performed in 0.01 M citrate buffer, pH 6.0, at 97 °C for 20 min. After washing with PBST, tissues were blocked in 3% goat serum (Sigma) diluted in PBS for 30 min at RT, then primary antibodies diluted in 1% goat serum in PBST were applied for overnight incubation at 4 °C, followed by PBST washing. Secondary antibodies used were goat anti-mouse Alexa Fluor 488 and goat anti-rabbit Alexa Fluor 546 (Invitrogen, Life Technologies), or donkey anti-rabbit conjugated with CY5 (Jackson Immunoresearch) diluted 1:500–1:2000 in 1% goat serum in PBST containing 0.5 µg/ml DAPI and incubated for 1 h at RT, followed by PBST washes. Sections were mounted in ProLong Gold antifade reagent (Invitrogen, Life Technologies). Gonads were analysed using Olympus FV1000 confocal microscope. 3D models of labelled gonads were generated using Imaris 6.2.1 software (Bitplane).
3D immunofluorescent staining (Immunofluorescent staining of whole tissues)
For 3D immunofluorescent staining we used rabbit pАТ ab8898 against H3K9Me3 (Abcam) and mouse pАТ 06-866 against H4Ac (Up-state). 3D immunofluorescent staining was performed on whole gonads dissected from tadpoles of P. lessonae (N = 4), diploid (N = 9) and triploid (N = 2) P. esculentus. Dissection was carried out under observation on stereomicroscope Leica MZ16. Tissues were fixed in 2% PFA prepared on 1× PBS during 1.5 hours at RT. Tissue fragments were stored in PBS with 0.02% Na azide. Before hybridization tissue fragments were permeabilized in 1% Triton X-100 in PBS for 4–5 hours at RT. Tissues were washed in PBS, blocked in PBS containing 1% blocking reagent (Roche) for 1 hour at RT and incubated with primary antibody (1:200 dilution) overnight at RT. Tissues were washed in PBS, and incubated in corresponding secondary antibodies (Alexa-488-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch Laboratories), Alexa-488-conjugated goat anti-mouse IgG (Molecular Probes) for 8 h at RT. Tissues were washed in PBS, and incubated in PBS containing 1 mg/ml DAPI overnight at RT.
TUNEL assay
In paraffin sections, apoptosis was assessed by TUNEL technique, using Click-iT Plus TUNEL Assay for in situ apoptosis detection with Alexa Fluor 647 dye (Molecular Probes, Life Technologies). The procedure was performed according to manufacturer’s instructions. In four gonads, TUNEL assay was followed by immunofluorescence staining for LC3 according to the procedure described above. Each gonad was evaluated in several sections for presence of apoptotic cells, autophagy and micronuclei in gonocytes. Documentation of results was done using Olympus FV1000 confocal microscope.
Measurements of micronuclei size and immunofluorescence signal intensity
Measurements of micronuclei diameter and immunofluorescence signal intensity were done in digital confocal images using Fiji software101 or LAS software (Leica) (for Nup and LC3 specimens or for histone modifications, respectively). Line tool was used to measure distance between the edges of DAPI signal in micronuclei on z-section showing the maximal diameter and DAPI staining. Results are presented in micrometers.
Intensity of DAPI fluorescence was measured in main nuclei and micronuclei using manual free line drawing tool to mark the region of interest (ROI) within the border of DAPI signal on z-section showing the maximal diameter of the measured structure. Collected data represented the values of relative fluorescence mean grey value from the area selected by ROI. To assess whether micronuclei possessed stronger DAPI signal (reflecting the DNA density in the structure) than main nuclei we calculated the ratio of micronucleus mean fluorescence value to main nucleus mean fluorescence value. Micronuclei were grouped arbitrally within the ranges: up to 0.79 – MN have lower fluorescence than nuclei; 0.8–1.2 – fluorescence levels are similar in MN and nuclei; higher than 1.21 – MN have distinctly stronger DAPI signal than main nuclei.
Nup fluorescence intensity measurements were done in borderline of chromatin area reflecting nuclear envelope Nup signal both in MN and main nuclei. The 2pixel-thick ROI lines were created after transformation of the ROI line set previously for DAPI, and program measured intensities of all pixels in the marked area. Raw data represented the mean grey value, minimum and maximum values of fluorescence intensity in the nuclear envelope area, and they were standardized to the perimeter of the structure by means of FIJI measurements settings. The results are represented as ratio of relative Nup fluorescence of micronuclei to main nuclei. We have compared data sets for groups set previously on basis of qualitative image analysis: (1) Nup staining present in rim area of micronuclei nuclear envelope, (2) Nup staining present as single dots or patches aggregated on one side of MN, (3) no distinct Nup signals detected visually in nuclear envelope.
LC3 fluorescence intensity measurements were done in the same ROI area set initially for DNA measurement in main nuclei and micronuclei. Additionally, LC3 intensity was measured in ROI of LC3 aggregates around micronucleus. The results are presented as ratio of relative LC3 fluorescence of micronuclei to main nuclei. We have compared data sets for groups set previously on basis of qualitative image analysis: (1) strong signal in LC3 aggregates surrounding micronuclei, (2) weak LC3 signals present as single dots or patches aggregated on one side of MN, (3) no distinct LC3 signals around micronuclei.
Estimation of signal intensiveness in the main nuclei and micronuclei after immunofluoresce staining with antibodies against H3K9me3 and H4Ac was made using Leica LasAF software. ROI was selected manually for each micronucleus and corresponding main nuclei. Signal intensity was measured for DAPI channel and channel with fluorescence staining for micronuclei (value IF signal MN; value DAPI signal MN) and main nuclei (value IF signal N; value DAPI signal N). To detect changes in fluorescence intensity for histone marks we used relative staining value of the antibody staining fluorescence (either H3K9me3 or H4Ac) to fluorescence of DAPI (value IF signal MN/value DAPI signal MN; value IF signal N/value DAPI signal N). The comparison of this relative indices between micronuclei and main nuclei allowed to assess fluctuation of the fluorescent staining in the micronuclei comparatively to main nucleus: (value IF signal MN/value DAPI signal MN) – (value IF signal N/value DAPI signal N).
Statistical analysis
Statistical analysis was performed using STATISTICA 12.5 software (StatSoft, Poland). Groups of data (groups of micronuclei displaying different distributions of NPC and LC3 signal) were compared using the non-parametric Kruskal-Wallis test. Statistical significance was defined when P ≤ 0.05.
Image processing
Any brightness and contrast adjustments were performed in Adobe Photoshop and Corel Draw.
Data Availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information files).
References
Kloc, M. & Zagrodzinska, B. Chromatin elimination - An oddity or a common mechanism in differentiation and development? Differentiation 68, 84–91 (2001).
Wang, J. & Davis, R. E. Programmed DNA elimination in multicellular organisms. Curr. Opin. Genet. Dev. 27, 26–34 (2014).
Stanley, H. P., Kasinsky, H. E. & Bols, N. C. Meiotic chromatin diminution in a vertebrate, the holocephalan fish Hydrolagus collie (Chondrichthyes, Holocephali). Tissue Cell 16, 203–15 (1984).
Pigozzi, M. I. & Solari, A. J. Meiotic recombination in the ZW pair of a tinamid bird shows a differential pattern compared with neognaths. Genome 48, 286–290 (2005).
Goday, C. & Pigozzi, M. I. Heterochromatin and histone modifications in the germline-restricted chromosome of the zebra finch undergoing elimination during spermatogenesis. Chromosoma 119, 325–336 (2010).
Mendes-Bonato, A. B., Pagliarini, M. S. & Valle, C. Bdo Abnormal spindle orientation during microsporogenesis in an interspecific Brachiaria (Gramineae) hybrid. Genet. Mol. Biol. 29, 122–125 (2006).
Lamatsch, D. & Stöck, M. In Lost sex. The Evolutionary Biology ofParthenogenesis (eds Schön, I., Martens, K. & Dijk, P.) 399–432 (Springer Netherlands,). 10.1007/978-90-481-2770-2 (2009).
Schultz, R. J. Hybridization, Unisexuality, and Polyploidy in the Teleost Poeciliopsis (Poeciliidae) and Other Vertebrates. Am. Nat. 103, 605–619 (1969).
Schmidt, D. J., Bond, N. R., Adams, M. & Hughes, J. M. Cytonuclear evidence for hybridogenetic reproduction in natural populations of the Australian carp gudgeon (Hypseleotris: Eleotridae). Mol. Ecol. 20, 3367–3380 (2011).
Kimura-Kawaguchi, M. R. et al. Identification of hemiclonal reproduction in three species of Hexagrammos marine reef fishes. J. Fish Biol. 85, 189–209 (2014).
Scali, V. In Lost sex. The Evolutionary Biology ofParthenogenesis (eds Schön, I., Martens, K. & Dijk, P.) 317–345 (Springer Netherlands,). 10.1007/978-90-481-2770-2 (2009).
Tinti, F. & Scali, V. Genome exclusion and gametic DAPI-DNA content in the hybridogenetic Bacillus rossius-grandii benazzii complex (Insecta Phasmatodea). Mol. Reprod. Dev. 33, 235–242 (1992).
Marescalchi, O. & Scali, V. New DAPI and FISH findings on egg maturation processes in related hybridogenetic and parthenogenetic Bacillus hybrids (Insecta, Phasmatodea). Mol. Reprod. Dev. 60, 270–276 (2001).
Scali, V., Passamonti, M., Marescalchi, O. & Mantovani, B. Linkage between sexual and asexual lineages: genome evolution in Bacillus stick insects. Biol. J. Linn. Soc. 79, 137–150 (2003).
Alves, M. J., Coelho, M. M. & Collares-Pereira, M. J. Diversity in the Reproductive Modes of Females of the Rutilus alburnoides Complex (Teleostei, Cyprinidae): A Way to Avoid the Genetic Constraints of Uniparentalism. Mol. Biol. Evol. 15, 1233–1242 (1998).
Goddard, K. A. & Schultz, R. J. Aclonal Reproduction by Polyploid Members of the Clonal Hybrid Species Phoxinus eos-neogaeus (Cyprinidae). Copeia 1993, 650–660 (1993).
Saitoh, K., Kim, I.-S. & Lee, E.-H. Mitochondrial Gene Introgression between Spined Loaches via Hybridogenesis. Zoolog. Sci. 21, 795–798 (2004).
Morishima, K., Yoshikawa, H. & Arai, K. Meiotic hybridogenesis in triploid Misgurnus loach derived from a clonal lineage. Heredity (Edinb). 100, 581–586 (2008).
Arias-Rodriguez, L., Yasui, G. S. & Arai, K. Disruption of normal meiosis in artificial inter-populational hybrid females of Misgurnus loach. Genetica 136, 49–56 (2009).
Zhang, Q., Arai, K. & Yamashita, M. Cytogenetic mechanisms for triploid and haploid egg formation in the triploid loach Misgurnus anguillicaudatus. J. Exp. Zool. 281, 608–619 (1998).
Stöck, M. et al. A Vertebrate Reproductive System Involving Three Ploidy Levels: Hybrid Origin Of Triploids In A Contact Zone Of Diploid And Tetraploid Palearctic Green Toads (Bufo viridis Subgroup). Evolution (N. Y). 64, 944–959 (2009).
Berger, L., Roguski, H. & Uzzell, T. Triploid F2 progeny of water frogs (Rana esculenta complex). Folia Biol. 26, 135–152 (1978).
Vinogradov, A. E., Borkin, L. J., Günther, R. & Rosanov, J. M. Genome elimination in diploid and triploid Rana esculenta males: cytological evidence from DNA flow cytometry. Genome 33, 619–627 (1990).
Berger, L. Systematics and Hybridization in European Green Frogs of Rana esculenta Complex. J. Herpetol. 7, 1–10 (1973).
Uzzell, T. & Berger, L. Electrophoretic Phenotypes of Rana ridibunda, Rana lessonae, and Their Hybridogenetic Associate, Rana esculenta. Proc. Acad. Nat. Sci. Philadelphia 127, 13–24 (1975).
Rybacki, M. & Berger, L. Types of water frog populations (Rana esculenta complex) in Poland. Zoosystematics Evol. 77, 51–57 (2001).
Plötner, J. D. W. Wasserfrösche. Beih. Zeitschrift für Feldherpetologie 9, 1–160 (2005).
Christiansen, D. G. Gamete types, sex determination and stable equilibria of all-hybrid populations of diploid and triploid edible frogs (Pelophylax esculentus). BMC Evol. Biol. 9, 135 (2009).
Hermaniuk, A., Pruvost, N. B. M., Kierzkowski, P. & Ogielska, M. Genetic and Cytogenetic Characteristics of Pentaploidy in Water Frogs. Herpetologica 69, 36–45 (2013).
Graf, J. D. & Müller, W. P. Experimental gynogenesis provides evidence of hybridogenetic reproduction in the Rana esculenta complex. Experientia 35, 1574–6 (1979).
Bucci, S. et al. Lampbrush and mitotic chromosomes of the hemiclonally reproducing hybrid Rana esculenta and its parental species. J. Exp. Zool. 255, 37–56 (1990).
Dedukh, D. et al. Optional endoreplication and selective elimination of parental genomes during oogenesis in diploid and triploid hybrid European water frogs. PLoS One 10, e0123304, https://doi.org/10.1371/journal.pone.0123304 (2015).
Graf, J. & Polls-Pelaz, M. In Evolution and ecology of unisexual vertebrates (eds Dawley, R. & Bogart, J.) 289–302 (New York State Museum Publications, 1989).
Ogielska, M. In Reproduction of Amphibians (ed Ogielska, M.) 343–410 (Science Publishers, 2009).
Tunner, H. G. & Heppich, S. Premeiotic genome exclusion during oogenesis in the common edible frog. Rana esculenta. Naturwissenschaften 68, 207–208 (1981).
Tunner, H. G. & Heppich-Tunner, S. Genome exclusion and two strategies of chromosome duplication in oogenesis of a hybrid frog. Naturwissenschaften 78, 32–34 (1991).
Vogt, A., Goldman, A. D., Mochizuki, K. & Landweber, L. F. Transposon Domestication versus Mutualism in Ciliate Genome Rearrangements. PLoS Genet. 9, e1003659, https://doi.org/10.1371/journal.pgen.1003659 (2013).
Sánchez, L. Sex-determining mechanisms in insects based on imprinting and elimination of chromosomes. Sex. Dev. 8, 83–103 (2014).
Herrick, G. & Seger, J. Imprinting and paternal genome elimination in insects. Results Probl. Cell Differ. 25, 41–71 (1999).
Ravi, M. & Chan, S. W. L. Haploid plants produced by centromere-mediated genome elimination. Nature 464, 615–618 (2010).
Sanei, M., Pickering, R., Kumke, K., Nasuda, S. & Houben, A. Loss of centromeric histone H3 (CENH3) from centromeres precedes uniparental chromosome elimination in interspecific barley hybrids. Proc. Natl. Acad. Sci. USA 108, E498–505 (2011).
Camenzind, R. & Fux, T. Dynamics and Ultrastructure of Monocentric Chromosome Movement. Caryologia 30, 127–150 (1977).
Kubai, D. F. Meiosis in Sciara coprophila: structure of the spindle and chromosome behavior during the first meiotic division. J. Cell Biol. 93, 655–69 (1982).
Kubai, D. F. Nonrandom chromosome arrangements in germ line nuclei of Sciara coprophila males: the basis for nonrandom chromosome segregation on the meiosis I spindle. J. Cell Biol. 105, 2433–46 (1987).
Cimino, M. C. E.-P. Polyploidization andEvolution in a Diploid All-Female Fish of the Genus Poeciliopsis. Evolution (N. Y). 26, 294–306 (1972).
Terradas, M., Martín, M., Tusell, L. & Genescà, A. Genetic activities in micronuclei: Is the DNA entrapped in micronuclei lost for the cell? Mutat. Res. - Rev. Mutat. Res. 705, 60–67 (2010).
Huang, Y., Fenech, M. & Shi, Q. Micronucleus formation detected by live-cell imaging. Mutagenesis 26, 133–138 (2011).
Utani, K., Okamoto, A. & Shimizu, N. Generation of micronuclei during interphase by coupling between cytoplasmic membrane blebbing and nuclear budding. PLoS One 6, e27233, https://doi.org/10.1371/journal.pone.0027233 (2011).
Gernand, D. et al. Uniparental Chromosome Elimination at Mitosis and Interphase in Wheat and Pearl Millet Crosses Involves Micronucleus Formation. Progressive Heterochromatinization, and DNA Fragmentation. 17, 2431–2438 (2005).
Gernand, D., Rutten, T., Pickering, R. & Houben, A. Elimination of chromosomes in Hordeum vulgare×H. bulbosum crosses at mitosis and interphase involves micronucleus formation and progressive heterochromatinization. Cytogenet. Genome Res. 114, 169–174 (2006).
Perondini, A. L. P. & Ribeiro, A. F. Chromosome elimination in germ cells of Sciara embryos: involvement of the nuclear envelope. Invertebr. Reprod. Dev. 32, 131–141 (1997).
Timoshevskiy, V. A., Herdy, J. R., Keinath, M. C. & Smith, J. J. Cellular and Molecular Features of Developmentally Programmed Genome Rearrangement in a Vertebrate (Sea Lamprey: Petromyzon marinus). PLoS Genet. 12, 1–20 (2016).
Luzhna, L., Kathiria, P. & Kovalchuk, O. Micronuclei in genotoxicity assessment: From genetics to epigenetics and beyond. Front. Genet. 4, 1–17 (2013).
Ogielska, M. Nucleus-like bodies in gonial cells of Rana esculenta [Amphibia, Anura] tadpoles—a putative way of chromosome elimination. Zool. Pol. 39, 461–474 (1994).
Dedukh, D., Litvinchuk, S., Rosanov, J., Shabanov, D. & Krasikova, A. Mutual maintenance of di- and triploid Pelophylax esculentus hybrids in R-E systems: Results from artificial crossings experiments. BMC Evol. Biol. 17, 1–15 (2017).
Ogielska, M. & Kotusz, A. Pattern and rate of ovary differentiation with reference to somatic development in anuran amphibians. J. Morphol. 259, 41–54 (2004).
Ogielska, M. & Bartmańska, J. In Reproduction of Amphibians (ed Ogielska, M.) 153–272 (Science Publishers, 2009).
Ogielska, M. & Bartmańska, J. In Reproduction of Amphibians (ed Ogielska, M.) 34–99 (Science Publishers, 2009).
Ogielska, M. In Reproduction of Amphibians (ed Ogielska, M.) 1–33 (Science Publishers, 2009).
Ogielska, M., Kotusz, A., Augustyńska, R., Ihnatowicz, J. & Paśko, Ł. A. Stockpile of Ova in the Grass Frog Rana temporaria is Established Once for the Life Span. Do Ovaries in Amphibians and in Mammals Follow the Same Evolutionary Strategy? Anat. Rec. 296, 638–653 (2013).
Haczkiewicz, K., Rozenblut-Kościsty, B. & Ogielska, M. Prespermatogenesis and early spermatogenesis in frogs. Zoology 122, 63–79 (2017).
Gosner, K. L. A Simplified Table for Staging Anuran Embryos Larvae with Notes on Identification. Herpetologica 16, 183–190 (1960).
Mijaljica, D. & Devenish, R. J. Nucleophagy at a glance. J. Cell Sci. 126, 4325–30 (2013).
Haczkiewicz, K. & Ogielska, M. Gonadal sex differentiation in frogs: how testes become shorter than ovaries. Zoolog. Sci. 30, 125–34 (2013).
Szydłowski, P., Chmielewska, M., Rozenblut-Kościsty, B. & Ogielska, M. The frequency of degenerating germ cells in the ovaries of water frogs (Pelophylax esculentus complex). Zoomorphology 136, 75–83 (2016).
Chmielewska, M. et al. Embryonic and adult isoforms of XLAP2 form microdomains associated with chromatin and the nuclear envelope. Cell Tissue Res. 344, 97–110 (2011).
Dubińska-Magiera, M. et al. Xenopus LAP2β protein knockdown affects location of lamin B and nucleoporins and has effect on assembly of cell nucleus and cell viability. Protoplasma 253, 943–956 (2016).
Terradas, M., Martín, M., Hernández, L., Tusell, L. & Genescà, A. Nuclear envelope defects impede a proper response to micronuclear DNA lesions. Mutat. Res. - Fundam. Mol. Mech. Mutagen. 729, 35–40 (2012).
Hatch, E. M., Fischer, A. H., Deerinck, T. J. & Hetzer, M. W. Catastrophic Nuclear Envelope Collapse in Cancer Cell Micronuclei. Cell 154, 47–60 (2013).
Bongiorni, S., Pasqualini, B., Taranta, M., Singh, P. B. & Prantera, G. Epigenetic regulation of facultative heterochromatinisation in Planococcus citri via the Me(3)K9H3-HP1-Me(3)K20H4 pathway. J. Cell Sci. 120, 1072–80 (2007).
Liu, Y. et al. RNAi-dependent H3K27 methylation is required for heterochromatin formation and DNA elimination in Tetrahymena. Genes Dev. 21, 1530–1545 (2007).
Herdy, J. R. Small RNA Expression During Programmed Rearragement of a Vertebrate Genome. (University of Kentucky, 2014).
Schoenmakers, S., Wassenaar, E., Laven, J. S. E., Grootegoed, J. A. & Baarends, W. M. Meiotic silencing and fragmentation of the male germline restricted chromosome in zebra finch. Chromosoma 119, 311–324 (2010).
Escribá, M. C. & Goday, C. Histone H3 phosphorylation and elimination of paternal X chromosomes at early cleavages in sciarid flies. J. Cell Sci. 126, 3214–3222 (2013).
Del Priore, L. & Pigozzi, M. I. Histone modifications related to chromosome silencing and elimination during male meiosis in Bengalese finch. Chromosoma 123, 293–302 (2014).
Rello-Varona, S. et al. Autophagic removal of micronuclei. Cell Cycle 11, 170–176 (2012).
Wirth, M., Joachim, J. & Tooze, S. A. Autophagosome formation-The role of ULK1 and Beclin1-PI3KC3 complexes in setting the stage. Semin. Cancer Biol. 23, 301–309 (2013).
Sergin, I. & Razani, B. Self-eating in the plaque: What macrophage autophagy reveals about atherosclerosis. Trends Endocrinol. Metab. 25, 225–234 (2014).
Nakayama, Y. & Inoue, T. Antiproliferative fate of the tetraploid formed after mitotic slippage and its promotion; a novel target for cancer therapy based on microtubule poisons. Molecules 21, 663 (2016).
Lu, E. & Wolfe, J. Lysosomal enzymes in the macronucleus of Tetrahymena during its apoptosis-like degradation. Cell Death Differ. 8, 289–297 (2001).
Liu, M. L. & Yao, M. C. Role of ATG8 and Autophagy in programmed nuclear degradation in Tetrahymena thermophila. Eukaryot. Cell 11, 494–506 (2012).
Gadeberg, H. C. et al. Heterogeneity of T-Tubules in Pig Hearts. PLoS One 11, e0156862 (2016).
Yamamoto, A., Masaki, R. & Tashiro, Y. Characterization of the isolation membranes and the limiting membranes of autophagosomes in rat hepatocytes by lectin cytochemistry. J. Histochem. Cytochem. 38, 573–580 (1990).
Crasta, K. et al. DNA breaks and chromosome pulverization from errors in mitosis. Nature 482, 53–8 (2012).
Fujiwara, Y. et al. Direct uptake and degradation of DNA by lysosomes. Autophagy 9, 1167–1171 (2013).
Fujiwara, Y., Wada, K. & Kabuta, T. Lysosomal degradation of intracellular nucleic acids-multiple autophagic pathways. J. Biochem. 161, 145–154 (2017).
Utani, K. I., Kohno, Y., Okamoto, A. & Shimizu, N. Emergence of micronuclei and their effects on the fate of cells under replication stress. PLoS One 5, e10089, https://doi.org/10.1371/journal.pone.0010089 (2010).
Klionsky, D. J. et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition. Autophagy 12, 1–222 (2016).
Heppich, S., Tunner, H. G. & Greilhuber, J. Premeiotic chromosome doubling after genome elimination during spermatogenesis of the species hybrid Rana esculenta. Theor. Appl. Genet. 61, 101–104 (1982).
Ogielska, M. & Wagner., E. Oogenesis and development of the ovary in European green frog, Rana ridibunda (Pallas). I. Tadpole stages until metamorphosis. Zool. Jahrbücher Abteilung für Anat. und Ontog. der Tiere 120, 211–221 (1990).
Vorburger, C., Schmeller, D., Hotz, H., Guex, G.-D. & Reyer, H.-U. In Lost Sex: The Evolutionary Biology of Parthenogenesis (eds Schön, I., Van Dijk, P. & Martens, K.) 433–446 (Springer,). 10.1007/978-90-481-2770-2 (2009).
Ragghianti, M. et al. Molecular characterization of a centromeric satellite DNA in the hemiclonal hybrid frog Rana esculenta and its parental species. Chromosom. Res. 3, 497–506 (1995).
Dedukh, D. et al. Cytological maps of lampbrush chromosomes of European water frogs (Pelophylax esculentus complex) from the Eastern Ukraine. BMC Genet. 14, 26 (2013).
Ogielska, M., Kierzkowski, P. & Rybacki, M. DNA content and genome composition of diploid and triploid water frogs belonging to the Rana esculenta complex (Amphibia, Anura). Can. J. Zool. 82, 1894–1901 (2005).
Zaleśna, A. et al. Evidence for Integrity of Parental Genomes in the Diploid Hybridogenetic Water Frog Pelophylax esculentus by Genomic in situ Hybridization. Cytogenet. Genome Res. 134, 206–212 (2011).
Rybacki, M. & Berger, L. Distribution and ecology of water frogs in Poland. Zool. Pol. 39, 293–303 (1994).
Berger, L., Rybacki, M. & Hotz, H. Artificial fertilization of water frogs. Amphibia-Reptilia 15, 408–413 (1994).
Kierzkowski, P. Paśko, Ł., Rybacki, M., Socha, M. & Ogielska, M. Genome dosage effect and hybrid morphology - the case of the hybridogenetic water frogs of the Pelophylax esculentus complex. Ann. Zool. Fenn. 48, 56–66 (2011).
Hauswaldt, J. S. et al. A simplified molecular method for distinguishing among species and ploidy levels in European water frogs (Pelophylax). Mol. Ecol. Resour. 12, 797–805 (2012).
Kolenda, K., Pietras-Lebioda, A., Hofman, S., Ogielska, M. & Pabijan, M. Preliminary genetic data suggest the occurrence of the Balkan water frog, Pelophylax kurtmuelleri, in southwestern Poland. Amphibia-Reptilia 38, 187–196 (2017).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Acknowledgements
We would like to thank our Referees for valuable suggestion and criticism that have improved our manuscript substantially. Authors want to thank also Magda Dubińska-Magiera for the donation of WGA and CY5-anti-rabbit antibodies, Arnold Garbiec for the help in 3D imaging, Tomasz Wójtowicz for assistance in immunofluorescence measurements and Mariusz Rybacki for providing information about distribution of triploid individuals in Polish populations. The work of MO, MC, BR-K, and ES was supported by the grant from Polish National Science Centre no. 2012/07/B/NZ3/02563, MK and KK were supported by grants for PhD students from University of Wrocław 0420/1407/16 and 0420/2312/17, and 0420/1408/16, respectively. The work of AK and DD was funded by grant of the Russian Science Foundation (project no. 14-14-00131). The work of DD was additionally funded by RFBR research project no. 18–34–00514, and his work was partially performed in the frames of Academic and Scientific Exchange under bilateral agreement between the Saint Petersburg University and the University of Wrocław. The authors acknowledge resource centres “Environmental Safety Observatory” and “Molecular and Cell Technologies” (Saint-Petersburg State University) for the access to experimental equipment.
Author information
Authors and Affiliations
Contributions
M.O., A.K., M.C., B.R.-K. conceived the project. Crossings and gonad preparation: M.O., M.C., B.R.-K., D.D., K.H., M.K., K.K. Taxonomic identification: D.D., K.K., A.P.-L. TEM microscopy and documentation: M.O., K.H. and B.R.-K. Immunocytochemistry and TUNEL: M.C., E.S., A.K., D.D., M.K. Analysis of confocal immunofluorescence images and documentation: M.C., B.R.-K., A.K. and D.D. The manuscript was written by M.O. and M.C. with the help of D.D. and improved by all other authors. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chmielewska, M., Dedukh, D., Haczkiewicz, K. et al. The programmed DNA elimination and formation of micronuclei in germ line cells of the natural hybridogenetic water frog Pelophylax esculentus. Sci Rep 8, 7870 (2018). https://doi.org/10.1038/s41598-018-26168-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-26168-z
This article is cited by
-
A cyclical switch of gametogenic pathways in hybrids depends on the ploidy level
Communications Biology (2024)
-
Meiosis and gametogenesis in hybrid, polyploid, and clonal fishes: case studies in the dojo loach Misgurnus anguillicaudatus
Fisheries Science (2023)
-
Karyotypes of water frogs from the Pelophylax esculentus complex: results of cross-species chromosomal painting
Chromosoma (2023)
-
Population structure, mate choice, and genome transmission in naturally formed pairs in a Pelophylax lessonae–Pelophylax esculentus hybridogenetic system
Behavioral Ecology and Sociobiology (2023)
-
Micronuclei in germ cells of hybrid frogs from Pelophylax esculentus complex contain gradually eliminated chromosomes
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
