
Abstract
Obesity affects male fertility and maternal diabetes affects the offspring sperm epigenome. However, the effects of in utero exposure to maternal glucose intolerance in combination with postnatal high fat, high sucrose (HFHS) diet consumption on offspring spermatogenesis is not clear. The present study was designed to test these effects. One week before and during pregnancy, dams were fed either control or HFHS diet to induce gestational glucose intolerance, and returned to standard diet during lactation. Male offspring from each maternal group were split into control and HFHS-fed groups for eight weeks prior to sacrifice at 11, 19 or 31 weeks of age, and reproductive tissues were harvested for analysis of testicular germ cell apoptosis and sperm output. Postnatal HFHS diet suppressed spermatogonia apoptosis in all age groups and maternal HFHS diet reduced testosterone levels at 11 weeks. At 31 weeks of age, the postnatal HFHS diet increased body weight, and reduced epididymis weight and sperm count. The combination of in utero and postnatal exposure impacted sperm counts most significantly. In summary, HFHS diet during pregnancy puts male offspring at greater risk of infertility, particularly when combined with postnatal high fat diet feeding.
Similar content being viewed by others

Introduction
One in ten American couples of reproductive age is involuntarily infertile. Male infertility accounts for half of these cases. Body mass index (BMI) in men, an indicator of body fat, has been inversely associated with semen quality1,2 and fertility3,4,5. For example, the percentage of spermatozoa with abnormal morphology was doubled in men with BMI ≥35 kg/m2 compared to those with BMI in the normal range6. Total sperm count in overweight or obese men was lower compared with men with normal BMI7. The number of spermatozoa with DNA damage was also significantly higher in obese men than in normal weight men2. Accordingly, couples whose BMI was ≥35 kg/m2 have a longer time to pregnancy in comparison to leaner couples (BMI ≤25 kg/m2)8.
Paternal obesity and diabetes also impact the reproductive health of offspring. For example, in rodents, male obesity alters blastocyst development, embryo metabolism9,10, placental gene expression and methylation status11, and reduces semen quality12 and fertility of male offspring13. Paternal prediabetes induced by streptozotocin treatment alters global sperm cytosine methylation profiles and offspring insulin sensitivity14. Similarly, maternal obesity, and maternal type I and II diabetes have been shown to negatively impact the metabolic health of adult offspring15,16,17, as well as the sperm epigenome of adult offspring18,19.
Collectively, these studies show that exposure to an adverse maternal metabolic environment can impact male offspring, and this may in turn affect the metabolic and reproductive health of their offspring. However, the effect of in utero exposure to maternal diabetes on fertility of male offspring has not been studied. We have developed an animal model of gestational glucose intolerance, in which C57BL/6J dams are fed a high fat, high sucrose diet from 1 week pre-gestation through the end of pregnancy20. Under this acute HFHS exposure, dams fail to expand beta cell numbers during pregnancy, leading to an inadequate insulin response to glucose challenge, and glucose intolerance during mid-late pregnancy20. Here, using this model, we ask whether maternal glucose intolerance that begins after conception, and occurs in the absence of maternal obesity, impairs fertility of male offspring.
Children’s diets are influenced by parental diets, particularly those of the mother21,22,23. Thus, prenatal exposures to poor diet are often compounded by similar postnatal diets. Moreover, many of the effects of developmental exposures are exacerbated by postnatal diet, or only become apparent when the offspring are exposed to additional metabolic challenge24,25. Conversely, it has been proposed that developmental exposure to high fat may produce predictive adaptive responses in offspring, such that they may be protected from similar exposures encountered in adulthood26. Therefore, the second objective of this study was to determine whether postnatal high fat diet interacts with prenatal exposure to maternal glucose intolerance to affect male fertility. To test this, male offspring were challenged with the same high fat, high sucrose diet that was fed to mothers during pregnancy.
One potential mechanism by which both pre- and postnatal exposure to high fat diet may affect male fertility is germ cell apoptosis. In male rats, there have been conflicting studies on the effects of diet on apoptosis, with postnatal high fat feeding having been shown to both suppress27 and increase cell death28. Also in the rat, maternal high fat feeding reduces oocyte apoptosis in prepubertal offspring, but ultimately leads to increased apoptosis at 17 weeks of age25. Thus, germ cell apoptosis was measured in male offspring shortly after puberty, and at 19 and 31 weeks following exposures to maternal and postnatal high fat diets.
Results
11 Weeks
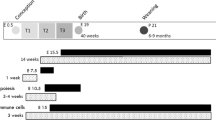
The effects of in utero and post-weaning HFHS diet consumption were first examined in male offspring at 11 weeks of age. There were two maternal diet groups during pregnancy, a standard chow control diet (CD) and a defined high fat, high sucrose diet (HFHS). At weaning at three weeks of age, one male pup from each litter was randomly assigned to the CD, and one to the HFHS diet. The combination of two maternal diet groups and two postnatal pup diet treatment groups resulted in four total groups of offspring, i.e., the CD-CD, CD-HFHS, HFHS-CD, and HFHS-HFHS.
Glucose metabolism and body weights in male offspring
Metabolic data for 11 week old male offspring are summarized in Table 1. Postnatal HFHS diet increased fasting glucose concentrations (pup CD group: 102.5 ± 8.3 vs pup HFHS group: 136.0 ± 9.1 mg/dL, P < 0.05), but did not affect the response to a glucose challenge. In pairwise comparisons, fasting blood glucose concentration was lower in the CD-CD group than in the CD-HFHS group, but not in the HFHS-CD group compared to the HFHS-HFHS group (Table 1). In contrast, insulin concentrations were significantly lower in the HFHS-CD group than in the HFHS-HFHS group, but were not different between the CD-CD and the CD-HFHS groups, suggesting that a slight increase in insulin compensated for insulin resistance in the HFHS-HFHS group (Table 1). Post-weaning feeding of HFHS diet to male offspring significantly increased body weights compared to the CD-CD group, with HFHS-CD offspring at an intermediate weight (Table 1).
Testicular gene expression
Real-time semi-quantitative RT-PCR was performed to examine the steady-state mRNA levels in the testis (Fig. 1). Inflammation-associated genes Tnfa, Il1 and Il6 were not significantly expressed above negative control levels in any group (data not shown). The transcription factors and master metabolic regulators, peroxisome proliferator-activated receptor -alpha and –gamma were also examined. Ppara transcript levels were not affected by either prenatal or postnatal HFHS. Pparg also was not affected by maternal diet, but was significantly increased by postnatal HFHS diet feeding (P < 0.01). There was a significant interaction between maternal and postnatal diets in the levels of insulin receptor (Insr) mRNA (P < 0.01). Within the maternal CD groups, postnatal HFHS significantly decreased Insr levels. Maternal HFHS also decreased Insr expression, but postnatal HFHS had no further effect on pups exposed to maternal HFHS. To examine molecular correlates of apoptosis, Fas and Fas ligand transcript levels were examined, but were not different among the treatment groups. In addition, compared to the CD-CD control, the overall Insr level in all HFHS diet feeding groups was lower (P < 0.05).

Gene expression of Ppara, Pparg, Insr, Fas and Fasl in the testis at 11 weeks of age (a and b: means with different letters were different, P < 0.05).
Case of azoospermia in a male exposed to HFHS diet during pregnancy and post-weaning
When testis sections were examined under the microscope for apoptosis analysis (summarized below), it was observed that those from one of the males (animal# 199) from the HFHS-HFHS group did not contain any round or elongating spermatids or spermatozoa (Fig. 2). Spermatocytes were found, and the structure of the seminiferous tubules was normal, although their diameter was reduced (57.7 ± 1.9 µm vs. 100.0 ± 1.8 µm for his sibling, mouse #198). The spacing of Sertoli cells along the tubule basement membrane appeared normal (Fig. 2E). This male’s body weight and serum testosterone (3.3 ng/ml) were normal, falling well within the range of the other animals. The number of TUNEL-positive germ cells per 20 seminiferous tubules was second highest in the HFHS-HFHS group and the TUNEL positive cells (Fig. 2E) were spermatogonia and spermatocytes, like in other males. All other males displayed normal spermatogenesis.

Cross sections of normal (animal #198) and azoospermic (animal #199) testes (A,B) and epididymides (C,D). (E) Immunostaining of SOX9 (Sertoli cell marker), DNA counterstaining by DAPI, and TUNEL labeling of apoptotic germ cells in the seminiferous tubules of male #199. The spacing of Sertoli cells (arrowheads) along tubule basement membrane appears normal, and the TUNEL positive cells are spermatogonia and spermatocytes, as in other males. Mice #198 and #199 were brothers exposed to HFHS in utero. After weaning, mouse 198 was on CD and 199 on HFHS diet.
19 and 31 Weeks
The four treatment groups were replicated in order to examine sperm counts and testicular morphology in fully mature adult males.
At 19 weeks of age
Data on male offspring body weight, testis weight, epididymis weight (whole and cauda only), and sperm numbers recovered from epididymides in the CD-CD, CD-HFHS, and HFHS-CD groups are summarized in Table 2. Body weight in the CD-HFHS group was heavier than the CD-CD and HFHS-CD groups (P < 0.05). No difference was found between the CD-CD and HFHS-CD groups. Testis weight did not differ among the three groups. Cauda and whole epididymis weight in the HFHS-CD group tended to be lower than the CD-CD group (P = 0.1). Adult diet tended to affect the numbers of spermatozoa recovered from epididymides, with some evidence of lower numbers in the CD-HFHS group compared to the CD-CD control (P = 0.08).
At 31 weeks of age
Mouse body, testis and epididymis weights, and total sperm numbers recovered from epididymides at 31 weeks of age are summarized in Table 2. Dam diet alone had no effects on body weights of 31 week old offspring (dam HFHS group: 32.6 ± 0.4 vs CD: 33.2 ± 0.3 g), but offspring diet did (pup HFHS group: 36.6 ± 0.4 vs CD: 29.3 ± 0.4, P < 0.01). Interestingly, there was a significant interaction between dam diet and offspring diet (P < 0.01), resulting in the lowest weights in the HFHS-CD mice, and heaviest in the HFHS-HFHS mice (Table 2). For testis weight, postnatal HFHS diet did not have any effect, while maternal HFHS diet tended to increase testis weight (P = 0.07). For epididymis, maternal diet did not have any effect on weight, but postnatal HFHS diet significantly reduced both whole epididymis (including caput, corpus, and cauda) and cauda epididymis weights (P < 0.05). Both maternal and postnatal HFHS diets significantly reduced the total number of spermatozoa recovered from epididymides (P < 0.01). As shown in Table 2, sperm number was highest in the CD-CD control group, while the HFHS-HFHS group had the lowest sperm number and the CD-HFHS and HFHS-CD groups fell in between them. The total number of spermatozoa recovered from epididymides was correlated with cauda epididymis weight (r = 0.57, r2 = 0.32; P < 0.05) as shown in Fig. 3.

Correlation between the total sperm number recovered from cauda epididymides and the weight of cauda epididymides.
Number of TUNEL-positive testicular germ cells at 11, 19 and 31 weeks of age
Apoptosis of germ cells was evaluated by the TUNEL assay on testis sections from all three ages (Fig. 4). Representative images from each diet group at 11 weeks of age are presented in Fig. 5. The in utero HFHS exposure did not affect the number of apoptotic germ cells in any age group, whereas postnatal feeding of HFHS diet for 8 weeks, about two rounds of spermatogenesis29, regardless of starting age, significantly reduced the number of apoptotic germ cells (P < 0.05). At 11 weeks, the number of TUNEL-positive germ cells in the CD-HFHS and HFHS-HFHS groups was lower (P < 0.05) than in CD-CD and HFHS-CD groups, respectively. When apoptosis was calculated as a percentage of all spermatogonia, the values of 1.10 ± 0.14, 0.61 ± 0.06, 0.92 ± 0.08, and 0.53 ± 0.05% for the CD-CD, CD-HFHS, HFHS-CD, and HFHS-HFHS groups, respectively, yielded the same statistical results as when calculating the number of TUNEL-positive germ cells per 20 seminiferous tubules. At 19 weeks, the number of TUNEL-positive germ cells in the CD-HFHS group was again lower (P < 0.05) than those in the CD-CD control and HFHS-CD groups. Similarly, at 31 weeks of age, it was lower in the CD-HFHS and HFHS-HFHS groups than in the CD-CD and HFHS-CD groups (P < 0.05); but there were no differences based on in utero diets.

Number of TUNEL-positive germ cells per 20 cross sections of seminiferous tubules in the CD-CD, CD-HFHS, HFHS-CD, and HFHS-HFHS groups at 11, 19, and 31 weeks of age (a,b, and c indicate statistical difference between diets within each age group; means with different letters differ at P < 0.05).

Representative images of TUNEL assay on seminiferous tubule cross-sections from four animals per treatment (A: CD-CD, B: CD-HFHS, C: HFHS-CD, and D: HFHS-HFHS diet group). (E) Identification of Sertoli cells by anti-SOX9 antibody in red, a Sertoli cell-specific marker (indicated by arrows in E’). SOX9 labeling does not overlap with green fluorescence of TUNEL probe, indicating germ cells, rather than Sertoli cells are apoptotic in the examined tissues. (F) Identification of spermatogonia by anti-UCHL1 antibody in red, a specific spermatogonial marker (arrows). Inset F’ shows the blue and red channel detail of two large, green-labeled TUNEL-positive cells visible in panel F. Colocalization of TUNEL labeling and UCHL1 (F) indicates that apoptosis occurs in the spermatogonia, close to basement membranes (see also white arrows in panel A’).
Age had a significant effect on the number of TUNEL-positive germ cells (P < 0.01). More specifically, this number was higher at 19 (21.9 ± 2.4) and 31 (22.0 ± 2.4) weeks of age compared to the 11 week age group (6.7 ± 1.5). Co-staining for the Sertoli cell marker SOX9 and spermatogonial marker UCHL1 showed that apoptosis occurred largely in spermatogonia, and a few spermatocytes, but not in Sertoli cells (Fig. 5E,F).
Serum testosterone concentrations of 11, 19, and 31 week old animals
Serum testosterone (T) concentrations were not different among age groups (Table 3), though there was an interaction between maternal diet and age (P < 0.05). The T concentrations in the older age groups (19 and 31 weeks) were less variable while they were highly variable in the 11 week age group. There were no maternal diet effects, postnatal diet effects or interactions between them in the 19 and 31 week old animals. However, in the 11 week age group, a significant effect of maternal diet on T levels was detected, with maternal HFHS diet reducing T levels in male offspring (the CD maternal offspring: 3.63 ± 1.09 vs. the HFHS maternal offspring: 0.95 ± 0.82 ng/ml, P < 0.05). No correlation was found between serum T concentrations, and body weight, testicular weight or sperm count (not shown).
Discussion
The main goal of the present study was to test the hypothesis that in utero exposure to HFHS-induced maternal glucose intolerance, postnatal HFHS diet and the interaction between them alter spermatogenesis and reduce sperm output in male offspring. We found that feeding of HFHS diet for 8 weeks after weaning suppressed germ cell apoptosis in testes in all age groups studied. While 8 weeks of postnatal feeding of HFHS diet alone significantly increased body weight at all three age points, it had a greater effect on the offspring of glucose intolerant dams than on the offspring of dams fed control diet at 31 weeks of age. High fat diet feeding from 23–31 weeks of age significantly reduced epididymis weight and sperm count, with the lowest count in the HFHS-HFHS group, intermediate counts in the CD-HFHS and HFHS-CD groups, and the highest sperm count in the CD-CD controls. Taken together, these data support the hypothesis that postnatal feeding of high fat diet alters spermatogenesis and reduces sperm production; exposure to glucose intolerance in utero exacerbates the effects of an adult high fat diet on weight gain and male fertility.
In utero exposure to the HFHS diet reduced the sperm count in 31 week old animals, and seemed to have slightly reduced apoptosis in the 11 week old animals. The combination of postnatal and in utero high fat feeding had the most dramatic effects on male offspring fertility. At 31 weeks, HFHS-HFHS had the lowest sperm counts, and testicular and cauda epididymal weights. Although it was an isolated observation, one of seven mice in the HFHS-HFHS exposure group was azoospermic at 11 weeks of age. These data indicate that in utero exposure to HFHS diet has negative effects on male offspring fertility, which to our knowledge has not been reported previously and is consistent with prior observations of sperm epigenetic alterations in offspring of obese diabetic mothers18.
The cause for the single case of azoospermia at 11 weeks is not clear. Determining whether this was a true effect of the HFHS-HFHS treatment or a chance observation would require a much larger sample size. Body weight, the number of TUNEL-positive germ cells, and the spacing of Sertoli cells along tubule basement membrane in the azoospermic animal were normal and close to those of other animals. Thus, none of these endpoints explain the observed azoospermia.
Direct exposure to HFHS diet after weaning alone affected the weight of epididymides, sperm count and body weight. This is consistent with observations in both human and animal studies. In humans, overweight or obese men with female partners of normal body mass index have increased time to pregnancy interval8, and are more likely to suffer from male infertility4,5. Furthermore, HFHS-induced paternal obesity impairs embryo development, and decreases pregnancy rates and live birth outcomes4,30. In rodents, paternal obesity affects gene expression in male placenta and global DNA methylation status in female placenta11,and reduces the fertility of his offspring13. Similarly, embryos produced by males fed an HFHS diet have reduced cell numbers9,10 and altered metabolism9.
These data inspired us to study blood testosterone (T) concentrations as a possible mediator of diet effects on male reproductive system. Overall the testosterone concentrations in the current study were similar to those of the same mouse strain, C57BL/6 J reported by others31. T was not abnormally low in the single azospermic male. Though testosterone is so pulsatile32 that it is seldom possible to see differences between treatment groups from a single blood sample, we were able to detect significantly lower T in the 11 week old offspring of HFHS-fed dams. There was some indication, though not significant due to the high inter-individual variation, of higher T in the 11 week old males with postnatal HFHS treatment. T concentrations in 19 and 31 weeks old animals were less variable than those in 11 week old animals, and did not differ among diet groups. Thus, the peripubertal period may be especially critical for T in the offspring exposed to glucose intolerance in utero, but differences in T do not explain the effects of postnatal diet, or the low sperm counts in the mature offspring of HFHS-fed dams.
Apoptosis is a genetically-controlled, programmed form of cell death required for normal spermatogenesis33. During spermatogenesis, apoptosis maintains the size of the germ cell population, optimizes germ cell–Sertoli cell ratio34, and eliminates defective germ cells that carry damaged DNA, thus playing a key role in maintaining normal sperm output35. In agreement with previous reports36,37, apoptosis was present in all animals in this study, and levels were lower in 11 week old group compared to 19 and 31 weeks of age, consistent with previous observations that apoptosis levels increase from early puberty through 4 months of age and then plateau38. Most of the apoptotic germ cells were UCHL1+ spermatogonia close to the basement membrane. Apoptosis in spermatogonia at early stages of differentiation, specifically at A2, A3 and A4 generations of spermatogonia37, is thought to result in early elimination of defective germ cells39.
A small percentage of germ cells destined to be eliminated in the testis escape apoptosis and find their way into the ejaculate, a phenomenon called abortive apoptosis40. In the current study, we found that postnatal HFHS diet significantly suppressed apoptosis during spermatogenesis. It is possible that this increases the number of defective spermatozoa in ejaculate, but ejaculate was not tested here. A recent study likewise reported suppression of germ cell apoptosis in immature rats fed a high fat diet27, whereas others have found increased apoptosis in mature males fed high fat diet28. How HFHS diet might inhibit apoptosis in testis is not known. One possible mechanism is via peroxisome proliferator-activated receptor gamma (PPARG). We found that Pparg mRNA was upregulated by postnatal HFHS feeding, consistent with a previous study of a 60% fat diet41. Pparg is a key regulator of metabolism, and its inhibitors can both block and promote apoptosis, depending on cell type42. However, Pparg function in testicular apoptosis has not yet been studied. Additionally, Sertoli cells tightly regulate germ cell proliferation and differentiation, and control germ cell apoptosis by a FAS/FASL-mediated pathway43. Thus, we examined Fas and Fasl mRNA in the testes in the 11 week old males, but they were not significantly changed by either prenatal or postnatal HFHS.
The reduction in spermatagonial cell death by postnatal HFHS does not explain lower sperm counts in the HFHS-fed males, and while maternal HFHS consumption also decreased offspring sperm counts, it did not affect apoptosis rates. Thus, we examined insulin receptor expression as a potential mechanistic link between HFHS diet and sperm counts. Previous studies have found that insulin and insulin-related proteins are required for FSH-mediated immature Sertoli cell proliferation during the late fetal and early neonatal testicular period44. Insulin plays an important role in regulating the final number of Sertoli cells, testis size, and daily sperm output through both systemic and local actions45,46. Expression of the insulin receptor was reduced by HFHS diet, whether maternal or postnatal (all HFHS diet groups vs. control) and all HFHS diet groups had reduced sperm count. Therefore, reduced local sensitivity to insulin in the HFHS diet feeding animals may be one of the mechanisms for low sperm output.
In summary, the present study demonstrated that in utero high fat diet exposure, consumption of this diet after weaning, and the combination of the two had significant effects on body weight/obesity, reproductive organ weight, and sperm count in the male offspring. These data are consistent with the DOHaD concept that developmental exposures influence male reproductive health.
Materials and Methods
Mice
C57BL/6J mice were used for all experiments. Dams were purchased from Jackson Laboratories (Bar Harbor, Maine). Animal experiments were performed at the School of Medicine (experiment 1) and the Animal Sciences Research Center (experiment 2) of the University of Missouri, in compliance with NIH Guidelines for the Care and Use of Laboratory Animals, under protocols approved by the University of Missouri Animal Care and Use Committee.
Diets
The control diet (CD) was a 5008 chow purchased from LabDiets Purina Mills (PMI Nutrition International LLC, Bentwood, MO). The defined high fat, high sucrose diet (HFHS, D12451) was from Research Diets Inc. (New Brunswick, NJ). The caloric and relative fat contents of these diets are summarized in Table 4.
Experimental Design
Seven-week old females were assigned randomly to one of the two diet groups: standard control diet or the HFHS diet and mated to proven breeder males after 1 week of acclimation as previously described20. The day of copulatory plug visualization was noted as day 0.5 of gestation. After delivery, all dams were placed on the standard chow diet and maintained on this diet until weaning when pups reached 3 weeks old. All pups were sexed at weaning. Only males were kept for the experiments and fed either CD or HFHS diet depending on the experiment design. In experiment 1, at weaning, one male pup from each litter was randomly assigned to the control diet, and one to the HFHS diet. At 11 weeks of age, after a total of 8 weeks on the postnatal CD or HFHS diet, intraperitoneal glucose tolerance tests (GTT) were performed as previously described20 with duplicate measurements made using two ReliOn Prime Blood Glucose Monitoring System meters (Walmart, Bentonville, AR). Pups were then sacrificed for collection of testes and epididymides. Tissues were processed as described below. In experiment 2, fifteen dams each were randomly assigned to the CD and HFHS groups, with 12 CD and 6 HFHS dams delivering pups, following a four day breeding window. One male offspring from each litter was assigned to HFHS diet beginning at 23 weeks of age, and one male offspring continued on CD diet, until both were sacrificed at 31 weeks of age with 8 weeks on experimental diet as in experiment 1. Remaining male offspring from each litter were assigned to either continued CD or HFHS beginning at 11 weeks of age, and sacrificed at 19 weeks of age, again 8 total weeks on experimental diet.
Blood, Tissue and Sperm Collection
At the end of experiments, mice were euthanized by CO2 followed by cervical dislocation. Blood was collected by cardiac puncture in a syringe with a 27G needle, stored at 4°C and allowed to clot until serum separation the following day. For experiment 1, testes and epididymides were dissected, immediately placed on wet ice, weighed and fixed for TUNEL staining as described below. For experiment 2, testes and epididymides were collected and weighed. Testes were fixed for apoptosis assay by TUNEL. To recover the cauda-epididymal spermatozoa, epididymides from both sides were placed in 3 ml of warm TCM 199 medium (Corning Life Sciences, Tewksbury, MA), cut into small pieces and incubated at 37°C in a CO2 incubator for two periods of 30 min with fresh medium each time, to allow spermatozoa to swim out. The numbers of spermatozoa recovered were then counted by using a hemocytometer.
Histological and Apoptosis Assessment of Testis Sections by TUNEL
Testes were embedded in paraffin wax after fixation with 4% paraformaldehyde in PBS overnight at 4°C. Serial sections were cut at a thickness of 5 µm, and stained with hematoxylin and eosin at the pathology laboratory of IDEXX BioResearch, Columbia MO. Presumed apoptotic cells in seminiferous tubules on testis cross sections were identified by terminal deoxynucleotidyl transferase (TdT)-mediated nick end labeling (TUNEL) using the In Situ Cell Death Detection Kit, AP (Roche, Indianapolis, IN) according to the manufacturer’s instructions. After deparaffinization in xylene, tissue sections were rehydrated in a series of ethanol (from 100% to 50% in water), incubated with proteinase K (20 mg/ml), and permeabilized with freshly prepared 0.1% Triton X-100 in 0.1% sodium citrate. Immediately before use, the TUNEL reaction mixture was prepared by mixing 5 µl of enzyme solution with 45 µl of label solution. Fifty µl of reaction mixture was added on to section and incubated for 1 h at 37°C in a humidified atmosphere in the dark. Negative controls were obtained by the replacement of enzyme solution with PBS. For immunostaining of SOX9 and UCHL1 proteins, after rehydration in series of ethanol, testis sections were permeabilized in PBS containing 0.1% (v/v) Triton X-100, and blocked for 25 min in 0.1 M PBS containing 5% normal goat serum and 0.1% Triton X-100. Samples were incubated overnight at 4°C with primary antibody diluted at 1:200 in 0.1 M PBS containing 1% normal goat serum and 0.1% Triton X-100. On the next day, after a wash in PBS, the primary antibodies (anti-SOX9, catalog# ab185966, purchased from Abcam, Cambridge, MA, and anti-UCHL1 antibodies, catalog# ADI-905-520-1 from Enzo Life Sciences, Ann Arbor, MI) were detected by a mixture of goat anti-rabbit IgG-TRITC diluted at 1:100 and DAPI (4,6-diamidino-2-phenylindole, 2.5 μg/mL) incubated at room temperature for 40 min. Negative controls were obtained by the replacement of primary antibody with normal rabbit serum at the concentration identical to that of specific antibody. At the end of incubation, sections were washed 3 times with PBS and mounted with anti-fade mounting medium (Vector Laboratories Inc., Burlingame, CA). Tissue sections were examined for fluorescence labeling and the acquisition of multiple images of each cross section was performed under a Nikon Eclipse 800 epifluorescence microscope (Nikon Instruments, Melville, NY) with Cool Snap camera (Roper Scientific, Tucson, AZ, USA) and MetaMorph software (v7.1, Universal Imaging, Downington, PA). Images were edited with Adobe Photoshop CS5 (Adobe Systems, Mountain View, CA). Both total and TUNEL-positive spermatogonia were recorded blind to treatment groups. The number of TUNEL-positive germ cells per 20 seminiferous tubules and percentage of positive cells out of total spermatogonia number within tubule section was calculated and used for data analysis.
RNA isolation and quantitative RT-PCR
RNA was isolated from offspring testes samples collected at 11 weeks of age by homogenization in TRI Reagent (Sigma-Aldrich, St Louis, MO), using a General Laboratory homogenizer (OMNI International, Kennesaw, GA). RNA was purified using the Nucleospin RNA clean-up kit (Clontech Mancherey-Nagel, Mountain View, CA) according to the manufacturer’s protocol. Genomic DNA was eliminated from RNA samples using the Turbo DNA-free kit (ThermoFisher Scientific, Waltham, MA).
Reverse transcription was performed using the SuperScript First-Strand Synthesis System (ThermoFisher Scientific) according to the manufacturer’s protocol. Briefly, 1 µg of RNA was reverse transcribed using random hexamer primers. Relative mRNA levels were quantified by real-time PCR with SYBR Green Master Mix (Qiagen, Germantown, MD) for the following genes: Pparg, Ppara Insr, FasL and Fas. Gapdh and Actb were used as the internal reference genes for normalization of gene expression level. PCR was performed on a CFX Connect Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA) with the following cycling conditions: 95°C for 10 minutes (1 rep), and followed by 40 cycles of 95°C (15 s), 60°C (60 s) and 72°C (30 s). Melting curves were generated following RT-PCR to assess the specificity of amplicons. Each sample had only one peak. Thus, no samples were excluded for final analysis.
Insr primer sequences have previously been published47, and Actb, Pparg, Ppara, Fas and FasL primers were designed and synthesized by Integrated DNA Technologies (https://www.idtdna.com/scitools/Applications/RealTimePCR). Primer sequences for the genes were: Actb, forward (5′-GATGACCCAGATCATGTTTGAGACC-3′) and reverse (5′-AGATGGGCACAGTGTGGGTGA-3′); Insr, forward (5′-CCACCAAGAACTCGTGAAAGG-3′) and reverse (5′-TGCACGCAGGAAAGAACCT-3′); Gapdh, forward (5′-TGCACCACCAACTGCTTAGC-3′) and reverse (5′-GGCATGGACTGTGGTCATGAG-3′); Ppara, forward (5′-TGCAACTTCTCAATGTAGCCT-3′) and reverse (5′-AATGCCTTAGAACTGGATGACA-3′); Pparg, forward (5′-TGCAGGTTCTACTTTGATCGC-3′) and reverse (5′-CTGCTCCACACTATGAAGACAT-3′); Fas forward (5′-GTTTGTATTGCTGGTTGCTGT-3′) and reverse (5′-ACCAGACTTCTACTGCGATTC-3′); Fasl, forward (5′-ATATGTGTCTTCCCATTCCAGAG-3′) and reverse (5′-CACCAACCAAAGCCTTAAAGTATC-3′). Primer efficiencies were validated by using serial dilutions of the respective template. Reference genes did not differ in expression across the treatment groups. PCR was analyzed using the ΔΔcycle threshold method as described previously48, but with adjustment for PCR reaction efficiencies, calculated from dilution series for each gene of interest.
Serum Analyses
All animals were selected for testosterone (T) assays except the 19 weeks age group, in which only animals from CD-CD and HFHS-CD groups were available for reproductive organ collection at this age. Serum T levels were determined by using T ELISA kit (Immuno-Biological Laboratories Inc., Minneapolis, MN) according to manufacturer’s instructions. Each blood sample was assayed in duplicate. The intra assay coefficients of variation (CV) was 6.4% and inter assay CV was 8.3%.
Intraperitoneal glucose tolerance tests (GTT) were performed as previously described in 11 week old mice of experiment 120. Fasting serum insulin concentrations were measured just prior to glucose tolerance testing by using a Rat/Mouse Insulin ELISA kit (EMD Millipore) in duplicate according to the manufacturer’s instructions. All samples were assessed in one assay. The intra assay CV was 2.6%.
Statistical Procedures
Mouse glucose tolerance data were analyzed as repeated measures, reading at time 0, 15, 30, 60, and 90 minutes after glucose challenge. The ANOVA model contained the effects of diet, time (minutes after glucose challenge), and the interaction of diet with time (diet × time). The mouse served as the experimental unit. These data were analyzed by using the PROC MIXED procedure in SAS (v 9.4)49. Differences in blood glucose concentrations between diet groups over time were determined by the Fisher least-significant difference test.
Data for the body, testis, and epididymis weights, sperm count, number of apoptotic germ cells on testis sections, and serum insulin concentrations were analyzed for normality using the Wilk-Shapiro test49. The sperm count data were logarithmically transformed to approach a normal distribution. All dependent variables were analyzed using the general linear model (GLM) procedure of the SAS software, with maternal and pup diet as the main effect and paired dependent sample t-test as options for comparison of brothers on different diet49. Mouse body weight data at the end of each experiment were analyzed by using GLM procedure of SAS with the starting body weight at weaning as a covariate. Differences in testis weight, epididymis weight, sperm count, and number of apoptotic germ cells among diet groups were determined by the Fisher least-significant difference. Serum testosterone data and the numbers of TUNEL-positive germ cells per 20 seminiferous tubules from all three age groups were analyzed by using the PROC MIXED procedure in SAS (v 9.4)49. The ANOVA model contained the effects of pup age, maternal diet, offspring diet and interaction between them. Offspring were nested within dam, and maternal and offspring diet interactions were used as error term. Gene expression data were analyzed by nested, two-way ANOVA with maternal treatment and offspring diet as the fixed effects and with dam considered a random effect. Linear regression analyses49 were also performed to determine the associations between epididymis weight and sperm count. All data are expressed as least squares means ± the standard error of the least squares means.
References
Jensen, T. K. et al. Body mass index in relation to semen quality and reproductive hormones among 1,558 Danish men. Fertil Steril 82, 863–870, https://doi.org/10.1016/j.fertnstert.2004.03.056 (2004).
Chavarro, J. E., Toth, T. L., Wright, D. L., Meeker, J. D. & Hauser, R. Body mass index in relation to semen quality, sperm DNA integrity, and serum reproductive hormone levels among men attending an infertility clinic. Fertil Steril 93, 2222–2231, https://doi.org/10.1016/j.fertnstert.2009.01.100 (2010).
Sallmen, M., Sandler, D. P., Hoppin, J. A., Blair, A. & Baird, D. D. Reduced fertility among overweight and obese men. Epidemiology 17, 520–523, https://doi.org/10.1097/01.ede.0000229953.76862.e5 (2006).
Nguyen, R. H., Wilcox, A. J., Skjaerven, R. & Baird, D. D. Men’s body mass index and infertility. Hum Reprod 22, 2488–2493, https://doi.org/10.1093/humrep/dem139 (2007).
Ramlau-Hansen, C. H. et al. Subfecundity in overweight and obese couples. Hum Reprod 22, 1634–1637, https://doi.org/10.1093/humrep/dem035 (2007).
Andersen, J. M. et al. Body Mass Index Is Associated with Impaired Semen Characteristics and Reduced Levels of Anti-Mullerian Hormone across a Wide Weight Range. PLoS One 10, e0130210, https://doi.org/10.1371/journal.pone.0130210 (2015).
Sermondade, N. et al. BMI in relation to sperm count: an updated systematic review and collaborative meta-analysis. Hum Reprod Update 19, 221–231, https://doi.org/10.1093/humupd/dms050 (2013).
Sundaram, R., Mumford, S. L. & Buck Louis, G. M. Couples’ body composition and time-to-pregnancy. Hum Reprod 32, 662–668, https://doi.org/10.1093/humrep/dex001 (2017).
Binder, N. K., Mitchell, M. & Gardner, D. K. Parental diet-induced obesity leads to retarded early mouse embryo development and altered carbohydrate utilisation by the blastocyst. Reprod Fertil Dev 24, 804–812, https://doi.org/10.1071/RD11256 (2012).
Mitchell, M., Bakos, H. W. & Lane, M. Paternal diet-induced obesity impairs embryo development and implantation in the mouse. Fertil Steril 95, 1349–1353, https://doi.org/10.1016/j.fertnstert.2010.09.038 (2011).
Binder, N. K. et al. Paternal obesity in a rodent model affects placental gene expression in a sex-specific manner. Reproduction 149, 435–444, https://doi.org/10.1530/REP-14-0676 (2015).
Navya, H. & Yajurvedi, H. N. Obesity causes weight increases in prepubertal and pubertal male offspring and is related to changes in spermatogenesis and sperm production in rats. Reprod Fertil Dev 29, 815–823, https://doi.org/10.1071/RD15480 (2016).
Fullston, T. et al. Diet-induced paternal obesity in the absence of diabetes diminishes the reproductive health of two subsequent generations of mice. Hum Reprod 27, 1391–1400, https://doi.org/10.1093/humrep/des030 (2012).
Wei, Y. et al. Paternally induced transgenerational inheritance of susceptibility to diabetes in mammals. Proc Natl Acad Sci USA 111, 1873–1878, https://doi.org/10.1073/pnas.1321195111 (2014).
Rooney, K. & Ozanne, S. E. Maternal over-nutrition and offspring obesity predisposition: targets for preventative interventions. Int J Obes (Lond) 35, 883–890, https://doi.org/10.1038/ijo.2011.96 (2011).
Tamashiro, K. L. & Moran, T. H. Perinatal environment and its influences on metabolic programming of offspring. Physiol Behav 100, 560–566, https://doi.org/10.1016/j.physbeh.2010.04.008 (2010).
Williams, L., Seki, Y., Vuguin, P. M. & Charron, M. J. Animal models of in utero exposure to a high fat diet: a review. Biochim Biophys Acta 1842, 507–519, https://doi.org/10.1016/j.bbadis.2013.07.006 (2014).
Ge, Z. J. et al. Maternal obesity and diabetes may cause DNA methylation alteration in the spermatozoa of offspring in mice. Reprod Biol Endocrinol 12, 29, https://doi.org/10.1186/1477-7827-12-29 (2014).
Ding, G. L. et al. Transgenerational glucose intolerance with Igf2/H19 epigenetic alterations in mouse islet induced by intrauterine hyperglycemia. Diabetes 61, 1133–1142, https://doi.org/10.2337/db11-1314 (2012).
Pennington, K. A., van der Walt, N., Pollock, K. E., Talton, O. O. & Schulz, L. C. Effects of acute exposure to a high-fat, high-sucrose diet on gestational glucose tolerance and subsequent maternal health in mice. Biol Reprod 96, 435–445, https://doi.org/10.1095/biolreprod.116.144543 (2017).
Oliveria, S. A. et al. Parent-child relationships in nutrient intake: the Framingham Children’s Study. Am J Clin Nutr 56, 593–598 (1992).
Lahmann, P. H., Williams, G. M., Najman, J. M. & Mamun, A. A. Mother-adult offspring resemblance in dietary intake: a community-based cohort study in Australia. Am J Clin Nutr 105, 185–193, https://doi.org/10.3945/ajcn.116.137539 (2017).
Dickens, E. & Ogden, J. The role of parental control and modelling in predicting a child’s diet and relationship with food after they leave home. A prospective study. Appetite 76, 23–29, https://doi.org/10.1016/j.appet.2014.01.013 (2014).
Pennington, K. A. et al. Effect of food restriction and leptin supplementation on fetal programming in mice. Endocrinology 153, 4556–4567, https://doi.org/10.1210/en.2012-1119 (2012).
Tsoulis, M. W. et al. Maternal High-Fat Diet-Induced Loss of Fetal Oocytes Is Associated with Compromised Follicle Growth in Adult Rat Offspring. Biol Reprod 94, 94, https://doi.org/10.1095/biolreprod.115.135004 (2016).
Elahi, M. M. et al. Long-term maternal high-fat feeding from weaning through pregnancy and lactation predisposes offspring to hypertension, raised plasma lipids and fatty liver in mice. Br J Nutr 102, 514–519, https://doi.org/10.1017/S000711450820749X (2009).
Zhang, J. et al. A high-fat diet impairs reproduction by decreasing the IL1beta level in mice treated at immature stage. Sci Rep 7, 567, https://doi.org/10.1038/s41598-017-00505-0 (2017).
Mu, Y., Yan, W. J., Yin, T. L. & Yang, J. Curcumin ameliorates highfat dietinduced spermatogenesis dysfunction. Mol Med Rep 14, 3588–3594, https://doi.org/10.3892/mmr.2016.5712 (2016).
Oakberg, E. F. Duration of spermatogenesis in the mouse and timing of stages of the cycle of the seminiferous epithelium. Am J Anat 99, 507–516, https://doi.org/10.1002/aja.1000990307 (1956).
Bakos, H. W., Henshaw, R. C., Mitchell, M. & Lane, M. Paternal body mass index is associated with decreased blastocyst development and reduced live birth rates following assisted reproductive technology. Fertil Steril 95, 1700–1704, https://doi.org/10.1016/j.fertnstert.2010.11.044 (2011).
Nelson, J. F., Latham, K. R. & Finch, C. E. Plasma testosterone levels in C57BL/6J male mice: effects of age and disease. Acta Endocrinol (Copenh) 80, 744–752 (1975).
Coquelin, A. & Desjardins, C. Luteinizing hormone and testosterone secretion in young and old male mice. Am J Physiol 243, E257–263 (1982).
Kumar, V., Abbas, A. K., Aster, J. C. & Robbins, S. L. xii, 910 p. (Elsevier/Saunders,, Philadelphia, PA, 2013).
Aitken, R. J., Smith, T. B., Jobling, M. S., Baker, M. A. & De Iuliis, G. N. Oxidative stress and male reproductive health. Asian J Androl 16, 31–38, https://doi.org/10.4103/1008-682X.122203 (2014).
Koji, T., Hishikawa, Y., Ando, H., Nakanishi, Y. & Kobayashi, N. Expression of Fas and Fas ligand in normal and ischemia-reperfusion testes: involvement of the Fas system in the induction of germ cell apoptosis in the damaged mouse testis. Biol Reprod 64, 946–954 (2001).
Kwon, J. et al. Ubiquitin C-terminal hydrolase L-1 is essential for the early apoptotic wave of germinal cells and for sperm quality control during spermatogenesis. Biol Reprod 73, 29–35, https://doi.org/10.1095/biolreprod.104.037077 (2005).
Allan, D. J., Harmon, B. V. & Roberts, S. A. Spermatogonial apoptosis has three morphologically recognizable phases and shows no circadian rhythm during normal spermatogenesis in the rat. Cell Prolif 25, 241–250 (1992).
Jeyaraj, D. A., Grossman, G. & Petrusz, P. Dynamics of testicular germ cell apoptosis in normal mice and transgenic mice overexpressing rat androgen-binding protein. Reprod Biol Endocrinol 1, 48, https://doi.org/10.1186/1477-7827-1-48 (2003).
Rodriguez, I., Ody, C., Araki, K., Garcia, I. & Vassalli, P. An early and massive wave of germinal cell apoptosis is required for the development of functional spermatogenesis. EMBO J 16, 2262–2270, https://doi.org/10.1093/emboj/16.9.2262 (1997).
Sakkas, D. et al. Origin of DNA damage in ejaculated human spermatozoa. Rev Reprod 4, 31–37 (1999).
Ghanayem, B. I., Bai, R., Kissling, G. E., Travlos, G. & Hoffler, U. Diet-induced obesity in male mice is associated with reduced fertility and potentiation of acrylamide-induced reproductive toxicity. Biol Reprod 82, 96–104, https://doi.org/10.1095/biolreprod.109.078915 (2010).
Bandera Merchan, B., Tinahones, F. J. & Macias-Gonzalez, M. Commonalities in the Association between PPARG and Vitamin D Related with Obesity andCarcinogenesis. PPAR research 2016, 2308249, https://doi.org/10.1155/2016/2308249 (2016).
Lee, J., Richburg, J. H., Younkin, S. C. & Boekelheide, K. The Fas system is a key regulator of germ cell apoptosis in the testis. Endocrinology 138, 2081–2088, https://doi.org/10.1210/endo.138.5.5110 (1997).
Pitetti, J. L. et al. An essential role for insulin and IGF1 receptors in regulating sertoli cell proliferation, testis size, and FSH action in mice. Mol Endocrinol 27, 814–827, https://doi.org/10.1210/me.2012-1258 (2013).
Soder, O., Bang, P., Wahab, A. & Parvinen, M. Insulin-like growth factors selectively stimulate spermatogonial, but not meiotic, deoxyribonucleic acid synthesis during rat spermatogenesis. Endocrinology 131, 2344–2350, https://doi.org/10.1210/endo.131.5.1425434 (1992).
Schoeller, E. L., Albanna, G., Frolova, A. I. & Moley, K. H. Insulin rescues impaired spermatogenesis via the hypothalamic-pituitary-gonadal axis in Akita diabetic mice and restores male fertility. Diabetes 61, 1869–1878, https://doi.org/10.2337/db11-1527 (2012).
Talton, O. O. et al. Maternal Hyperleptinemia Improves Offspring Insulin Sensitivity in Mice. Endocrinology 157, 2636–2648, https://doi.org/10.1210/en.2016-1039 (2016).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods (San Diego, Calif.) 25, 402–408, https://doi.org/10.1006/meth.2001.1262 (2001).
SAS. SAS/STAT User’s Guide, Release 9.4. Cary, NC: SAS Institute Inc (2014).
Acknowledgements
The authors would like to thank Keenan A. Bates for help with animal husbandry and tissue collection. Funding was provided by Agriculture and Food Research Initiative Competitive Grant no. 2015-67015-23231 from the USDA National Institute of Food to PS, and supported by an American Diabetes Association Basic Science Award 1-14-BS-181 to LCS as well as seed funding from the Food for the 21st Century Program of the University of Missouri to PS.
Author information
Authors and Affiliations
Contributions
The authors’ contributions are as follows: J.M., L.C.S. and P.S. contributed to the study design, data analysis and interpretation of data, and writing of the manuscript. K.A.P., O.O.T., M.S. and Y.L. carried out the study, contributed to the sample analysis, data collection and analysis, and interpretation of data. All authors read and approved the final manuscript. L.C.S. and P.S. are the senior co-authors/team leaders.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mao, J., Pennington, K.A., Talton, O.O. et al. In Utero and Postnatal Exposure to High Fat, High Sucrose Diet Suppressed Testis Apoptosis and Reduced Sperm Count. Sci Rep 8, 7622 (2018). https://doi.org/10.1038/s41598-018-25950-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-25950-3
This article is cited by
-
Maternal energy insufficiency affects testicular development of the offspring in a swine model
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
