
Abstract
Little is known about levels and patterns of genetic diversity for the entire range of endangered orchids native to China, Korea, and Japan. In this study, we focus on Cypripedium japonicum and suggest three hypotheses: 1) that genetic drift has been a primary evolutionary force; 2) that populations in central and western China harbor higher levels of genetic variation relative to those from eastern China; and 3) that C. japonicum in China maintains the highest genetic variation among the three countries. Using ISSR and SCoT markers, we investigated genetic diversity in 17 populations to test the three hypotheses. As anticipated, we found low levels of genetic diversity at the species level with substantially high degree of genetic divergence, which can be mainly attributed to random genetic drift. Chinese populations harbor the highest within-population genetic variation, which tends to increase from east to west. We also found a close relationship between Korean populations and central/western Chinese populations. Historical rarity coupled with limited gene flow seems to be important factors for shaping genetic diversity and structure of C. japonicum. Our results indicate that the mountain areas in central and western China were likely refugia at the Last Glacial Maximum.
Similar content being viewed by others
Introduction
The lady’s slipper orchid genus Cypripedium L., comprising ca. 50 species of terrestrial herbs, is distributed throughout subtropical to temperate regions of the Northern Hemisphere with the exception of northern Africa1. Although East Asia and North America represent the two major centers of diversity for this genus, the former region harbors the majority of diversity with 32 species2. Cypripedium japonicum Thunb. belongs to section Flabellinervia2 and is distributed across East Asia in China, Korea, and Japan3. In China, it is the only slipper orchid that is distributed from western to eastern regions. Possessing two large fan-shaped leaves and highly appealing flowers, C. japonicum is of high horticultural value. In addition, its roots and rhizomes, and even the whole plant, are used in Traditional Chinese Medicine (TCM) to promote blood circulation for regulating menstruation, to dispel “wind” (in TCM, wind is one of the six “excesses” or external cases of disease) and to alleviate pain4,5,6. Although C. japonicum is widely distributed in East Asia, the size and number of populations have recently declined because of the destruction of their habitats2,5,7. It is found only on humus-rich soil under mature and successional deciduous forests on mountainous slopes partly as a result of narrow mycorrhizal specificity8. Although the species is not listed as threatened in the Chinese Red List, it will be included in the second batch of “National Key Protected Wild Plants” as a “Class I protected species”9. In Japan, it is categorized as “Vulnerable” (VU)10, whereas in Korea, where it is known to be extremely rare, is listed as “Critically Endangered” (CR)11. At the global level, C. japonicum is listed as “Endangered” (EN)5 and in need of protection and conservation. Under this situation, sound long-term scientific conservation strategies for protecting and conserving the wild populations of C. japonicum are badly needed; such strategies should be based on thorough knowledge of the species’ biology, especially its genetic diversity and structuring within the whole distribution area12.
Population genetic theory predicts that small size and spatial isolation of populations will lead to the random fixation of alleles (i.e., random genetic drift, RGD), promoting allele frequency divergence and inbreeding as well as a reduction in heterozygosity within populations. Overall, RGD reduces genetic diversity within populations and enhances genetic differentiation between populations. Chung et al.7 tried to test this prediction in six Korean populations of C. japonicum but, unfortunately, found no allozyme-based genetic diversity within and among populations. Qian et al.13, using amplified fragment length polymorphisms (AFLP), investigated six populations of C. japonicum in Tianmu Mts. of Zhejiang Province (eastern China) and found low polymorphism (21.0%) and high genetic differentiation (GST = 0.518). Qian et al.4, using inter-simple sequence repeats (ISSR), also found extremely low levels of within-population genetic variation (HeP = 0.042) and substantially high among-population genetic differentiation (GST = 0.671) in six populations from eastern and central China. Biological and ecological traits such as small population size and founder effects, coupled with limited pollen and seed dispersal across isolated fragmented landscapes, have been suggested as factors for shaping genetic diversity and structure of C. japonicum4,7. According to population genetic theory, RGD should affect levels and the distribution of genetic diversity within and among populations, but with a relatively little effect on the level of genetic diversity within the species as a whole. If RGD has operated in populations of C. japonicum as a major evolutionary force for a long period of time, we further expect extremely low levels of genetic variation at the population level but low to moderate levels of genetic diversity at the species level, and substantially high genetic divergence between populations recovered from the full geographic range of this orchid.
The current geographical distribution of plant taxa is assumed to be the result of both present and historical factors (e.g., Pleistocene climate oscillations), being the latter important drivers in shaping the genetic structure and phylogeographical patterns of plants14,15. High levels of genetic diversity are expected for those areas that are thought to be refugial16,17,18,19,20,21 (but see22), as plants would have endured the most adverse periods maintaining relatively large populations, and also because populations having persisted in refugia would have had a longer demographic history than those that are the result of post-glacial colonization. Mountains anywhere played a key role as refugia for biota during the Neogene and Quaternary global cooling23,24,25; reasons include their relative eco-environmental stability and the need of mountain species for moving considerably shorter distances to track climatic oscillations as compared with lowland species21.
Our study area in East Asia, the so-called “Sino-Japanese Floristic region” was not glaciated during the Last Glacial Maximum (LGM, ca. 21,000 yr before present) except for a few high-elevation mountains21,26. In China, several mountainous areas served as important East Asian Pleistocene refugia at the LGM for boreal and temperate species; such areas are mainly located in the western and the central parts of the country (e.g., the Hengduan Mts. and the central China Mountains, including the Qinling Mts., Daba Mts., and the Three Gorges Mountain Region; Fig. 1) but not exclusively (e.g., the Nanling Mts., Fig. 1; see also Fig. 3 in López-Pujol et al.25 and Fig. 4 in Qiu et al.26). If this scenario is also true for C. japonicum in China, we expect relatively higher levels of genetic variation in western populations compared to eastern ones. In addition, we can also anticipate that Chinese populations would harbor higher within-population genetic variation than Japanese and Korean ones, given that the main East Asian Quaternary refugia were located in China26; Chinese mountains usually reach higher altitudes (having, thus, larger altitudinal gradients), which allowed plants to track glacial-interglacial climate oscillations by means of vertical shifts16,17.

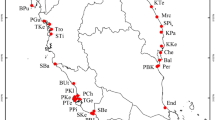
Sampled populations of Cypripedium japonicum in this study. The reconstructed Last Glacial Maximum coastlines are represented as thin dotted lines. All the geographical features quoted in the text are also indicated. The areas indicated by ①, ②, and ③ partitioned by thick dotted lines represent the “first”, the “second”, and “third” steps of the “three-step ladder” of Chinese geographic features58. The base map has been generated with ArcGIS 9.3 (ESRI, Redlands, CA, USA) from a 30 arc-sec layer downloaded from WorldClim Version 1 (http://worldclim.org/), and modified using Adobe Illustrator CS5.1 (Adobe Systems Incorporated, San Jose, CA, USA). Layers from WorldClim Version 1 are licensed under a Creative Commons Attribution-ShareAlike 4.0 International License (https://creativecommons.org/licenses/by-sa/4.0/).
Despite several previous population genetic studies of C. japonicum based on small regional scale sampling have generated considerable background population genetics knowledge of this species4,7,13, a comprehensive study based on a large number of populations across its whole range in East Asia is necessary for interpreting the dynamics of variation over the range of the species, which would allow to design more efficient conservation strategies at both regional and global scales. Therefore, a thorough understanding of population genetic diversity and structuring of C. japonicum across its full range is desirable.
The genetic variation and relationships among wild populations can be efficiently determined by using different molecular markers27, and they can be combined to reach more comprehensive conclusions. Neutral ISSR markers have been widely used for detection of DNA polymorphism in rare species28 due to their advantage of being fast, simple, and cost effective, with good reproducibility4,29. In addition, the start codon targeted (SCoT) polymorphisms, which were developed30 based on the translation start codon, are simple, highly polymorphic, and abundant in the genome27. As the SCoT loci are gene-based markers and are likely to be subject to selection (i.e., non-neutral)31, their use in population genetic studies may be compromised. However, many studies have shown that they are highly correlated with genuinely neutral markers such as ISSR, and are usually more informative and effective than the latter32,33,34.
Using ISSR and SCoT markers, in the present study we estimated the levels and distribution of genetic diversity of C. japonicum sampled from China, Korea, and Japan to test the three hypotheses presented above (i.e., low levels of genetic variation and high genetic divergence between populations for this orchid, higher levels of genetic variation in western Chinese populations compared to eastern ones, and higher levels of genetic variation in Chinese populations compared to Japanese and Korean ones). With the resulting genetic information, along with available demographic information, we tried to provide recommendations for efficient in situ conservation and ex situ preservation of this rare and endangered orchid.
Results
Genetic Diversity
With ISSR, a total of 78 bands were scored with an average of 7.8 bands per primer, and 45 polymorphic bands were obtained. At the population level, the mean PPBP was 9.7%, ranging from 1.3% (BT, LS) to 24.4% (FP), whereas the mean NaP, PA, NeP, HEP, and SI were 1.10, 1.88, 1.04, 0.027, and 0.042, respectively (see the Methods section or Table 1 for the definitions of abbreviations of genetic parameters). Population FP exhibited the highest HEP value (0.062), while population BT did the lowest HEP (0.004). At the species level, all these parameters exhibited higher mean values, with PPBS of 57.7%, NaS of 1.58, NeS of 1.16, HES of 0.104, and SI of 0.169 (Table 1). With SCoT, a total of 77 bands were observed with an average of 8.6 bands per primer, of which 47 were polymorphic. At the population level, the mean PPBP was 10.9%, ranging from 2.6% (Ka) to 24.7% (FP), and the mean NaP, PA, NeP, HEP, and SI were 1.11, 1.69, 1.05, 0.033, and 0.051, respectively (Table 1). FP exhibited the highest HEP value (0.074) again, but Ka had the lowest HEP (0.007). At the species level, like the ISSR, all those parameters exhibited higher mean values: PPBS = 61.0%, NaS = 1.61, NeS = 1.24, HES = 0.145, and SI = 0.224 (Table 1).
Mean genetic estimates (mean PPBP, NaP, NeP, HEP, and SI) from SCoT were higher than those from ISSR (Table 1), and these differences were significant (Wilcoxon signed-rank tests; for all genetic parameters P < 0.05; see Supplementary Table S1). However, we found a significant correlation between ISSR and SCoT datasets (Spearman’s rank correlation test; for all genetic parameters R2, coefficient of determination, ranged from 0.791 for HEP to 0.930 for PPBP and NaP, P < 0.000; Supplementary Table S1), suggesting these two datasets are highly compatible and, thus, that SCoT markers behaved as selectively (or nearly selectively) neutral in C. japonicum. For PA, however, we found neither significant difference nor correlation between the two datasets (Supplementary Table S1).
In China, levels of genetic diversity with the exception of PA and NeP tended to increase from east to west (Table 1); populations in western region harbored significantly higher levels of within-population genetic variation than those in eastern region (with ISSR, mean HEP = 0.049 vs. 0.018, P = 0.008; with SCoT, mean HEP = 0.058 vs. 0.028, P = 0.030). On the other hand, levels of genetic diversity of the six parameters tended to increase from Korea to China (Table 1); 11 Chinese populations exhibited marginally significantly higher levels of within-population genetic variation than two Korean populations (with ISSR, mean HEP = 0.031 vs. 0.008, P = 0.100; with SCoT, mean HEP = 0.040 vs. 0.015, P = 0.054).
Genetic differentiation between populations
Values of GST showed very small differences between the total set of populations and the set of populations excluding those with small sample sizes (n < 10), and this was applicable for the whole species range (e.g., for ISSR, GST = 0.749 and 0.737 including and excluding small populations, respectively) as well as within each country separately (e.g., for ISSR in China, GST = 0.636 vs. 0.599) and with all possible combinations of country pairs (e.g., for ISSR in China vs. Japan, 0.335 vs. 0.362). We consider, therefore, that including populations with small sample size would not significantly alter results and conclusions, and we conducted further data analyses with the total set of populations.
With ISSR, the total genetic diversity for the species (HT) was 0.106, while the mean heterozygosity within populations (HS) and between populations (DS) were 0.027 and 0.080, respectively. The degree of genetic differentiation between populations (GST) was 0.749. Similar results were found with the SCoT dataset: HT = 0.152, HS = 0.033, DS = 0.119, and GST = 0.779. Chinese populations showed higher GST (0.636 for ISSR and 0.697 for SCoT) than those from Japan (0.321 for ISSR and 0.406 for SCoT) and Korea (0.104 for ISSR and 0.145 for SCoT) (Supplementary Table S2). When populations in each country were combined, the highest GST value was found for “Korea vs. Japan” (0.672 for ISSR; 0.608 for SCoT), followed by “China vs. Japan” (0.335 for ISSR; 0.300 for SCoT), and “China vs. Korea” (0.120 for ISSR; 0.159 for SCoT) (Supplementary Table S2).
AMOVA analysis with ISSR revealed that variation among regions (China, Japan, and Korea) and among populations within regions contributed 48.1% and 30.9% to the total genetic variance, respectively (Table 2). Similar results were found with SCoT: 43.5% and 38.3% of the total variation among regions and among populations within regions, respectively (Table 2). Consistent with the hierarchical analyses for GST, separate AMOVA analyses for different subsets of the data showed that the highest genetic divergence between regions was found between Korea and Japan (80.2% for ISSR and 75.4% for SCoT), followed by China vs. Japan (52.6% for ISSR and 43.3% for SCoT; Table 2); in contrast, low degree of divergence was found between China and Korea (22.6% for ISSR and 32.2% for SCoT; Table 2).
We found a significant correlation between pairwise genetic distances and linear geographic distances (km) of populations (Mantel test for IBD, isolation by distance: r = 0.713, P = 0.001 for ISSR; r = 0.515, P = 0.001 for SCoT) when all populations were used (Supplementary Fig. S1). Except for SCoT in Japan (r = 0.478, P = 0.262, primarily due to low statistical power, only six pairs), results of the hierarchical test for IBD showed significant correlations within countries (China, r = 0.752 for ISSR and r = 0.669 for SCoT; Japan, r = 0.858 for ISSR), and also significant correlations were observed when countries were combined (China vs. Japan, r = 0.782 for ISSR and r = 0.600 for SCoT; Korea vs. Japan, r = 0.687 for ISSR and r = 0.747 for SCoT; China vs. Korea, r = 0.316 for ISSR and r = 0.362 for SCoT) (Supplementary Table S2).
Genetic structure and relationships between three countries
The UPGMA dendrograms constructed from ISSR (Fig. 2A) and SCoT (Fig. 2B) markers both showed that the populations of C. japonicum could be largely divided into two geographic clusters: the China + Korea cluster and the Japanese cluster. The main difference between the two datasets was that Korean populations clustered with western and central Chinese populations with ISSR (even when deleting population ZP; data not shown), whereas they clustered only with WX (a western Chinese population) and then with the remaining Chinese populations with SCoT.

UPGMA dendrograms showing the relationships between the populations with ISSR (A) and SCoT (B). Numbers below branches represent bootstrap support (BS) for 999 replicates; only BS values ≥ 50% are provided.
Principal coordinate analysis (PCoA) results were in good agreement with those from the UPGMA dendrograms (Fig. 3). In the ISSR PCoA (Fig. 3A), the first two components accounted for 76.5% (axis 1 = 47.2%; axis 2 = 29.3%) of the total genetic variance, in a similar way that in the SCoT PCoA (Fig. 3B) in which they accounted for 70.8% (axis 1 = 44.7%; axis 2 = 26.1%) of the total genetic variance. The main differences between the two distribution plots were BT and SN populations, which were placed between the eastern and western Chinese populations in the SCoT analysis but nested within western Chinese populations in the ISSR analysis.

The principal coordinate analysis (PCoA) plots based on the two principal axes of ISSR (A) and SCoT (B) analysis.
In the combined datasets (ISSR and SCoT), the ΔK statistics indicated K = 2 as the best grouping scheme (K = 5 was the second highest value; Fig. 4A), whereas the estimated log probability of data obtained with Structure reached a plateau when the number of clusters was set to 5 (Fig. 4B). Thus, we chose K = 5 as the most meaningful clustering. However, we also represented K = 2, K = 3, and K = 4 as these results might be informative. When K = 2, the Chinese and Korean populations clustered together and Japanese population had an independent genetic pool (Fig. 4C). However, when K = 5, the Japanese, Korean, eastern Chinese, western Chinese, and central Chinese populations represented independent genetic pools (Fig. 4C). Interestingly, at K = 3 the Korean populations were clustered together with central and western Chinese populations, and their own cluster were not assigned until K = 4 (Fig. 4C). The findings of Bayesian clustering analysis implemented in Structure were in broad agreement with the UPGMA and PCoA results, clearly depicting that 1) populations from China and Korea have a close relationship, and 2) Korean populations are genetically more related to central and western Chinese populations than to eastern Chinese populations.

Discussion
Comparisons between ISSR and SCoT
In this study, two dominant genetic markers, ISSR and SCoT, were employed to assess the levels and distribution of genetic diversity of C. japonicum in East Asia. The amplification products generated from the SCoT markers may be correlated to functional genes and their corresponding traits, while the ISSR markers detect polymorphisms in microsatellite regions that do not necessarily represent functional genes35. Each marker system targets different regions of the genome, which leads to different sources of the detected diversity. In the present study, both of them indicated low genetic diversity both at population and species levels, and showed a similar clustering of C. japonicum populations (see below for details). Although the values for the genetic parameters were significantly higher for SCoT compared to ISSR, overall a significant correlation was found between these two datasets. To sum up, using independent data from two different methods (as complementary analyses) can represent a more efficient and accurate way to survey the genetic variability of a given species.
Extremely low levels of within-population and high degree of between population genetic diversity
As expected, C. japonicum exhibits extremely low levels of within-population genetic variation (with ISSR, PPBP = 9.7% vs. 46.6%; HEP = 0.027 vs. 0.167) when compared with other orchids that have been screened with ISSR (Supplementary Table S3). At the species level, C. japonicum still harbors low levels of genetic variation (with ISSR, PPBS = 57.7% vs. 63.4%; HES = 0.104 vs. 0.263) compared to means found in orchids (Supplementary Table S3). The much greater estimates at the species level are due to the substantial genetic divergence detected between populations of C. japonicum (GST = 0.749 for ISSR; GST = 0.779 for SCoT). We found considerably high GST values within China (GST = 0.636 for ISSR; GST = 0.697 for SCoT) and Japan (GST = 0.321 for ISSR; GST = 0.406 for SCoT), which are likely to be caused primarily by RGD (see below) and limited gene flow. A very similar GST estimate was reported in the epiphytic or lithophytic orchid Dendrobium fimbriatum from Xishuangbanna (a region of ca. 19,700 km2 in Yunnan Province, South China); Ma and Yin36, using 12 ISSR markers, estimated high species-level genetic variation (PPBS = 89.7%; HES = 0.323) and high degree of population differentiation GST (0.744) from five populations, and, accordingly, low population-level variation (PPBP = 23.9%; HEP = 0.087). We believe that, like C. japonicum, RGD coupled with restricted pollen and seed dispersal would have shaped the genetic structure of D. fimbriatum in Xishuangbanna.
Chung et al.7 found no allozyme variation in six Korean populations of C. japonicum and argued that the species was historically rare compared to its congener C. macranthos on the Korean Peninsula, suggesting that RGD would be a major evolutionary force in the former. Consistent with this and more recently, Son and Son37 found low levels of microsatellite-based genetic variation in four Korean populations of C. japonicum (%PP = 35.0%; AP = 1.53; HeP = 0.109, based on 10 polymorphic loci) but a higher among-population divergence (ΦST = 0.810 including one Japanese population). Our finding of much lower levels of within-population genetic variation when compared to those at the species level, indirectly indicates the importance of RGD for C. japonicum. A recent meta-analysis of allozyme-based population genetic studies in orchids also revealed that “rare” taxa harbor significantly lower levels of genetic diversity than their “common” congeners (at the population level, percentage of polymorphic loci, %PP = 46.7% vs. 23.6%; HEP = 0.154 vs. 0.065 and at the species level, %PS = 63.8% vs. 30.4%; HES = 0.190 vs. 0.072)38. One of the exceptions was C. macranthos var. rebunense, endemic to the small Rebun Island close to Hokkaido (Japan), which harbors comparable levels of genetic diversity with its common North American congeners. This unexpected genetic result is probably due to the fact that the decline in habitat range and population size of this island’s endemic variety has been very recent (since 1950s)39; there have not been sufficient generations for the initial diversity to be substantially eroded by RGD40.
Other life-history or ecological traits associated with pollen and seed dispersal have a highly significant impact on genetic variability and are strong predictors for the levels and distribution of genetic diversity in plant species41,42. The three bumblebees Bombus remotus, B. imitator and B. picipes are the most effective primary pollinators of C. japonicum in China43, while B. ardens and B. diversus are the effective pollinators in Japan44. It is known that bumblebees have a flight range of less than 15 km45. Pollen dispersal is, therefore, likely to be relatively restricted among the highly isolated populations of C. japonicum. Moreover, non-rewarding C. japonicum flowers bloom in May43, when precipitation is high in most of its distribution range in China, which could dramatically reduce the foraging activity of bumblebees and indirectly limit pollen dispersal46. Pollination also suffers from visits by illegitimate visitors as these insects destroy the flowers47. All these factors might contribute to the low pollen dispersal observed in the species43,44.
Since orchids produce tiny, “dust-like” numerous seeds, they have the potential for long-distance seed movement once seeds are released and enter the air column48. However, direct and indirect (genetic) studies have documented that the majority of seeds are transported to very short distances, in the close vicinity of maternal plants, with events of long-distance dispersal being sporadic49,50,51. In addition, like other terrestrial orchids, the seeds of C. japonicum require mycorrhizal fungi for germination and seedling nutrition8,52,53. The limitation of pollen and seed flow across highly isolated populations would lead to low gene flow, increasing the genetic differentiation between populations of C. japonicum (see below for further details).
Levels of genetic diversity and population genetic structure: insights into phylogeographic history
Since historical events leave an “indelible” mark on patterns of genetic diversity found within plant species, understanding their current levels and distribution can provide valuable insights into the recent evolutionary and biogeographical history of these species16,17,19,26,54. As hypothesized, levels of genetic diversity (with the exception of PA and NeP) tend to increase from east to west in China. The finding that the central and western populations harbor relatively higher levels of genetic variation compared to eastern ones are consistent with the hypothesis that the former areas served as large LGM refugia for many species, mainly boreal and temperate plants, in China25,26. In particular, population FP, the highest in levels of genetic diversity among all Chinese populations examined, is located in the south Qinling Mts. (Fig. 1); this mountain region is considered one of the main refugia of western China, and was an area not heavily influenced by the LGM55. In general, large parts of central and southwestern China, including the Three Gorges Region, the Yungui Plateau, the Qinling Mts., and partially the Hengduan Mts. (Fig. 1), acted as plant refugia25,26 due to their relatively stable climatic conditions56, which allowed the persistence of even thermophilous, relict plant taxa (e.g., Davidia involucrata57). All the studied populations of C. japonicum occurring in these areas (with the only exception of population BT) are among the most genetically variable (Table 1). It should be noted that, although in eastern China the presence of LGM refugia has also been hypothesized, these were generally small (“microrefugia”), such as Tianmu Mts. (Fig. 1)25,26. Despite the levels of genetic variability for the eastern Chinese populations are smaller, their similar number of private alleles (PA) compared to western ones (Table 1) also suggests the occurrence of glacial refugia, albeit small, there.
The genetic clustering patterns of Chinese populations might, indeed, reflect the different nature of the terrain, which would have deeply influenced the occurrence of C. japonicum refugial areas. The two main genetic clusters detected in the species (the western/central China cluster and the eastern China cluster, clearly depicted by the UPGMA, PCoA, and Structure) roughly correspond to the “second” and “third” steps of the “three-step ladder” of Chinese geographic features58, respectively (see ② and ③ in Fig. 1, respectively). The second step approximately extends from the eastern margin of the Qinghai-Tibetan Plateau to Taihang Mts. (Fig. 1), and it is comprised of plateaus and middle-altitude mountains (of generally 1,000–2,000 m), including the Qinling Mts. and other mountain systems of central China (Daba Mts. and the Three Gorges area; Fig. 1), which would have served as the main glacial refugia for C. japonicum and many other species25,26; the third step, containing China’s largest plains, would have harbored, instead, populations that endured the LGM in small, favorable pockets in eastern China (in “microrefugia”, e.g., Tianmu Mts.; Fig. 1), and perhaps also newly-founded populations (i.e., populations of post-glacial origin).
As also hypothesized, the levels of genetic diversity of C. japonicum are higher for Chinese populations, followed by Japanese populations, and finally Korean ones, which further supports the species’ glacial refugial scenario for central and western China. Korean populations exhibit extremely low levels of genetic diversity, despite that the Baekdudaegan (BDDG, the main mountain system of the Korean Peninsula) was a glacial refugium for a large assemblage of boreal and temperate plants during the LGM21,59. Plants from the BDDG often harbor moderate to high levels of within-population genetic diversity (Table 1 in Chung et al.21), because these mountains might provide relatively stable habitats and enabled plants to track the climate changes by altitudinal movements, ensuring relatively large population sizes. For example, C. macranthos (a congener of C. japonicum), largely occurring along the BDDG ridge, harbors moderate levels of within-population genetic variation7. Populations of C. japonicum, however, occur on hillsides with elevations of less than 600 m on the Korean Peninsula. Likewise, in Japan, C. japonicum largely occurs on lower hillsides, and our samples were collected from hillsides at altitudes below 400 m. Like in Korea, lowlands and low-elevation hills in Japan hardly harbored large refugia (“macrorefugia”). Instead, contemporary populations would be descendants of populations that endured the LGM in microrefugia, where they suffered the effects of RGD.
The closer relationships between Korean and Chinese populations than between Korean and Japanese populations can be simply due to the fact that the Korean Peninsula is contemporarily connected with NE China, although such connection was even more straightforward at the LGM, when the Yellow Sea was completely exposed (cf. dotted lines for the reconstructed LGM coastlines; Fig. 1). The Japanese Archipelago, instead, was nearly isolated from the continent throughout the Quaternary. Unexpectedly, the two Korean populations are clustered with western and central Chinese populations rather than with the eastern Chinese populations (Figs 2–4), which are geographically closer to the Korean populations. An origin of the Korean populations from the mountains of western China instead of eastern China could explain such pattern of genetic clustering; since C. japonicum occurs in middle-low altitude mountains in China, the most straightforward migration routes were probably through the mountains of north China [Qinling, Taihang Mts., then the north Hebei Mts. (Yan Mts.); see Fig. 1]. Colonization of Korea from eastern Chinese mountains seems unlikely, as both regions are separated by large plains (e.g., the huge North China Plain; Fig. 1) with no suitable habitat for the species. Our ongoing study on phylogeography of C. japonicum in East Asia using cpDNA has revealed that the only haplotype found in Korean populations also occur in SN and ZP, which are located in Daba Mts. (L.X. Han et al. unpubl. data).
The results of AMOVA, UPGMA, PCoA, and Structure (but also the hierarchical analyses of GST and IBD) show that C. japonicum in East Asia can largely be grouped into two genetic clusters, namely the China + Korea cluster and the Japan cluster. The different chromosome number of C. japonicum in China (2n = 22 in Tianmu Mts.60) and Japan (2n = 20 based on cultivated plants61,62) suggests an ancient separation and a reproductive barrier between Chinese and Japanese populations, in case that these counts are confirmed for other populations within the species’ range in these two areas. Our genetic data that indicate a sharp genetic break between continental (China + Korea) and Japanese populations are in agreement with the cytogenetic divergence. Whether both genetic lineages (China + Korea and Japan) represent cryptic species, and merit any taxonomic (as species, subspecies or varieties) or evolutionary recognition (e.g., evolutionarily significant units), need further research including morphological, cytogenetic (e.g., its chromosome number in Korea is not known), and molecular phylogenetic studies.
Implications for conservation
Cypripedium japonicum is threatened with extinction because of its low levels of genetic variation and its high ornamental and medicinal value, particularly in China. The species is categorized as “Endangered” at global scale following the most recent criteria of IUCN63: B2ab(ii,iii,iv,v); C2a(i)5. Thus, in situ and ex situ conservation efforts should be of particular importance for this species. Some previously recorded populations in China might have disappeared already due to deforestation and/or illegal collection4,5.
For a short-time conservation strategy, C. japonicum should be considered as the priority subject of in situ conservation with an emphasis on maintaining the number of genetically distinct individuals. Designation of priority protected areas for natural habitats of C. japonicum could provide ideal protection for in situ conservation64. In Korea, fortunately, all known Korean populations are protected by laws11.
For a long-term conservation strategy, the four populations in central and western China with highest genetic diversity (FP, SZ, SN and JF) should be prioritized for protection, because such populations would provide the best source of propagules for ex situ conservation and in situ restoration efforts65. Although populations from Japan exhibit low within-population genetic variation, they should also be considered for in situ conservation and ex situ preservation because of their considerable divergence from the Chinese plus Korean populations. Indeed, the extinction of any population may lead to a considerable loss of genetic diversity because of the species’ high degree of between-population genetic differentiation, restricted distribution pattern and narrow habitat preferences. Thus, all populations are of great importance for short-term and long-term survival of C. japonicum in East Asia.
Since about 20 conservation actions have already been recommended for Korean, Chinese, and overall populations in East Asia4,5,7, we here further provide a few suggestions for the management and conservation of C. japonicum: 1) to use hand-pollination within or among populations in order to enhance the fruit set and even to accelerate the rejuvenation of small populations because of low rate of fruit set (5.2–7.7% in Hubei, China43; 14.9% in Chiba, Japan44); 2) to apply tissue culture techniques for propagation of different genotypes as well as for satisfying the demand of traditional medicine; 3) to identify detailed clonal structure and fine-scale genetic structure of typical or representative populations in the three countries where the species is present (China, Korea, Japan), in order to provide more scientific and efficient sampling for ex situ preservation; and 4) to develop comprehensive conservation strategies through international collaboration among concerned countries.
Conclusions
To our knowledge, this study is the first detailed examination of any East Asian orchid covering its entire distribution range to evaluate the levels of genetic diversity, to provide hypotheses about the past phylogeographic history, and to recommend conservation strategies. As expected, the endangered terrestrial orchid C. japonicum harbors extremely low genetic diversity at the population level across its whole distribution area. At the species level (samples as a whole), C. japonicum maintains low levels of genetic diversity with high degree of genetic divergence between populations. These genetic results suggest that random genetic drift (RGD) coupled with limited gene flow between populations have played a significant role in shaping levels of genetic diversity and structure in the species. Genetic data revealed that the populations are genetically arranged into two major groups: Chinese + Korean and Japanese groups. Although in situ conservation and ex situ preservation strategies should be primarily focused on populations with high genetic diversity in central and western China, conservation of Korean and Japanese populations is also desirable through close international cooperation among concerned countries.
Methods
Study species
Cypripedium japonicum grows in moist humus-rich soil in the understory of mature and successional deciduous forests on hillsides from China, Japan, and Korea. The plants can also propagate via buds that grow from the creeping underground rhizomes2. Most plants of C. japonicum have two leaves and a 35–55 cm tall stalk bearing a single, large, pendent flower with sac-like labellum (4–5 × 3–3.5 cm). Cypripedium japonicum is self-compatible with a non-rewarding, deceptive pollination system and requires effective pollinators43,44,47. Five species of the genus Bombus have been reported as effective pollinators both in China and Japan with a low fruit set of 5.2–14.9%43,44. Seeds of C. japonicum have a low germination rate, which could restrict their survival and development9,53.
Population sampling
The number of samples collected per population was in proportion to the size of that population. In order to avoid clonal interference for estimating genetic variation in populations, we collected only one sample per small patch (as in small patches all individuals may be ramets of the same genet); we did our best to collect at least 20–30% of all shoots per population. A total of 287 samples from 17 natural populations of C. japonicum were collected from China, Japan and Korea during four years (2010–2013), including small populations with only a few individuals (n = 3 in LS; Fig. 1 and Table 1). The collecting sites were chosen to approximately represent the complete distribution range of C. japonicum; the mean distance among sampled populations was 1,553.6 km, with a range of 44.2–3,834.5 km (in China, mean = 652.5 km, range = 44.2–1,443.5 km; in Japan, mean = 774.1 km, range = 175.3–1,279.1 km; the two Korean populations are separated by 74.2 km). We further divided the 11 Chinese populations into three groups according to geographic and physiographic criteria: DB, HS, LS and TM formed the “eastern” group, BT and SN the “central” group, and FP, JF, SZ, WX and ZP the “western” group. Population SZ, although geographically located in Hunan (province often included in central China according to Chinese geographers) was, instead, merged with the group of western populations because it occurs within Wuling Mts. (that actually constitute the eastern tip of the Yungui Plateau; Fig. 1). For each sampled individual, a small piece of fresh leaf was collected from a single plant and kept in a paper bag with silica gel until DNA extraction.
DNA isolation
Total genomic DNA was extracted by using the modified CTAB method66 and dissolved in ultrapure water for future use. DNA quality was checked by electrophoresis in 1% agarose gels. Nanodrop was used to test its concentration and optical density value. For the ISSR experiment, working DNA was diluted to a concentration of 40–100 ng/μL in ultrapure water and stored at −20 °C before subsequent use. For the SCoT experiment, the most appropriate concentration of working DNA was 100–160 ng/μL. The materials used for ISSR analysis (n = 285) were slightly different from SCoT analysis (n = 265) due to technical constraints. In particular, samples of ZP could not be used for the SCoT analysis due to the degradation of materials (Table 1).
ISSR and SCoT amplification and optimum primer selection
To ensure the reproducibility of both ISSR and SCoT markers, we first conducted many preliminary experiments to optimize PCR amplification (by trying several concentrations of DNA, primers, dNTPs, etc.), and then we chose primers with high polymorphism and good repeatability for subsequent manipulations. About 45% (ISSR) and 35% (SCoT) of the samples were re-run with all the primers, respectively. For ISSR analysis (n = 285), 10 primers were chosen from 100 ISSR primers supplied by University of British Columbia (http://www.biotech.ubc.ca/services/naps/primers.html) for PCR research (Supplementary Table S4). For SCoT analysis (n = 265), nine SCoT primers (Supplementary Table S5) were selected from 80 primers according to Collard and Mackill30 and Luo et al.35. Amplification with these primers was carried out in Takara TP600 and commenced with initial denaturation of 2 min at 94 °C, followed by 35 cycles of 35 s at 94 °C, 30 s annealing at 51–56.8 °C (SCoT: 51–53.4 °C), 90 s of elongation at 72 °C, and ending with a 7 min extension at 72 °C. The 15 μL reaction mixture contained 1 μL DNA, 7.5 μL 2 × Es Taq Master Mix (3 mM Mg2+, 0.4 mM dNTPs, Taq DNA polymerase and 2 × Es Taq PCR Buffer) (Biomiga, China), 40–100 ng DNA template and 0.9 μL (10 pmol/μL) primers. Amplification products were electrophoresed on a 2% agarose gel stained with 1% Goldview at 100 V for 30 min along with 100 bp DNA Ladder Marker, and photographed using the Bio-Rad Gel Documentation System. Reproducibly distinguishable bands were recorded and used in further analysis.
Statistical analyses
Visible and clear DNA bands obtained from each ISSR and SCoT primers were scored as absent (0) or present (1). Bands length between 200–1,200 bp (ISSR) and 400–1,500 bp (SCoT) were scored. Using the program Popgene 1.3267, and assuming that populations were in Hardy–Weinberg equilibrium, we estimated the following genetic diversity parameters: percentage of polymorphic bands (PPB), observed number of alleles (Na), number of “private” alleles (PA), average effective number of alleles per locus (Ne), Nei’s gene diversity index (HE)68, and Shannon’s information index (SI).
To assess the correspondence of genetic diversity between the two datasets (ISSR vs. SCoT), we performed Wilcoxon signed-rank tests on 16 population pairs (with the exception of population ZP) for five within-population genetic variation parameters. We further conducted a Spearman’s rank correlation analysis between ISSR and SCoT for each genetic measure; the higher correlation between ISSR and SCoT for each genetic measure, the smaller the differences in the measurements between the two datasets. Thus, this correlation analysis can be viewed as an indirect way to check the suitability of the SCoT markers in surveying the genetic variability in C. japonicum.
Although we found significant differences in the distribution of values of HE and SI (Mann-Whitney U-test or Wilcoxon rank-sum test for both ISSR and SCoT, P = 0.000), the order of values of those two genetic parameters were nearly the same (Spearman’s rank correlation analysis for both ISSR and SCoT, RS2 = 0.995 and 0.985, respectively, P = 0.000), suggesting that using either HE or SI would be appropriate. We used a Mann-Whitney U-test or Wilcoxon rank-sum test to assess the significance of differences in HE (a summary statistic of within-population genetic variation) between the pair of regions with the highest and the lowest estimates in China. In addition, we further conducted Mann-Whitney U-test to determine any significant difference in HE between the pair of countries with the highest and the lowest estimates in East Asia.
To estimate the degree of population genetic differentiation, we further calculated total genetic diversity (HT), genetic diversity within populations (HS), and genetic differentiation among populations (GST) using Popgene. As the combination of dominant markers and small sample sizes could artificially inflate genetic structure (e.g., GST), we estimated GST values with and without the four populations with small sample sizes [n < 10; LS (3 individuals), DB (5), Ha (7), and JF (8)] to see how much these small populations may be influencing our statistics. To test for the influence of individuals within populations, populations within regions (countries), and regions on the observed genetic variation, we conducted a hierarchical analysis of molecular variance (AMOVA) using GenAlEx 6.569. To determine the relative importance of gene flow and RGD at regional scale70, we conducted a correlation analysis between pairwise genetic distances71 and linear geographic distances (km) and ran a Mantel test for IBD (with 999 permutations) using GenAlEx. A positively significant linear relation suggests that populations are at regional equilibrium between gene flow and RGD70. As reproductive barriers would exist between Chinese and Japanese populations due to different chromosome number (2n = 22 in China60; 2n = 20 in Japan61,62) and geographical isolation, we could be dealing with two cryptic species or subspecies. In order to avoid such potential “phylogenetic” biases in our genetic results, we performed further analyses of GST and IBD in a hierarchical fashion: within each country separately and then with all possible combinations of country pairs (i.e., “China vs. Korea”, “China vs. Japan”, and “Korea vs. Japan”).
We conducted unweighted pair-group method with arithmetic means (UPGMA) based on Nei’s unbiased genetic distances72 between populations using the Tfpga 1.3 program73. Bootstrap values for nodes were estimated based on 999 replications. As a complementary analysis, we performed a principal coordinate analysis (PCoA) with GenAlEx69, on the basis of Nei’s genetic distances74, to investigate the relationships among the populations, and the two principal coordinates were used to visualize the dispersion of accessions in a two-dimensional array of eigenvectors. To deeply explore the population structure and unravel genetic admixture, we used Structure 2.3.475 to analyze the combined dataset (16 populations with ZP exclusion due to lack of SCoT data) of two markers with a Bayesian approach. Posterior probabilities of the data for each K were obtained for K = 1 to K = 18 clusters using the Admixture Model. Fifteen runs were completed for each K, with a Markov Chain Monte Carlo (MCMC) of 200,000 iterations, following a burn-in period of 1,000,000 iterations. We inferred the most likely value of K by the ΔK statistics76, with the aid of Structure Harvester77. Since the ΔK method tends to identify K = 2 as the top level of hierarchical structure78, we combined it with the method of selecting the smallest K after the log probability of data [ln Pr(X|K)] values reached a plateau79. Programs Clumpp 1.1.280 and Distruct 1.181 were used to combine the results of the 15 replicates of the best K and to visualize the results produced by Clumpp, respectively.
References
Chen, X. Q. & Cribb, P. J. Cypripedium Linnaeus in Flora of China, Vol. 25 (Orchidaceae) (eds Wu, Z. Y., Raven, P. H. & Hong, D. Y.) 22–33 (Science Press, Beijing and Missouri Botanical Garden Press, St. Louis, 2009).
Li, J. H. et al. Molecular phylogeny of Cypripedium (Orchidaceae: Cypripedioideae) inferred from multiple nuclear and chloroplast regions. Mol. Phylogenet. Evol. 61, 308–320 (2011).
Kitamura, S., Murata, G. & Koyama, T. Colored illustrations of herbaceous plants of Japan (Choripetalae) (Hoikusha Publishing Co., Osaka, 1986).
Qian, X. et al. Conservation genetics of an endangered Lady’s Slipper Orchid: Cypripedium japonicum in China. Int. J. Mol. Sci. 15, 11578–11596 (2014).
Rankou, H. Cypripedium japonicum. The IUCN Red List of Threatened Species 2014: e.T13188414A16672875. Available from http://dx.doi.org/10.2305/IUCN.UK.2014-1.RLTS.T13188414A16672875.en (2014).
Xie, Z. W., Fan, C. S. & Zhu, Z. Y. The national assembly of Chinese herbal medicine (People’s Medical Publishing House, Beijing, 1975).
Chung, J. M. et al. Contrasting levels of genetic diversity between the historically rare orchid Cypripedium japonicum and the historically common orchid Cypripedium macranthos in South Korea. Bot. J. Linn. Soc. 160, 119–129 (2009).
Shefferson, R. P., Weiß, M., Kull, T. & Taylor, D. High specificity generally characterizes mycorrhizal association in rare lady’s slipper orchids, genus. Cypripedium. Mol. Ecol. 14, 613–626 (2005).
Qiao, Y. B. Mycorrhizal microstructure and fungal endophyte diversity of Cypripedium japonicum Thunb. MS thesis, Southwest University, Chongqing, China (2011).
MOE, Japan (Ministry of the Environment, Japan). Red Data Book, 2014-Threatened Wildlife of Japan–Vol. 8 Vascular Plants. Gyosei, Tokyo, pp. 646 (2015).
MOE, Republic of Korea (Ministry of Environment, Republic of Korea). Korean red list of threatened species. Second edition. National Institute of Biological Resources, Incheon, pp. 242 (2014).
Reed, D. H. & Frankham, R. Correlation between fitness and genetic diversity. Conserv. Biol. 17, 230–237 (2003).
Qian, X., Li, Q. J., Lian, J. J., Wang, C. X. & Tian, M. Genetic diversity of endangered wild Cypripedium japonicum populations: An AFLP analysis. Acta Ecol. Sinica 32, 1445–1450 (2013a).
Cheng, Y. P., Hwang, S. Y. & Lin, T. P. Potential refugia in Taiwan revealed by the phylogeographical study of Castanopsis carlesii Hayata (Fagaceae). Mol. Ecol. 14, 2075–2085 (2005).
Tao, X. Y. Genetic diversity and phylogeographic study of the endangered species Platycrater arguta (Hydrangaceae) endemic to the Sino-Japanese region. MS thesis, Zhejiang University, Hangzhou, China (2008).
Hewitt, G. M. Some genetic consequences of ice ages, and their role in divergence and speciation. Biol. J. Linn. Soc. 58, 247–276 (1996).
Hewitt, G. M. The genetic legacy of the Quaternary ice ages. Nature 405, 907–913 (2000).
Shen, L., Chen, X. Y. & Li, Y. Y. Glacial refugia and postglacial recolonization patterns of organisms. Acta Ecol. Sinica 22, 1983–1990 (2002).
Provan, J. & Bennett, K. D. Phylogeographic insights into cryptic glacial refugia. Trends Ecol. Evol. 23, 564–571 (2008).
Nieto-Feliner, G. Southern European glacial refugia: a tale of tales. Taxon 60, 365–372 (2011).
Chung, M. Y., López-Pujol, J. & Chung, M. G. The role of the Baekdudaegan (Korean Peninsula) as a major glacial refugium for plant species: A priority for conservation. Biol. Conserv. 206, 236–248 (2017).
Petit, R. J. et al. Glacial refugia: hotspots but not melting pots of genetic diversity. Science 300, 1563–1565 (2013).
Fjeldså, J. & Lovett, J. C. Geographical patterns of old and young species in African forest biota: the significance of specific montane areas as evolutionary centres. Biodiver. Conserv. 6, 325–346 (1997).
Médail, F. & Diadema, K. Glacial refugia influence plant diversity patterns in the Mediterranean Basin. J. Biogeogr. 36, 1333–1345 (2009).
López-Pujol, J., Zhang, F. M., Sun, H. Q., Ying, T. S. & Ge, S. Centres of plant endemism in China: places for survival or for speciation? J. Biogeogr. 38, 1267–1280 (2011).
Qiu, Y. X., Fu, C. X. & Comes, H. P. Plant molecular phylogeography in China and adjacent regions: tracing the genetic imprints of Quaternary climate and environmental change in the world’s most diverse temperate flora. Mol. Phylogenet. Evol. 59, 225–244 (2011).
Alikhani, L., Rahmani, M. S., Shabanian, N., Badakhshan, H. & Khadivi-Khub, A. Genetic variability and structure of Quercus brantii assessed by ISSR, IRAP and SCoT markers. Gene 552, 176–183 (2014).
Lin, L., Hu, Z. Y., Ni, S., Li, J. Y. & Qiu, Y. X. Genetic diversity of Camellia japonica (Theaceae), a species endangered to East Asia, detected by inter-simple sequence repeat (ISSR). Biochem. Syst. Ecol. 50, 199–206 (2013).
Sarwat, M. ISSR: a reliable and cost-effective technique for detection of DNA polymorphism. Methods Mol. Biol. 862, 103–121 (2012).
Collard, B. C. & Mackill, D. J. Start codon targeted (SCoT) polymorphism: a simple, novel DNA marker technique for generating gene-targeted markers in plants. Plant Mol. Biol. Rep. 27, 86–93 (2009).
Xiong, F. Q. et al. Start codon targeted polymorphism for evaluation of functional genetic variation and relationships in cultivated peanut (Arachis hypogaea L.) genotypes. Mol. Biol. Rep. 38, 3487–3494 (2011).
Pakseresht, F., Talebi, R. & Karami, E. Comparative assessment of ISSR, DAMD and SCoT markers for evaluation of genetic diversity and conservation of landrace chickpea (Cicer arietinum L.) genotypes collected from north-west of Iran. Physiol. Mol. Biol. Plants 19, 563–574 (2013).
Tsaballa, A. et al. Molecular characterization of Greek pepper (Capsicum annuum L.) landraces with neutral (ISSR) and gene-based (SCoT and EST-SSR) molecular markers. Biochem. Syst. Ecol. 59, 256–263 (2015).
Igwe, D. O. et al. Assessment of genetic diversity in Vigna unguiculata L. (Walp) a cessions using inter-simple sequence repeat (ISSR) and start codon targeted (SCoT) polymorphic markers. BMC Genetics 18, 98 (2017).
Luo, C. et al. Genetic diversity of mango cultivars estimated using SCoT and ISSR markers. Biochem. Syst. Ecol. 39, 676–684 (2011).
Ma, J. M. & Yin, S. H. Genetic diversity of Dendrobium fimbriatum (Orchidaceae), an endangered species, detected by inter-simple sequence repeat (ISSR). Acta Bot. Yunnanica 31, 35–41 (2009).
Son, O. & Son, S. W. Development and characterization of EST-SSR markers for the endangered species Cypripedium japonicum in Abstract Book of the 7th EAFES International Congress. pp. 373 (Inter-Burgo Daegu, 2016).
Chung, M. Y. et al. Patterns of genetic diversity in rare and common orchids focusing on the Korean Peninsula: implications for conservation. Bot. Rev. 84, 1–25 (2018).
Izawa, T., Kawahara, T. & Takahashi, H. Genetic diversity of an endangered plant, Cypripedium macranthos var. rebunense (Orchidaceae): background genetic research for future conservation. Conserv. Genet. 8, 1369–1376 (2007).
Chung, M. Y., Nason, J. D. & Chung, M. G. Implications of clonal structure for effective population size and genetic drift in a rare terrestrial orchid. Cremastra appendiculata. Conserv. Biol. 18, 1515–1524 (2004).
Hamrick, J. L. & Godt, M. J. W. Allozyme diversity in plant species in Plant population genetics, breeding and genetic resources (eds. Brown, A. H. D., Clegg, M. T., Kahler, A. L. & Weir, B. S.) 43–63 (Sinauer Associates, 1989).
Hamrick, J. L. & Godt, M. J. W. Effects of life history traits on genetic diversity in plant species. Philos. Trans. R. Soc. Lond. B Biol. Sci. 351, 1291–1298 (1996).
Sun, H. Q., Cheng, J., Zhang, F. M., Luo, Y. B. & Ge, S. Reproductive success of non-rewarding Cypripedium japonicum benefits from low spatial dispersion pattern and asynchronous flowering. Ann. Bot. 103, 1227–1237 (2009).
Suetsugu, K. & Fukushima, S. Pollination biology of the endangered orchid Cypripedium japonicum in a fragmented forest of Japan. Plant Species Biol. 29, 294–299 (2014).
Goulson, D. & Stout, J. C. Homing ability of the bumblebee Bombus terrestris (Hymenoptera: Apidae). Apidologie 32, 105–111 (2001).
Pyke, G. H., Inouye, D. W. & Thomson, J. D. Activity and abundance of bumble bees near Crested Butte, Colorado: diel, seasonal, and elevation effects. Ecol. Entomol. 36, 511–521 (2011).
Liu, F., Li, Q. J., Wang, C. X., Lian, J. J. & Tian, M. Floral Characteristics and breeding systems of an endangered species Cypripedium japonicum. Sci. Silvae Sin. 49, 53–60 (2013).
Trapnell, D. W. & Hamrick, J. L. Partitioning nuclear and chloroplast variation at multiple spatial scales in the neotropical epiphytic orchid, Laelia rubescens. Mol. Ecol. 13, 2655–2666 (2004).
Chung, M. Y., Nason, J. D. & Chung, M. G. Spatial genetic structure in populations of the terrestrial orchid Cephalanthera longibracteata (Orchidaceae). Amer. J. Bot. 91, 52–57 (2004).
Jacquemyn, H. et al. A spatially explicit analysis of seedling recruitment in the terrestrial orchid. Orchis purpurea. New Phytol. 176, 448–459 (2007).
Jersáková, J. & Malinová, T. Spatial aspects of seed dispersal and seedling recruitment in orchids. New Phytol. 176, 237–241 (2007).
Rasmussen, H. N. & Rasmussen, F. N. Orchid mycorrhiza: implications of a mycophagous life style. Oikos 118, 334–345 (2009).
Liu, F., Tian, M., Wang, C. X., Gong, M. J. & Li, Q. J. Observation on fruit growth dynamics and embryo development process of Cypripedium japonicum. J. Plant Res. Environ. 21, 28–35 (2012).
Godt, M. J. W. & Hamrick, J. L. Genetic diversity in rare southeastern plants. Nat. Areas J. 21, 61–70 (2001).
Bai, W. N., Liao, W. J. & Zhang, D. Y. Nuclear and chloroplast DNA phylogeography reveal two refuge areas with asymmetrical gene flow in a temperate walnut tree from East Asia. New Phytol. 188, 892–901 (2010).
Tian, Z. P. & Jiang, D. B. Revisiting last glacial maximum climate over China and East Asian monsoon using PMIP3 simulations. Palaeogeogr. Palaeoclimatol. Palaeoecol. 453, 115–126 (2016).
Tang, C. Q. et al. Potential effects of climate change on geographic distribution of the Tertiary relict tree species Davidia involucrata in China. Sci. Rep. 7, 43822 (2017).
Zheng, D. & Dai, E. F. Physical geography in Plants of China–A companion to the Flora of China (eds Hong, D. Y. & Blackmore, S.) 35–53 (Science Press, 2015).
Chung, M. Y., López-Pujol, J. & Chung, M. G. Is the Baekdudaegan “the Southern Appalachians of the East”? A comparison between these mountain systems, focusing on their role as glacial refugia. Korean J. Pl. Taxon. 46, 337–347 (2016).
Qian, X. et al. Karyotype analysis of Cypripedium japonicum Thunb. and Calanthe tsoongiana T. Tang et F. T. Wang. J. Trop. Subtrop. Bot. 21, 414–419 (2013b).
Karasawa, K. & Aoyama, M. Karyomorphological studies on Cypripedium in Japan and Formosa. Bull. Hiroshima Bot. Gard. 8, 1–22 (1986).
Kondo, K., Hoshi, Y. & Tanaka, R. Somatic chromosome differentiation in Cypripedium segawai Masamune and C. japonicum Thunberg. Cytologia 59, 115–120 (1994).
IUCN. IUCN Red List Categories: Version 3.1. Prepared by the IUCN Species Survival Commission. IUCN, Gland, Switzerland and Cambridge, UK. Available from http://www.iucn.org/themes/ssc (2001).
Wan, J. Z., Wang, C. J., Han, S. J. & Yu, J. H. Planning the priority protected areas of endangered orchid species in northeastern China. Biodiver. Conserv. 23, 1395–1409 (2014).
Godt, M. J. W., Hamrick, J. L. & Bratton, S. Genetic diversity in a threatened wetland species, Helonias bullata (Liliaceae). Conserv. Biol. 9, 596–604 (1995).
Doyle, J. J. & Doyle, J. L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15 (1987).
Yeh, F. C. et al. POPGENE, the user friendly shareware for population genetic analysis. Molecular Biology and Biotechnology Center. University of Alberta, Edmonton (1997).
Nei, M. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 70, 3321–3323 (1973).
Peakall, R. & Smouse, P. E. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research – an update. Bioinformatics 6, 2537–2539 (2012).
Hutchison, D. W. & Templeton, A. R. Correlation of pairwise genetic and geographic distance measures: Inferring the relative influences of gene flow and drift on the distribution of genetic variability. Evolution 53, 1898–1914 (1999).
Huff, D. R., Peakall, R. & Smouse, P. E. RAPD variation within and among natural populations of outcrossing buffalo grass Buchloe dactyloides (Nutt) Engelm. Theor. Appl. Genet. 86, 927–934 (1993).
Nei, M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89, 583–590 (1978).
Miller, M. P. Tools for population genetic analyses (TFPGA) 1.3: A Windows program for the analysis of allozyme and molecular population genetic data. Utah State University, Logan, UT, USA (1997).
Nei, M. Genetic distance between populations. Am. Nat. 106, 283–392 (1972).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure from multilocus genotype data. Genetics 155, 945–959 (2000).
Evanno, G., Regnaut, S. & Goudet, J. K. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Earl, D. A. & vonHoldt, B. M. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361 (2012).
Janes, J. K. et al. The K = 2 conundrum. Mol. Ecol. 26, 3594–3602 (2017).
Pritchard, J. K., Wen, X. & Falush, D. Documentation for structure software: Version 2.3. Department of Human Genetics, University of Chicago, Chicago, USA. Available from http://pritch.bsd.uchicago.edu/structure_software/release_versions/v2.3.4/structure_doc.pdf (2010).
Jakobsson, M. & Rosenberg, N. A. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 23, 1801–1806 (2007).
Rosenberg, N. A. DISTRUCT: a program for the graphical display of population structure. Mol. Ecol. Notes. 4, 137–138 (2004).
Acknowledgements
The authors thank Xianyun Mu, Gaodi Dang, Dejun Pan, Takao Aoki, and Hiroshi Igarashi for their help during the samples collection; Jae Min Chung for providing the Korean samples; David Baines, Constantijn B. Mennes, and Aiqun Hu for improving the English and their constructive comments. This study was supported by Natural Science Foundation of China (No. 30900077) to H.Z.T. and in part by Basic Science Program through the National Research Foundation of Korea (NRF-2017R1A2B4012215) funded by the Ministry of Education, Science and Technology to M.G.C.
Author information
Authors and Affiliations
Contributions
H.Z.T., J.L.Z., X.L.L., L.X.H., T.K., and T.Y. collected samples; J.L.Z., X.L.L., and L.X.H. conducted the experiments; H.Z.T., J.L.Z., X.L.L., L.X.H., J.L.-P., M.G.C., and M.Y.C. analyzed the data; H.Z.T., J.L.Z., X.L.L., and L.X.H. designed the study; H.Z.T., M.G.C., and M.Y.C. led the writing of the manuscript; H.Z.T., J.L.-P., P.K., M.G.C., and M.Y.C. contributed to the writing. All authors read and reviewed the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tian, H.Z., Han, L.X., Zhang, J.L. et al. Genetic diversity in the endangered terrestrial orchid Cypripedium japonicum in East Asia: Insights into population history and implications for conservation. Sci Rep 8, 6467 (2018). https://doi.org/10.1038/s41598-018-24912-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-24912-z
This article is cited by
-
Phylogenetic relationships and genetic diversity of the Korean endemic Phedimus latiovalifolius (Crassulaceae) and its close relatives
Scientific Reports (2024)
-
Investigation of genetic diversity among autochthonous grape cultivars grown in Türkiye using molecular primers
Genetic Resources and Crop Evolution (2024)
-
Sorghum [Sorghum bicolor (L.) Moench] breeding enrichment potential through genetic comparison of Hungarian and East African lines
Cereal Research Communications (2024)
-
Genetic relationships of salep orchid species and gene flow among Serapias vomeracea × Anacamptis morio hybrids
Plant Biotechnology Reports (2023)
-
Start codon targeted (SCoT) polymorphism marker in plant genome analysis: current status and prospects
Planta (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
