
Abstract
Wnt1-inducible signaling pathway protein-1 (WISP1) is a novel target of the Wnt pathway for modulating osteogenesis and improving bone strength. However, it is not clear if genetic variants in the WISP1 region are associated with bone mineral density (BMD) in human. The aim of this study is to investigate the role of genetic variation in WISP1 gene as a determinant of BMD in 1,510 Old Order Amish (OOA). We performed regional association analysis of 58 tag variants within 5 kb upstream and downstream to WISP1 with BMD and found 5 variants that were associated with BMD at multiple skeletal sites (P values from 2.89 × 10−6 to 1.62 × 10−2), with some significant associations even after adjustment for multiple comparisons. To replicate these results in an independent dataset, we performed a look-up of BMD associations with these variants in European ancestry subjects from the large GEFOS Consortium and observed the nominal associations of two of these variants with BMD (P values: 0.031 to 0.048). In conclusion, we have demonstrated that genetic variants surrounding WISP1 are associated with BMD at multiple skeletal sites in the OOA, thus influencing osteoporosis risk. These results support a role for the WISP1 gene on influencing variation in BMD.
Similar content being viewed by others
Introduction
Osteoporosis is a common disorder affecting hundreds of millions of people and is one of the leading causes of fractures in the world. This disease accounts for approximately 1.5 million new fracture cases each year, representing a huge economic burden on health care systems, with annual costs estimated to be $17 billion in the United States alone and expected to rise 50% by the year 20251,2. Osteoporosis is mainly characterized by bone fragility (most individuals have a low BMD) and it is a highly heritable trait with heritability ranging from 0.5 to 0.83. Recent studies have shown that multiple genetic variants in Wnt pathway components including AXIN1, CTNNB1, DKK1, LRP4, LRP5, MEF2C, RSPO3, SFRP4, SOST, WLS, WNT3, WNT4, WNT5B and WNT16 are associated with BMD4,5,6,7,8,9,10,11. Identifying new genetic variants that influence BMD could lead to new strategies to treat osteoporosis.
Within the components of the Wnt signaling pathway, WISP1, also known as CCN4, has been found to be a novel target for modulating osteogenesis and improving bone strength. The WISP1 gene is located on human chromosome 8q24.22 and contains six exons and five introns. Expression of WISP1 has been observed in the developing skeleton and later in both osteoblast precursors and osteoblastic cells12, specifically at sites of new bone formation during development or fracture healing13. Moreover, mice with WISP1 knocked out have lower BMD, trabecular bone volume/total volume, and cortical bone thickness than wild-type mice14, and transgenic mice with human WISP1 over-expressed have increased BMD, trabecular thickness, and bone volume/total volume over wild-type controls15. In humans, genetic variants of WISP1 have been associated with increasing the risk of these diseases, such as spinal osteoarthritis, scirrhous gastric carcinoma, lung cancer, and myocardial infarction16,17,18,19. These findings indicate that WISP1 is necessary for bone formation and regulation of skeletogenesis. Therefore, we hypothesized that polymorphic changes around WISP1 loci are associated with BMD.
The aim of this study was to evaluate the role of polymorphisms around WISP1 locus on BMD at total hip, hip femoral neck, hip intertrochanter, hip trochanter and lumbar spine in 1,510 OOA individuals.
Results
Clinical characteristics of BMD measures and other anthropometrical traits
This study recruited 1,510 OOA who were measured for BMD using Dual-energy X-ray Absorptiometry (DXA). Participant clinical characteristics were shown in Table 1. This study included 715 males (mean age 50.73 years, range 20–95 years) and 795 females (mean age 51.90 years, range 18–93 years). There was no difference in mean age between males and females. On average, females showed higher body mass index (BMI) than males (28.65 ± 5.73 kg/m2 vs 26.66 ± 4.01 kg/m2, p < 0.001). However, the mean levels of BMD in the hip total, femoral neck, hip intertrochanter, trochanter and lumbar spine were significantly higher in males than in females (p < 0.001).
Heritability of the BMD at multiple skeletal sites in the OOA
The heritability of each of the densitometric phenotypes was shown in Table 2 and was based on the most parsimonious model of variance component analysis for each phenotype, including only significant sources of variation. All of the results were statistically significant (p < 0.05). In the whole group consisting of males and females, the heritabilities of the BMD measurements in specific sites were high with variations from 0.66 (lumbar spine BMD) to 0.58 (hip femoral neck BMD). In the sex-stratified model, we found that heritabilities of BMD at multiple skeleton sites were generally greater in females than in males. The heritability of hip intertrochanter BMD (h2 = 0.56) was the highest in the male group, whereas the heritability of lumbar spine BMD (h2 = 0.67) was the highest in female group.
Association of variants in WISP1 with BMD in the OOA
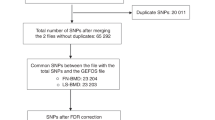
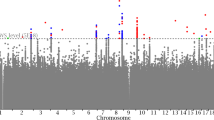
We performed association analyses of 58 tag variants in WISP1 gene region with BMD in total hip, femoral neck, hip intertrochanter, hip trochanter and lumbar spine, adjusted for family structure, study, age, sex, age*sex (model 1). We found 5 variants that were significantly associated with all BMD traits (P values range from 2.89 × 10−6 to 1.62 × 10−2, Table 3 and Fig. 1). The SNP (rs72731533) located in intron 2 was the most significantly associated variant with all the phenotypes (total hip BMD, p = 3.85 × 10−6; hip femoral neck BMD, p = 1.66 × 10−4; hip intertrochanter BMD, p = 2.89 × 10−6; hip trochanter BMD, p = 1.67 × 10−5 and lumbar spine BMD, p = 7.84 × 10−4; Table 3 and Fig. 1). After Bonferroni correction for multiple testing (p < 1.72 × 10−4), rs72731533 was still significantly associated with total hip, intertrochanter and trochanter BMD (Table 3 and Fig. 1). To determine if WISP1 influencing variation in BMD was independent of BMI, we adjusted BMI as a covariant (model 2) and found that these variants remained significant association with BMD traits (Table 3).

Regional association plots for the WISP1 (+/−5 kb) region for BMD in (A) total hip, (B) femoral neck, (C) hip intertrochanter, (D) hip trochanter and (E) lumbar spine. Genetic variants in and around the WISP1 gene (+/−5 kb) are depicted on x axis, and the corresponding association p value (−log10) on y axis. The top SNP, i.e. rs72731533, is denoted by a purple color. Variants are color coded according to their LD (r2) with the lead SNP (1000 Genomes Project Nov 2014 EUR population). The recombination rate (grey line) and position of gene, its exons and direction of transcription are also indicated.
Replication study in GEFOS
We further looked for associations of the 5 lead SNPs in the published meta-analysis data in Caucasian20. We found that one SNP (rs11778573) were nominally associated with BMD at both femoral neck (p = 4.82 × 10−2) and lumbar spine (p = 3.20 × 10−2), and another SNP (rs116873248) showed suggestive level of association with BMD at lumbar spine (p = 3.10 × 10−2) (Table 3). We compared the minor allele frequencies of these 5 polymorphisms in the OOA with minor allele frequencies in the GEFOS Caucasian population and found that OOA minor allele frequencies varied little from GEFOS Caucasian population allele frequencies (Table 3).
Conditional analyses
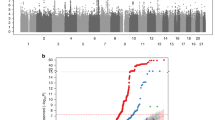
To further determine independent variants, we performed a conditional analysis using the top variant rs72731533 as a covariate and no signal in this region remained (p > 0.01) (Fig. 2). This result indicated that only one association signal in this region was associated with BMD at multiple skeletal sites. We performed linkage disequilibrium (LD) analysis for the five variants associated with BMD using Haploview and found that rs35513885 (exon 4, A118S) was in high LD with rs72731533 (r2 = 0.70, Fig. 3). Using rs35513885 as a covariate, conditional association analysis showed that rs72731533 was still associated with BMD traits (Table 4). This result suggested that rs72731533 may be a leading variant to regulate BMD.

Conditional association plots for the WISP1 (+/−5 kb) region for BMD in (A) total hip, (B) femoral neck, (C) hip intertrochanter, (D) hip trochanter and (E) lumbar spine. Genetic variants within the WISP1 (+/−5 kb) are depicted (x axis) along with their association p value (−log10). The top SNPs, i.e. rs6992383, are denoted by a purple color. Variants are color coded according to their LD (r2) with the lead SNP (1000 Genomes Project Nov 2014 EUR population). The recombination rate (grey line) and position of gene, its exons and direction of transcription are also indicated.

Linkage disequilibrium(D’ top, r2 bottom) structure for all associated SNPs. For both (D’ and r2, values range from 0 (no LD) to 100 (complete LD). The thick white line represents a strand of a chromosome. The black bars on the white line of the chromosome are SNPs that have been identified and sequenced. These SNP locations are labeled in this picture from 1 to 5. Each of these SNPs has a name with corresponding numeric code. Each SNP is represented by a labeled grey triangle below the thick white line. Value in each box indicates (D’ or r2 value between 2 SNPs (intersection). For (D’ figures, no value indicates complete LD (100).
Bioinformatics analysis
Based on the UCSC Genome Bioinformatics website (http://genome.ucsc.edu/), we annotated all five BMD association variants in regulatory elements catalogued in Encyclopedia of DNA Elements (ENCODE) project. As shown in Fig. 4C, rs11778573 and rs72731533 are situated near the active enhancer elements on STRM.MRW.MSC (Bone Marrow Derived Mesenchymal Stem Cell Cultured Cells), M.CHON.MRW.DR.MSC (Chondrocytes from Bone Marrow Derived Mesenchymal Stem Cell Cultured Cells) and BONE.OSTEO (primary osteoblast). As shown in Fig. 4D, rs72731533 is situated near the enhancer elements (H3K4Me1 and H3K27Ac marks) and also near the promoter elements (H3K4Me3 and H3K9Ac marks) on the BMDMSC (Bone Marrow Derived Mesenchymal Stem Cell Cultured Cells) and cfBMDMSC (Chondrocytes from Bone Marrow Derived Mesenchymal Stem Cell Cultured Cells) from ENCODE, which indicated that rs72731533 was probably involved in the regulation of gene expression. The SNP, rs72731533, also fell into a DNase Hypersensitivity site in primary osteoblasts (Fig. 4E,F). The accessible chromatin zone is functionally related to transcriptional activity, since this remodeled state is necessary for the binding of proteins such as transcription factors. We further examined whether the 5 significant association SNPs influence gene expression using public databases such as the Genotype-Tissue Expression (GTEx) project and ExSNP. However, we did not find significant eQTL for these SNPs that may be due to lack of bone tissue information in the databases. We further conduct cis-eQTL analyses on the SNPs in WISP1 gene region with transcripts in primary osteoblasts (obtained from bone biopsies)21. We found that a SNP, rs144161059, in high LD (linkage disequilibrium) with rs72731533 (D’ = 1) was significantly associated with WISP1 gene expression (P = 2.17 × 10−3). The SNP, rs144161059, with low minor allele frequency (MAF = 0.003 in 1000 Genomes) failed to be imputed in this study.

UCSC bioinformatics view of the WISP1 region (chr8:134214753–134241422, GRCh37/hg19). The light green, long vertical line indicates the position of SNPs. (A) The Base Position feature; (B) The WISP1 gene prediction using Ensembl gene prediction; (C) the chromatin state segmentation for the cell types using the imputed ChromHMM (Hidden Markov Model), orange: active enhancer, yellow: weak enhancer or enhancer acetylation, PaleTurquoise: Het (Heterochromatin), Silver: ReprPC (Repressed PolyComb); (D) four histone markers modification map in two cell lines generated by Broad Institute using ChIP-Seq. (E) shows the density signal of DNaseI HS and (F) shows the peaks of DNaseI HS in the osteoblast from ENCODE/OpenChrom(Duke University). HMM (Hidden Markov Model), STRM.MRW.MSC and BMDMSC (Bone Marrow Derived Mesenchymal Stem Cell Cultured Cells; M.CHON.MRW.DR.MSC and cfBMDMSC (Chondrocytes from Bone Marrow Derived Mesenchymal Stem Cell Cultured Cells); BONE.OSTEO (primary osteoblast); DS (density signal); PK (peaks).
Discussion
The Wnt signaling pathway is one of the most important signaling pathways in bone regulation because of its essential role in development, particularly in tissue specification and in cellular migration22. This signaling pathway influences all types of bone cells (osteoblasts, osteoclasts and osteocytes) and has showed to be important in skeletal development, maintenance of skeletal homeostasis and in bone remodeling23. Recently, several independent studies, with the goal to detect candidate genes underlying osteoporosis, revealed that many genes in the Wnt signal pathway are associated with lumbar spine, hip femoral neck and whole body BMD, bone strength, cortical bone thickness, and fracture risk10,24. Within the components of the Wnt signaling pathway, the gene coding for WISP1 has been found as a novel target for modulating osteogenesis and improving bone strength. The importance of WISP1 gene in the regulation of BMD and bone strength had been also confirmed by WISP1 knockout (WISP1−/−) mice14 and human WISP1 transgenic mice15. In this study, we have investigated whether the variants in the WISP1 gene are associated with BMD at multiple skeletal sites in the OOA. Our finding is the first report that shows the significant association of the variant surrounding WISP1 gene on BMD in the OOA.
The heritability of BMD is significantly high (h2 >0.5) reported by several studies [6, 17, 27, 30]. We calculated heritability of BMD at multiple skeletal sites in 1,510 OOA subjects and compared the difference of heritability between males and females. The heritability of lumbar spine BMD (h2 = 0.66) was the highest and the heritability of hip femoral neck BMD (h2 = 0.58) was the lowest in the whole group of males and females combined suggesting a greater genetic determinant of BMD in the lumbar spine than in proximal femur that was similar to previous studies25. Variance in the BMD heritability of different skeletal sites is possibly due to dissimilar external forces placed on certain bones of the skeleton. Previous studies had successfully noted site-specific variations of BMD heritability that are gender-dependent26. In this study, we observed that both female and male groups have strong genetic correlations of BMD and we found that heritability of BMD was partly different between females (h2:0.59–0.69) and males (h2:0.53–0.56). It was similar to our previous result that the heritability in BMD was larger in women than in men27,28. However, our results do not support some previous studies that males tend to have higher heritability values than females29,30. Amish males perform farm work and Amish females perform housekeeping work. There is higher physical activity in Amish males than in Amish females that may contribute the difference of heritability in BMD. Tse and colleagues observed that the high degree of BMD heritability may reflect a preservation of genetic influence in the relative absence of external forces26. Our results revealed that 53–69% of variance in BMD of OOA is contributed by genetic factors.
WISP1 is a member of the CCN family growth factors and its variants have been confirmed to be associated with various pathologies. Tao J. et al.17 found that the WISP1 rs16893344 variant allele (T) was associated with a significantly increased risk of myocardial infarction. Chen J. et al.19 found that WISP1 rs16893344, rs2977530, rs2977537, and rs62514004 (P < 0.05) polymorphisms were related to susceptibility of lung cancer; and WISP1 rs11778573, rs16893344, rs2977536, rs2977549 and rs62514004 polymorphisms were significantly associated with platinum-based chemotherapy response in lung cancer patients. Interestingly, Tomohiko et al.16 evaluated the association of a SNP (rs2929970) in the WISP1 3′-UTR region with the presence of osteophytes, endplate sclerosis, and narrowing of disc spaces for spinal osteoarthritis in 304 postmenopausal Japanese women and found strong associations of rs2929970 with endplate sclerosis. Several GWAS identified BMD SNPs are nominally associated with prevalent radiographic knee osteoarthritis (OA)31. The previous studies suggested that the loci, associated with osteoarthritis, might be also association with BMD. In the present study, we examined the association between polymorphisms around the WISP1 gene and BMD at the total hip, femoral neck, hip intertrochanter, hip trochanter and lumbar spine to investigate a possible contribution of WISP1 to human bone metabolism.
In this study, we identified 58 tag genetic variations with a minor allele frequency (MAF) of at least 1% surrounding the WISP1 gene (+/−5 kb) and found 5 variants significantly associated with all BMD traits. The top associated variant was a SNP (rs72731533) that was located in intron 2 of the WISP1 gene. We further investigated whether the 5 SNPs are independently associated with BMD and found that only one association signal in this region is associated with BMD at multiple skeletal sites. A previous study reported that an intergenetic SNP, rs7839059, located on chromosome 8q24.12 near to WISP1 gene was significantly associated with cortical vBMD (P = 1.2 × 10−10)32. We found that rs7839059 was associated with all the phenotypes (total hip BMD, p = 5.32 × 10−5; hip femoral neck BMD, p = 2.88 × 10−4; hip intertrochanter BMD, p = 1.52 × 10−4; hip trochanter BMD, p = 6.06 × 10−5 and lumbar spine BMD, p = 3.53 × 10−2). We performed a conditional analysis that showed rs7839059 was independent of the associated signal in WIPS1 gene (Table 5). To replicate our results in larger sample sets, we checked the published meta-analysis data in Caucasians20 and found that two SNPs (rs11778573 and rs116873248) were nominally associated with BMD at multiple skeleton sites (p = 0.031–0.0482). We think the following two reasons may cause nominal replications. First, we used chips for genotyping that just included common variants. The significantly associated variants may be partially link to causal variants. Second, the significantly associated variants may be population specific. The most significant SNP, rs72731533, was involved in regulatory elements (such as enhancer and promoter around WISP1 gene) in both MSCs and OPCs. We did not find significant eQTL for rs72731533. However, the SNP, rs144161059, in high LD with rs72731533 was significantly associated with WISP1 gene expression in primary osteoblasts, although, we did not imputed rs144161059 in this study. Thus, a denser fine-mapping study using sequencing of the WISP1 locus will provide a better resolution to identify potential causal variant(s) and will be helpful for future functional validation. Those reports suggested that variants around the WISP1 region were actively involved in regulation of multiple phenotypes. This combined evidence suggests that polymorphisms around the WISP1 are associated with BMD at multiple skeletal sites.
In conclusion, we performed a regional analysis for 5 kb upstream and downstream WISP1 with specific BMD adjusted for study, sex and age in 1,510 OOA. We confirmed that genetic variation at the WISP1 locus is significantly associated with BMD at multiple skeletal sites. These results identify that genetic variants in WISP1 gene region are associated with BMD levels. Bioinformatics analyses suggest that this feasible association is partly caused by regulatory effects on the enhancer or promoter of WISP1 gene. The results suggest that WISP1 gene could be important for bone health in humans, as has already been shown in vitro and vivo. The denser fine-mapping, replication, and functional validation will be necessary to understand the mechanisms underlying these associations.
Methods
Subjects
The OOA of Lancaster Pennsylvania are relatively homogenous in terms of both genetic ancestry and lifestyle characteristics. Subjects (n = 1,510) included in this study were adults aged 18 years or older, who participated in the Amish Family Osteoporosis Study (AFOS), the Amish Family Longevity Study (AFLS) and Pharmacogenomics of Anti-Platelet Intervention (PAPI). The protocols and procedures for those studies were approved by the Institutional Review Boards of the University of Maryland and all subjects gave written informed consent. All methods were performed in accordance with the relevant guidelines and regulations. Details of these studies design, recruitment, and phenotyping had been described previously33,34,35,36. Briefly, the AFOS was started in March 1997 to study genes that are important for bone health. This was a population-based study to identify individuals with osteoporosis. After the identification of individuals with osteoporosis by BMD, family members were recruited including first-degree relatives aged 20 years and older. Spouses were also invited to participate in the study. The goal of AFLS is to identify genes related to a long and healthy life and to understand the function of these genes. The goal of the PAPI study was to understand the reason why some people do not respond to commonly used medications to prevent myocardial infarction, including aspirin and clopidogrel (Plavix). The subjects in the three studies had bone mineral density (BMD) measured by DXA.
Body composition and bone mineral density
Examinations were conducted at the Amish Research Clinic in Strasburg, PA31,33,37,38. Height was measured by using a stadiometer. Height and weight were recorded without shoes. Body mass index (BMI) was calculated as weight in kilograms divided by height in meters squared. Areal BMD (grams per square centimeter) was measured by DXA at the total hip, hip femoral neck, hip intertrochanter, hip trochanter bone and lumbar spine (L1–L4), using a Hologic 4500 W (Bedford, MA, USA) by a technician certified in bone densitometry. Our study focused on multiple densitometric phenotypes that we considered clinically relevant. For femoral neck, there were 1510 OOA examined (male n = 715 and female n = 795), but for spine, five subjects were excluded, due to either prior spine surgery or structural abnormalities, leaving male n = 713 and female n = 792 participants.
Genotyping and single nucleotide polymorphism (SNP) Selection
Study participants were genotyped using either the Affymetrix 500k or Affymetrix v6.0 SNP chips by the Genomics Core Laboratory at the University of Maryland. SNPs with a minor allele frequency (MAF) ≥1%, a call rate exceeding 95% and conforming to the expectations of Hardy-Weinberg equilibrium (p > 10−6) were used for imputation with IMPUTE-2 using 1000 G CEU reference sample phase2. SNPs with Imputation quality score (INFO) ≥30% were considered. Selection of the tagSNPs was performed based on the OOA genotyping data. Using the aggressive tagger mode of Haploview version 4.2 (http://www.broadinstitute.org/haploview/), we selected 58 tagSNPs which cover all common genetic variation within 5 kb upstream and downstream to WISP1 gene (Chr8: 134198282–134248933, GRCh37.p13) (Table 6). Association analysis including these 58 tagSNPs was performed.
Statistical Analysis
Summary statistics of baseline clinical characteristics were expressed as unadjusted means ± standard deviations (SD) using the SPSS statistics version 23 (IBM Corporation, N.Y., NY, USA). The association analyses were carried out using in-house software called MMAP (https://mmap.github.io/). The polygenic component was modeled using the relationship matrix derived from the complete 14-generation Amish pedigree structure. We included family structure, study, age, sex, age*sex, as covariates in the association models. BMI was associated with BMD on univariate analysis and was therefore included as a covariate in model 2. Subgroup analyses to determine whether there were differences in gender were performed. Estimation of the additive genetic heritability follows basic quantitative genetic theory, which models the phenotypic covariance (conditional upon covariate effects) between two individuals in a pedigree as a function of their degree of biologic relatedness. Maximum likelihood methods were used to estimate the values of the parameters, such as heritability, that resulted in highest likelihood obtained across all of the pedigrees. Covariates for BMD heritability analysis were study, sex and age. P-values less than 0.05 were considered as significant. Correction for multiple testing was performed using the Bonferroni method for the number of SNPs and traits tested (P = 0.05/(58 × 5) = 1.72 × 10−4).
Bioinformatics analysis
Analysis of linkage disequilibrium (LD) statistics (r2) surrounding variants of interest was performed using Haploview version 4.2 (http://www.broadinstitute.org/haploview/). Prediction of histone marks and DNAse hypersensitivity sites was performed using HaploReg v4.139, and the five SNPs were annotated in regulatory elements cataloged in Encyclopedia of DNA Elements (ENCODE) project according to UCSC Genome Bioinformatics website (http://genome.ucsc.edu/). The eQTL analyses were performed in GTEx (https://www.gtexportal.org/home/), ExSNP (http://www.exsnp.org/), and primary osteoblasts (obtained from bone biopsies)21.
References
Ensrud, K. E. & Crandall, C. J. Osteoporosis. Annals of internal medicine 167, ITC17–ITC32, https://doi.org/10.7326/AITC201708010 (2017).
Burge, R. et al. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005–2025. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research 22, 465–475, https://doi.org/10.1359/jbmr.061113 (2007).
Park, J. H. et al. Genetic influence on bone mineral density in Korean twins and families: the healthy twin study. Osteoporosis international: a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA 23, 1343–1349, https://doi.org/10.1007/s00198-011-1685-z (2012).
Piters, E. et al. Common genetic variation in the DKK1 gene is associated with hip axis length but not with bone mineral density and bone turnover markers in young adult men: results from the Odense Androgen Study. Calcified tissue international 86, 271–281, https://doi.org/10.1007/s00223-010-9334-7 (2010).
Lee, H. J. et al. Association of a RUNX2 promoter polymorphism with bone mineral density in postmenopausal Korean women. Calcified tissue international 84, 439–445, https://doi.org/10.1007/s00223-009-9246-6 (2009).
Velazquez-Cruz, R. et al. WNT3A gene polymorphisms are associated with bone mineral density variation in postmenopausal mestizo women of an urban Mexican population: findings of a pathway-based high-density single nucleotide screening. Age 36, 9635, https://doi.org/10.1007/s11357-014-9635-2 (2014).
Grundberg, E. et al. Large-scale association study between two coding LRP5 gene polymorphisms and bone phenotypes and fractures in men. Osteoporosis international: a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA 19, 829–837, https://doi.org/10.1007/s00198-007-0512-z (2008).
Hendrickx, G. et al. Variation in the Kozak sequence of WNT16 results in an increased translation and is associated with osteoporosis related parameters. Bone 59, 57–65, https://doi.org/10.1016/j.bone.2013.10.022 (2014).
Hsu, Y. H. & Kiel, D. P. Clinical review: Genome-wide association studies of skeletal phenotypes: what we have learned and where we are headed. The Journal of clinical endocrinology and metabolism 97, E1958–1977, https://doi.org/10.1210/jc.2012-1890 (2012).
Estrada, K. et al. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nature genetics 44, 491–501, https://doi.org/10.1038/ng.2249 (2012).
Gori, F., Lerner, U., Ohlsson, C. & Baron, R. A new WNT on the bone: WNT16, cortical bone thickness, porosity and fractures. BoneKEy reports 4, 669, https://doi.org/10.1038/bonekey.2015.36 (2015).
Parisi, M. S., Gazzerro, E., Rydziel, S. & Canalis, E. Expression and regulation of CCN genes in murine osteoblasts. Bone 38, 671–677, https://doi.org/10.1016/j.bone.2005.10.005 (2006).
French, D. M. et al. WISP-1 is an osteoblastic regulator expressed during skeletal development and fracture repair. The American journal of pathology 165, 855–867, https://doi.org/10.1016/s0002-9440(10)63348-2 (2004).
Maeda, A. et al. WNT1-induced Secreted Protein-1 (WISP1), a Novel Regulator of Bone Turnover and Wnt Signaling. The Journal of biological chemistry 290, 14004–14018, https://doi.org/10.1074/jbc.M114.628818 (2015).
Ono, M., Inkson, C. A., Kilts, T. M. & Young, M. F. WISP-1/CCN4 regulates osteogenesis by enhancing BMP-2 activity. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research 26, 193–208, https://doi.org/10.1002/jbmr.205 (2011).
Urano, T. et al. Association of a single nucleotide polymorphism in the WISP1 gene with spinal osteoarthritis in postmenopausal Japanese women. Journal of bone and mineral metabolism 25, 253–258, https://doi.org/10.1007/s00774-007-0757-9 (2007).
Tao, J. et al. Association of genetic variations in the Wnt signaling pathway genes with myocardial infarction susceptibility in Chinese Han population. Oncotarget, https://doi.org/10.18632/oncotarget.10401 (2016).
Tanaka, S. et al. A novel variant of WISP1 lacking a Von Willebrand type C module overexpressed in scirrhous gastric carcinoma. Oncogene 20, 5525–5532, https://doi.org/10.1038/sj.onc.1204723 (2001).
Chen, J. et al. Association of Wnt-Inducible Signaling Pathway Protein 1 Genetic Polymorphisms With Lung Cancer Susceptibility and Platinum-Based Chemotherapy Response. Clinical lung cancer, https://doi.org/10.1016/j.cllc.2014.12.008 (2014).
Zheng, H. F. et al. Whole-genome sequencing identifies EN1 as a determinant of bone density and fracture. Nature 526, 112–117, https://doi.org/10.1038/nature14878 (2015).
Grundberg, E. et al. Population genomics in a disease targeted primary cell model. Genome research 19, 1942–1952, https://doi.org/10.1101/gr.095224.109 (2009).
Gough, N. R. Focus issue: Wnt and beta-catenin signaling in development and disease. Science signaling 5, eg2, https://doi.org/10.1126/scisignal.2002806 (2012).
Costantini, A. & Makitie, O. Value of rare low bone mass diseases for osteoporosis genetics. BoneKEy reports 5, 773, https://doi.org/10.1038/bonekey.2015.143 (2016).
Rivadeneira, F. et al. Twenty bone-mineral-density loci identified by large-scale meta-analysis of genome-wide association studies. Nature genetics 41, 1199–1206, https://doi.org/10.1038/ng.446 (2009).
Pocock, N. A. et al. Genetic determinants of bone mass in adults. A twin study. The Journal of clinical investigation 80, 706–710, https://doi.org/10.1172/JCI113125 (1987).
Tse, K. Y., Macias, B. R., Meyer, R. S. & Hargens, A. R. Heritability of bone density: regional and gender differences in monozygotic twins. Journal of orthopaedic research: official publication of the Orthopaedic Research Society 27, 150–154, https://doi.org/10.1002/jor.20721 (2009).
Shen, H. et al. Relationship between vascular calcification and bone mineral density in the Old-order Amish. Calcified tissue international 80, 244–250, https://doi.org/10.1007/s00223-007-9006-4 (2007).
Brown, L. B. et al. Assessment of sex-specific genetic and environmental effects on bone mineral density. Genetic epidemiology 27, 153–161, https://doi.org/10.1002/gepi.20009 (2004).
Yang, T. L. et al. Genetic and environmental correlations of bone mineral density at different skeletal sites in females and males. Calcified tissue international 78, 212–217, https://doi.org/10.1007/s00223-005-0267-5 (2006).
Boudin, E., Fijalkowski, I., Hendrickx, G. & Van Hul, W. Genetic control of bone mass. Molecular and cellular endocrinology 432, 3–13, https://doi.org/10.1016/j.mce.2015.12.021 (2016).
Yerges-Armstrong, L. M. et al. Association analysis of BMD-associated SNPs with knee osteoarthritis. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research 29, 1373–1379, https://doi.org/10.1002/jbmr.2160 (2014).
Paternoster, L. et al. Genetic determinants of trabecular and cortical volumetric bone mineral densities and bone microstructure. PLoS genetics 9, e1003247, https://doi.org/10.1371/journal.pgen.1003247 (2013).
Streeten, E. A. et al. Quantitative trait loci for BMD identified by autosome-wide linkage scan to chromosomes 7q and 21q in men from the Amish Family Osteoporosis Study. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research 21, 1433–1442, https://doi.org/10.1359/jbmr.060602 (2006).
Mitchell, B. D. et al. The genetic response to short-term interventions affecting cardiovascular function: rationale and design of the Heredity and Phenotype Intervention (HAPI) Heart Study. American heart journal 155, 823–828, https://doi.org/10.1016/j.ahj.2008.01.019 (2008).
McArdle, P. F. et al. Does having children extend life span? A genealogical study of parity and longevity in the Amish. The journals of gerontology. Series A, Biological sciences and medical sciences 61, 190–195 (2006).
Wang, H. et al. Effect of Two Lipoprotein (a)-Associated Genetic Variants on Plasminogen Levels and Fibrinolysis. G3 (Bethesda), https://doi.org/10.1534/g3.116.034702 (2016).
Liu, J. et al. A functional haplotype in EIF2AK3, an ER stress sensor, is associated with lower bone mineral density. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research 27, 331–341, https://doi.org/10.1002/jbmr.549 (2012).
Hoppman, N. et al. A common variant in fibroblast growth factor binding protein 1 (FGFBP1) is associated with bone mineral density and influences gene expression in vitro. Bone 47, 272–280, https://doi.org/10.1016/j.bone.2010.04.607 (2010).
Ward, L. D. & Kellis, M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic acids research 40, D930–934, https://doi.org/10.1093/nar/gkr917 (2012).
Acknowledgements
We gratefully acknowledge the support of the families participating in these studies and the Amish Research Clinic Staff for their great efforts in study subject recruitment and characterization. We acknowledge Dr. Elin Grundberg and Dr. Bing Ge for performing some of the eQTL analysis reported in this study. This work was supported by research grants HL69313, DK54261, AG1872801, HL088119, AR046838, HL72515 from the National Institutes of Health, American Diabetes Association grant [7-12-CT-26], American Heart Association grant [17GRNT33440151], and the program of China Scholarships Council (No. 1410260006).
Author information
Authors and Affiliations
Contributions
E.S., A.R.S. and M.F. contributed to the design of study. X.W. S.S., J.P., K.R., D.L., Z.D. and Z.L. conducted experiment and performed data analysis. X.W., E.S. and M.F. drafted manuscript. All authors participated in the revision of the manuscript. All authors read, and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
Dr. Alan R. Shuldiner, in addition to his part-time appointment at the University of Maryland School of Medicine, is Vice President and Co-Head of the Regeneron Genetics Center, LLC, a fully owned subsidiary of Regeneron Pharmaceuticals, Inc. The other authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, X., Salimi, S., Deng, Z. et al. Evaluation of WISP1 as a candidate gene for bone mineral density in the Old Order Amish. Sci Rep 8, 7141 (2018). https://doi.org/10.1038/s41598-018-25272-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-25272-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
