
Abstract
Light-driven nano/micromotors are attracting much attention, not only as molecular devices but also as components of bioinspired robots. In nature, several pathogens such as Listeria use actin polymerisation machinery for their propulsion. Despite the development of various motors, it remains challenging to mimic natural systems to create artificial motors propelled by fibre formation. Herein, we report the propulsion of giant liposomes driven by light-induced peptide nanofibre growth on their surface. Peptide-DNA conjugates connected by a photocleavage unit were asymmetrically introduced onto phase-separated giant liposomes. Ultraviolet (UV) light irradiation cleaved the conjugates and released peptide units, which self-assembled into nanofibres, driving the translational movement of the liposomes. The velocity of the liposomes reflected the rates of the photocleavage reaction and subsequent fibre formation of the peptide-DNA conjugates. These results showed that chemical design of the light-induced peptide nanofibre formation is a useful approach to fabricating bioinspired motors with controllable motility.
Similar content being viewed by others


Introduction
Molecular robotics is a recently emerged concept for the construction of bioinspired robots composed of nano- and micro-scale devices, such as sensors, logic circuits and actuators1,2,3. By using these components, a molecular robot responds to its surrounding environment and makes decisions autonomously. For instance, Nomura et al. recently developed an amoeba-like molecular robot consisting of a body (liposome), motor (kinesin, microtubules) and clutch (DNA)3. When the kinesin was attached to a liposomal inner membrane in response to a signal molecule composed of complementary DNA, the robot exhibited a continuous shape change caused by microtubules gliding on the membrane. Some of the important components of molecular robots are self-propelled nano- and micro-scale motors, which are propelled by converting external energy to mechanical motion4,5,6,7,8,9,10,11. Compared with the above kinesin-microtubule system, a simple strategy for propelling a motor is the induction of an energy gradient between its two sides4,5,6,7,8,9,10,11. One of the major driving forces of the propulsion is the Marangoni effect, caused by a surface tension gradient. This gradient propels the motor from its low-surface-tension side to its high-surface-tension side4,12,13. Based on these design principles, various nano/microparticles with asymmetric catalytic sites have been developed to convert chemical energy to mechanical motion14,15,16,17,18,19,20,21,22. In the presence of substrates, a catalytic motor generates an asymmetric distribution of products, which causes diffusiophoresis14,15,16,17,18,19,20, thermophoresis21 and pH phoresis22. In addition to catalytic motors, light-driven motors have been recently developed because light is an attractive energy source for modulating the movement of motors by switching the on/off timing and orientation of light23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41. For instance, Ren et al. reported the construction of TiO2, BiOI and carbon WO3-based Janus micromotors, which were propelled by photocatalytic electrophoresis23,24 or diffusiophoresis25. A photothermal effect has been utilised to propel a gold-based motor by the light-induced formation of a thermal gradient across the motor, resulting in thermophoresis26,27,28. Sugawara et al. constructed oil droplets in a metal-free system. This was done by decomposing photolabile 2-nitrobenzyl oleate by UV light irradiation to generate a partial oleic acid distribution on the surface, resulting in the propulsion of oil droplets by the Marangoni effect29.
To design new light-driven motors, we focused on the self-propelled motility of several bacterial pathogens driven by nanofibre growth. Actin polymerisation on the surface of Listeria monocytogenes and Shigella flexneri forms a local network of actin filaments called an ‘actin comet tail’, which provides a powerful force to propel the bacteria through host cell cytoplasm42. The moving mechanisms of Listeria have been investigated by the activation of actin polymerisation on the surface of liposomes and polystyrene microspheres, which has been achieved by coating them with the Listeria transmembrane protein ActA43,44. Thus, light-induced nanofibre growth can be used as a new propulsion force for light-driven motors. Peptides are promising building blocks for this purpose because (1) light-responsive amino acids can be introduced in peptides and (2) nanofibre-forming peptide sequences can be designed. Amyloidogenic, β-sheet and β-hairpin-forming peptides have been conjugated with light-responsive moieties such as the azobenzene, nitrobenzyl and spiropyran groups for light-induced fibre formation45,46,47,48.
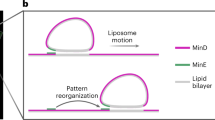
Herein, we constructed light-driven motors that were propelled by light-induced fibre formation on their surfaces (Fig. 1a). A peptide nanofibre-forming unit with a photocleavage unit was asymmetrically conjugated on the surface of giant liposomes by DNA hybridisation of the addressing unit. By light irradiation, the peptide bonds were cleaved and nanofibre-forming peptides were released around the liposome. The released peptides subsequently self-assembled to form nanofibres, which could induce propulsion of the liposome by a mechanism similar to actin polymerisation-induced motility. We previously reported a light-induced peptide nanofibre growth system using conjugate 1, consisting of β-sheet-forming FKFEFKFE peptide and addressing single-stranded DNA (dA20), which are linked by a photo-cleavable amino acid X (Fig. 1b)49. Ultraviolet (UV) irradiation to conjugate 1 releases free FKFEFKFE peptide, which self-assembles to form peptide nanofibre. In this work, we newly synthesised conjugate 2 to increase the cleavage rates of the peptide bond compared with conjugate 1 (Fig. 1c). Conjugates 1 and 2 were introduced onto the surfaces of the liposomes, and the light-induced translational movements of the liposomes were analysed.

Design of a light-driven motor using photo-cleavable peptide-DNA conjugates. (a) Light-induced propulsion of a phase-separated liposome driven by local peptide nanofibre growth. The photocleavage reaction and peptide nanofibre formation of (b) conjugate 1 and (c) conjugate 2.
Results
Kinetics of light-induced fibre formation of conjugates 1 and 2
Conjugate 2 was designed by replacing the photoresponsive amino acid X of conjugate 1 with amino acid Z for a faster photocleavage reaction. Upon light irradiation of conjugate 1, the unprotected phenolic intermediate was generated and then cleaved into dA20 and peptide moieties by intramolecular cyclisation (Fig. 1b). The intramolecular cyclisation was expected to be slow, especially when the sterically hindered residue was introduced to the position adjacent to X50. By contrast, Z could be cleaved by light without the intramolecular cyclisation that took place in the case of X (Fig. 1c). 9-Fluorenylmethyloxycarbonyl (Fmoc) derivatives of Z are commercially available and have been used for light-induced self-assembly45, conformational change51, binding to the epitopes of peptides52, and control of DNA binding53. Conjugates 1 and 2 were synthesised by our reported procedure (see Methods and Supplementary Scheme 1).
The photocleavage reactions of conjugates 1 and 2 were monitored using reverse phase high-performance liquid chromatography (RP-HPLC). The processing products were characterised by matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry (MALDI-TOF-MS) (refer to Supplementary Information). Upon UV irradiation (365 nm, 4 W cm−2) into conjugate 1, the phenolic intermediate was immediately generated. The reaction showed a first-order rate dependence, and the kinetic constant k1 and half-life t1/2 were determined as 0.42 sec−1 and 1.7 sec, respectively (Supplementary Fig. 3). By UV irradiation into conjugate 1 for 30 sec and subsequent incubation at 25 °C for 0–130 min, the intermediate was gradually cleaved. In this cleavage reaction, k2 and t1/2 were determined as 0.17 × 10−3 sec−1 and 4380 sec, respectively (Supplementary Fig. 5). Thus, the intramolecular cyclisation of the phenolic intermediate was extremely slow, as expected. By contrast, UV irradiation of conjugate 2 caused immediate cleavage with a first-order rate dependence. In the reaction, k3 and t1/2 were determined as 0.05 sec−1 and 14 sec, respectively (Supplementary Fig. 7). These results showed that the cleavage reaction of conjugate 2 was obviously faster than that of conjugate 1.
The increment of the turbidity (optical density at 400 nm) of conjugates 1 and 2 caused by to light-induced fibre formation was monitored (refer to Supplementary Information). Upon UV irradiation for 3 min, the turbidity of conjugate 2 was dramatically increased because of the fibre formation, whereas conjugate 1 showed only a slight change (Supplementary Fig. 9). This result indicated that conjugate 2 was cleaved by UV irradiation, resulting in the release of the peptide moiety to form the nanofibres immediately. However, the cleavage reaction of conjugate 1 was too slow to produce nanofibres from 3 min of UV irradiation. The light-induced fibre formation of conjugate 2 was observed by transmission electron microscopy (TEM) (Supplementary Fig. 10). These results indicated that the rates of fibre formation could be modulated by changing the photocleavage moiety.
Light-induced translational movement of giant liposomes
Because the asymmetric distribution of actin filaments is important for the propulsion of the actin-based motors43,44, phase-separated giant liposomes (PS-liposomes) were prepared for the asymmetric modification of conjugates 1 and 2 (Supplementary Fig. 11). Liposomes composed of a saturated phospholipid 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), an unsaturated lipid 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and cholesterol (Chol) are known to phase-separate into coexisting liquid phases by optimising the mixing ratio of the three components54,55. DPPC and Chol form a liquid-ordered (Lo) domain, and DOPC forms a liquid-disordered (Ld) domain in a single liposome. 1,2-Dioleoyl-sn-glycero-3-phosphoethanolamine-N-(cap biotinyl) (sodium salt) (DOPE-biotin) was used for the localisation of biotin on the Ld domains of PS-liposomes. Using a natural swelling method, we prepared typically sized 10 to 30-μm PS-liposomes consisting of DPPC/DOPC/DOPE-biotin/Chol with a molar ratio of 5:4:1:4.5. The 1- and 2-modified PS-liposomes (1-PS and 2-PS) were prepared by the selective conjugation of 1 and 2 on the Ld domain of the PS-liposomes, which was achieved by the sequential conjugation of 1 and 2 with dT20-biotin molecules (dA20-dT20 complementary hybridisation), streptavidin (streptavidin-biotin interaction) and the DOPE-biotin of PS-liposomes (streptavidin-biotin interaction) (Supplementary Fig. 11). To facilitate the fibre formation under light irradiation, the excess amounts of conjugates 1 and 2 (1.5 equivalent to dT20-biotin) were used because the amount of the released peptide moiety is important for fibre formation. For the comparison with PS-liposomes, we also prepared homogeneous liposomes (H-liposomes) consisting of DOPC/DOPE-biotin/Chol with a molar ratio of 9:1:4.5. The phase separation and homogeneity of the PS- and H-liposomes, respectively, were confirmed by the localisation of FITC-labelled streptavidin to the DOPE-biotin on the liposomes (Fig. 2). The localised fluorescence of FITC-streptavidin on PS-liposomes indicates the binding of FITC-streptavidin to biotin on Ld domain of PS-liposomes (Fig. 2a). In contrast, the homogeneous fluorescence of FITC-streptavidin on H-liposomes indicates the binding of FITC-streptavidin to homogeneously-distributed biotin on H-liposomes (Fig. 2b). By light irradiation of 1-PS, the formation of peptide nanofibres surrounding the liposomes was observed by TEM (Supplementary Fig. 12).

Phase separation and homogeneity of liposomes. CLSM images of (a) phase-separated (PS) and (b) homogeneous (H) liposomes stained with FITC-streptavidin (green) and Nile red (red).
The light-induced translational movements of 1-PS and 2-PS were monitored by optical microscopy. As a control, we prepared PS-liposomes, which were modified with a dA20 without a peptide moiety (dA20-PS). In the microscopic observation, we selected similar size of liposomes (13–16 μm) for comparison (Supplementary Table 1). In the light-irradiation experiments, the movement of liposomes was monitored under darkness for 10 min, followed by UV light irradiation for 10 min, then again under darkness for 40 min with the settings shown in Supplementary Fig. 13. Snapshots of 2-PS show the enhanced propulsion of 2-PS after light irradiation (Fig. 3, captured from Supplementary Video 1). The trajectories of 1-PS, 2-PS and dA20-PS are plotted in Fig. 4 and Supplementary Fig. 14. The movement of 2-PS exhibits a typical random walk caused by Brownian motion (Fig. 4). The moving distance of 2-PS was much longer under light irradiation (Fig. 4a) than under darkness (Fig. 4b), suggesting the enhanced diffusion of 2-PS by light-induced fibre formation. The distances of 1-PS, 2-PS and dA20-PS from their initial positions were calculated from the trajectories (Fig. 5). The light irradiation promptly induced the translational movement of 2-PS (Fig. 5a), whereas no specific movement was observed under darkness (Fig. 5b). The effect of light irradiation on 1-PS seemed to be less than that of 2-PS (Fig. 5c,d). The movement of dA20-PS was not changed by light irradiation (Fig. 5e), indicating the requirement of conjugates 1 and 2 for light-induced translational movement. The comparison of 1-PS, 2-PS and dA20-PS indicated that the local heating by the light irradiation was not the reason for the enhanced diffusion of 2-PS because 1-PS and dA20-PS showed no enhanced diffusion during the light irradiation (Fig. 5c,e). Comparison of the averages of the moving distances clearly showed that light irradiation immediately induced the translational movement of 2-PS, whereas the response of 1-PS to the light was slow (Fig. 5f). This difference reflected the fast fibre formation of conjugate 2 compared with conjugate 1 under light irradiation (Supplementary Fig. 9), indicating the importance of the nanofibre growth for propulsion.

Snapshots of 2-PS with UV light irradiation. White and blue circles represent the initial and final positions of the liposomes, respectively. The UV light irradiation lasted for 10–20 min of recording.

Tracking trajectories of 2-PS. The trajectories were recorded during 60 min (a) with and (b) without UV light. Irradiation lasted for 10–20 min of recording for (a). Each colour represents a different liposome (N = 20).

Time dependence of the distance of the PS-liposomes from the initial position. The distance was analysed from the trajectory tracking: (a,b) 2-PS; (c,d) 1-PS; (e) dA20-PS and (f) average of (a–e) under UV light irradiation (orange zones) and non-irradiation (white zones). In (a–e), each colour represents a different liposome (N = 20).
Discussion
Kinetics of photocleavage reaction of conjugate 1 and 2 were correlated with kinetics of fibre formation and the movement of 1- and 2-conjugated liposomes. Although the photolysis of 1 was fast (t1/2 = 1.7 sec), the intramolecular cyclisation of the intermediate to generate the free peptide was slow (t1/2 = 4380 sec) (Supplementary Figures 3 and 5). The overall slow cleavage of 1 induced slow fibre formation (Supplementary Figure 9), driving slight movement of 1-PS (Fig. 5c). In contrast, light irradiation to conjugate 2 readily generated the peptide moiety (t1/2 = 14 sec) (Supplementary Figure 7) to induce the fast fibre formation (Supplementary Figure 9). The fast fibre formation was efficient to induce prompt movement of 2-PS upon light irradiation (Fig. 5a). Thus, the chemical design of the photocleavage moiety is an efficient strategy to modulate the light-induced movement of liposomes.
To clarify whether the movement of the liposomes was Brownian motion, the mean square displacement (MSD) of each liposome was calculated by MSD(Δt) = [(r(t + Δt) − r(t)]2, where Δt is time interval and r(t) = (x(t), y(t)) is the vector position of the liposome at time t. The plots of MSD versus Δt are shown in Fig. 6. All of the liposomes can be linearly fitted, indicating that the enhanced diffusion of 2-PS by light-induced fibre formation was caused by non-directional Brownian motion, similar to the reported catalytic motors that generate an asymmetric distribution of products for diffusiophoresis15,17,19,20. The diffusion coefficient (D) was calculated from the fitted line, using D = MSD/i·Δt, where i is the dimensional index, which is equal to 4 in the case of a two-dimensional analysis. The diffusion coefficients and velocities are summarised in Table 1. The diffusion coefficient of 2-PS under darkness was 0.56 ± 0.06 μm2/min. With light irradiation, the value increased to 1.29 ± 0.18 μm2/min. In the case of 1-PS, the diffusion coefficient was slightly increased by light irradiation (0.78 ± 0.10 μm2/min) compared with that under darkness (0.55 ± 0.06 μm2/min). The value of dA20-PS with light (0.58 ± 0.07 μm2/min) was similar to that of 1-PS and 2-PS under darkness. These results indicated that light irradiation of 1-PS and 2-PS enhanced their diffusion, which was dependent on the rates of fibre formation. The velocity of 2-PS had a 5-fold increased under light irradiation (0.31 ± 0.04 μm/min) compared with that under darkness (0.06 ± 0.02 μm/min). Although it is difficult to compare directly, previous motility assays reported that the velocities of Listeria and ActA (Listeria transmembrane protein)-modified liposomes propelled by actin polymerisation were 1.9 ± 0.3 and 0.8 ± 0.2 μm/min, respectively43. The slow movement of 2-PS compared with the actin-based motors is thought to be caused by the lower density of FKFEFKFE nanofibre in our motors compared with that of the actin filaments used by Listeria, as the high density of actin filaments has corresponded to a large force generation56. The different density of the two fibres is due to the differences in size and flexibility of the fibres. In addition, the formed FKFEFKFE nanofibre may not be bound to the liposomes after the propulsion, resulting in a reduction of the propulsive force, as suggested in a review42. A possible driving force for the propulsion of 1-PS and 2-PS is the Marangoni effect caused by the surface tension gradient between the nanofibre-forming side and the other side of a motor. The surface tension gradient caused by nanofibre growth might be small, which could explain the slow movement of our motors.

Mean square displacement (MSD) plots of PS-liposomes. Fitting plots of MSD versus time interval (Δt) were analysed from the trajectory tracking of 20 liposomes during 10–60 min of measurement.
To evaluate the effect of asymmetrical modification of conjugate 2 on PS-liposomes, the movement of a 2-modified homogeneous liposome (2-H) was tracked. The propulsion of the 2-H was induced by light irradiation (Supplementary Figs 15 and 16). However, the moving distance of 2-H was shorter than that of 2-PS (Supplementary Fig. 17). A comparison of the MSD plots also showed that the diffusion enhancement of 2-H by light was not great compared to that of 2-PS (Supplementary Fig. 18). In fact, the calculated diffusion coefficient of 2-H (0.87 ± 0.11 μm2/min) under light irradiation was smaller than that of 2-PS (1.29 ± 0.18 μm2/min). These results suggest that asymmetrical fibre formation is important for the propulsion of the liposomes.
In conclusion, we developed novel light-driven motors propelled by peptide nanofibre growth on their surfaces, inspired by natural motors that use actin fibre formation for propulsion. The responsiveness of the motors to light was dependent on the rates of the photocleavage reaction and subsequent fibre formation, indicating that the movement of the motors could be tuned by modifying the photocleavage moiety. These results demonstrated the proof of concept for light-driven motors based on the chemical design of the light-induced peptide nanofibre formation. Elucidation of the detailed mechanism of the propulsion, visualization of the nanofibre formation during the movement of the motors, and modulation of our motors for directional motion are under investigation.
Method
Equipment and materials
High-performance liquid chromatography (HPLC) was performed using a Shimadzu LC-6AD liquid chromatograph with GL Science Inertsil WP300 C18 columns (4.6 × 250 mm for analysis and 20 × 250 mm for purification). Matrix-assisted laser desorption/ionisation time-of-flight (MALDI-TOF) mass spectra were taken using a Bruker Daltonics Autoflex TII with α-cyano-4-hydroxycinnamic acid as matrix for CZFKFEFKFE and 3-hydroxypicolinic acid as matrix for dA20-maleimide and conjugate 2. UV-vis spectra were obtained using a Jasco V-630. Transmission electron microscope (TEM) was measured using a Jeol JEM 1400 Plus with a grid (C-SMART Hydrophilic TEM grid, ALLIANCE Biosystems Inc.). Confocal laser scanning microscopy (CLSM) measurement was conducted using a FluoView FV10i (Olympus). Giant liposomes were imaged under an inverted microscope (Eclipse TS100, Nikon) equipped with a digital camera (MC120 HD, Leica). UV irradiation was performed using an LED lump, ULEDN 102-CT (365 nm, 4 W/cm2, NS Lighting Co., Ltd.). The reagents were purchased from Watanabe Chemical Ind., Ltd., Tokyo Chemical Industry Co., Dojindo Laboratories Co., Ltd., Avanti Polar Lipids Co. and Wako Pure Chemical Industries. Chemically modified DNAs, dA20-NH2 (20 mer of deoxyadenosine monophosphate having an amino group at the 5′ end, Supplementary Fig. 1a) and dT20-biotin (20 mer of thymidine monophosphate having a biotin group at the 5′ end, Supplementary Fig. 1b) were purchased from Gene Design Inc. All the chemicals were used without further purification.
Synthesis of conjugates 1 and 2
Conjugate 1 was synthesized according to our previous methods49. Conjugate 2 was synthesized by the same methods (Supplementary Scheme 1).
Synthesis of photoresponsive peptide moiety CZFKFEFKFE: CZFKFEFKFE (Z is D-3-amino-3-(2-nitrophenyl)-propionic acid) was synthesized by 9-fluorenylmethyloxycarbonyl (Fmoc) solid phase peptide synthesis. The amino group of each amino acid was protected with Fmoc group. The mercapto, amino, and carboxy groups of C, K, and E were protected with trityl, tert-butoxycarbonyl, and tert-butyl groups, respectively. Fmoc-D-β-Phe(2-NO2)-OH (Watanabe Chemical Ind., Ltd.) was used for Z. To the resin having free amino groups as reaction sites (Fmoc-Glu(OtBu)-Alko PEG Resin, 0.25 mmol/g, 224 mg) were added Fmoc amino acid (4 equiv. for C, Z, K, E and 8 equiv. for F), 1-[(1-(Cyano-2-ethoxy-2-oxoethylideneaminooxy)-dimethylamino-morpholinomethylene)] methanaminium hexafluorophosphate (COMU, 4 equiv. for C, Z, K, E and 8 equiv. for F), and diisopropylethylamine (DIPEA, 4 equiv. for C, Z, K, E and 8 equiv. for F) in N-methylpyrrolidone (NMP). Each condensation reaction was performed at room temperature for 2 h. Removal of Fmoc groups from the resin was performed using 40% and 20% piperidine N,N-dimethylformamide (DMF) solution. Both introduction of Fmoc amino acids and removal of Fmoc groups were checked by a 2,4,6-trinitrobenzene sulfonic acid (TNBS) test kit (Tokyo Chemical Industry Co., Ltd.) after washing the resin with NMP (2 mL × 5). Cleavage of the 10-mer peptide and deprotection at side chains were performed with a cleavage cocktail (trifluoroacetic acid (TFA)/thioanisole/water/ethanedithiol/triisopropylsilane = 81.5/5/5/1/1, v/v/v/v/v). The mixture was stirred at room temperature for 3 h. After filtration, the solution was evaporated, followed by trituration with tert-butylmethylether (25 mL × 3) to give the product in a 29% yield (crude) as white solid. Purification was performed by RP-HPLC with water/acetonitrile (both containing 0.1% TFA, 75/25 to 40/60, v/v for 75 min, linear gradient, 10 mL/min, detected at 220 nm) to give the purified product. MALDI-TOF-MS: m/z found: 1419 ([M + H]+), calcd. 1418.
Synthesis of dA20-maleimide
To a 1 mM aqueous solution of dA20-NH2 (40 nmol, 40 μL) were added N-(4-maleimidobutyryloxy)-sulfosuccinimide sodium salt (Sulfo-GMBS, 120 equiv., 1.8 mg, 4.8 μmol) and 0.1 M NaHCO3 aqueous solution (120 μL). The mixture was incubated at 25 °C for 4 h. The reaction mixture was dialyzed using a dialysis membrane (Spectra/por7, cutoff Mw: 1,000, Spectrum Laboratories, Inc.) with water for 20 h to remove the excess Sulfo-GMBS. MALDI-TOF-MS: m/z found: 6547 ([M + H]+), calcd. 6547.
Synthesis of conjugate 2
CZFKFEFKFE (0.43 mg, 300 nmol) in acetonitrile (500 μL) was added to 0.2 M sodium phosphate buffer pH 7.0 (520 μL). Then 100 mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP-HCl) aqueous solution (30 μL) was added to the solution. The dA20-maleimide solution (40 nmol, 450 μL) was added and the mixture was incubated at 40 °C for 48 h. Purification was performed by RP-HPLC with 0.1 M ammonium formate aqueous solution/acetonitrile (95/5 to 0/100, v/v for 95 min, linear gradient, 10 mL/min, detected at 260 nm) to give the purified product. MALDI-TOF-MS: m/z found: 7964 ([M + H]+), calcd. 7964 (Supplementary Fig. 2).
Preparation of giant liposomes
Phase-separated giant liposome (PS-liposome, Supplementary Fig. 11) was prepared by a natural swelling method using D-glucose57. DPPC/DOPC/DOPE-biotin/Chol with the molar ratios of 5:4:1:4.5 were mixed with D-glucose (1 equiv. to the lipids) in chloroform/methanol (2/1, v/v). The solution in a glass test tube (i.d. ~1 cm) was dried in vacuo for 1–2 days. The dried film was then hydrated at 50 °C with water (500 μL) for 1 h. The final concentration of lipids was 0.1 mM. Homogeneous liposome (H-liposome) was prepared by the same method using the lipids consisting of DOPC/DOPE-biotin/Chol with the molar ratios of 9:1:4.5. The liposomes were stored under a nitrogen atmosphere at 4 °C and used within a week.
Observation of movement of liposomes
To conjugates 1, 2 or dA20-NH2 (15 μM, 1 μL) were sequentially added dT20-biotin (10 μM, 1 μL), streptavidin (10 μM, 1 μL), D-glucose (400 μM, 1 μL), and the liposomes (1 μL, containing 10 μM DOPE-biotin). The setting of the observation system is shown in Supplementary Fig. 13. To close off the sample from its surroundings, a square, a double-sided piece of tape (0.05 mm-thick, ESCO, Japan) with the center removed was adhered onto a cover glass (24 mm × 60 mm, Matsunami, Japan) to create a well (ca. 12 mm × 12 mm). The liposome solution (2 μL) was added to the well, then a cover glass (18 mm × 18 mm, Matsunami, Japan) was adhered to the top of the tape. After standing for 2 h, the images were recorded as a video for 60 min. For light-irradiation experiment, the samples were kept under dark for 10 min, irradiated with UV light (365 nm, 4 W cm−2) for 10 min with 4 cm distance from the center of the well, then again kept under dark for 40 min. Because the distance between the light source and sample was little longer (4 cm) compared to in vitro uncaging experiments (3 cm), we irradiated the UV light for 10 min for complete uncaging. The trajectory of the liposomes was tracked by analysing the images by a software ImageJ (n ≥ 10).
References
Hagiya, M., Konagaya, A., Kobayashi, S., Saito, H. & Murata, S. Molecular robots with sensors and intelligence. Acc. Chem. Res. 47, 1681–1690 (2014).
Lund, K. et al. Molecular robots guided by prescriptive landscapes. Nature 465, 206–210 (2010).
Sato, Y., Hiratsuka, Y., Kawamata, I., Murata, S. & Nomura, S.-I. M. Micrometer-sized molecular robot changes its shape in response to signal molecules. Sci. Rob. 2, eaal3735 (2017).
Wang, J. Nanomachines: Fundamentals and Applications (Wiley-VCH, Weinheim, Germany, 2013).
Tu, Y. et al. Mimicking the cell: bio-inspired functions of supramolecular assemblies. Chem. Rev. 116, 2023–2078 (2016).
Katuri, J., Ma, X., Stanton, M. M. & Sánchez, S. Designing micro- and nanoswimmers for specific applications. Acc. Chem. Res. 50, 2–11 (2017).
Wu, Z., Lin, X., Si, T. & He, Q. Recent progress on bioinspired self-propelled micro/nanomotors via controlled molecular self-assembly. Small 12, 3080–3093 (2016).
Gao, W. & Wang, J. The environmental impact of micro/nanomachines: a review. ACS Nano 8, 3170–3180 (2014).
Moo, J. G. S. et al. Nano/microrobots meet electrochemistry. Adv. Funct. Mater. 27, 1604759 (2017).
Sánchez, S., Soler, L. & Katuri, J. Chemically powered micro- and nanomotors. Angew. Chem. Int. Ed. 54, 1414–1444 (2014).
Wang, H. & Pumera, M. Fabrication of micro/nanoscale motors. Chem. Rev. 115, 8704–8735 (2015).
Ikezoe, Y., Washino, G., Uemura, T., Kitagawa, S. & Matsui, H. Autonomous motors of a metal-organic framework powered by reorganization of self-assembled peptides at interfaces. Nat. Mater. 11, 1081–1085 (2012).
Scriven, L. E. & Sternling, C. V. The Marangoni effects. Nature 187, 186–188 (1960).
Howse, J. R. et al. Self-motile colloidal particles: from directed propulsion to random walk. Phys. Rev. Lett. 99, 048102 (2007).
Muddana, H. S., Sengupta, S., Mallouk, T. E., Sen, A. & Butler, P. J. Substrate catalysis enhances single-enzyme diffusion. J. Am. Chem. Soc. 132, 2110–2111 (2010).
Wilson, D. A., Nolte, R. J. M. & Van Hest, J. C. M. Autonomous movement of platinum-loaded stomatocytes. Nat. Chem. 4, 268–274 (2012).
Lee, T.-C. et al. Self-propelling nanomotors in the presence of strong Brownian forces. Nano Lett. 14, 2407–2412 (2014).
Yamamoto, D. et al. Micromotors working in water through artificial aerobic metabolism. Nanoscale 7, 13186–13190 (2015).
Ma, X., Hahn, K. & Sánchez, S. Catalytic mesoporous Janus nanomotors for active cargo delivery. J. Am. Chem. Soc. 137, 4976–4979 (2015).
Ma, X. et al. Enzyme-powered hollow mesoporous Janus nanomotors. Nano Lett. 15, 7043–7050 (2015).
Qin, W. et al. Catalysis‐driven self‐thermophoresis of Janus plasmonic nanomotors. Angew. Chem. Int. Ed. 56, 515–518 (2017).
Dey, K. K., Bhandari, S., Bandyopadhyay, D., Basu, S. & Chattopadhyay, A. The pH taxis of an intelligent catalytic microbot. Small 9, 1916–1920 (2013).
Dong, R., Zhang, Q., Gao, W., Pei, A. & Ren, B. Highly efficient light-driven TiO2–Au Janus micromotors. ACS Nano 10, 839–844 (2016).
Dong, R. et al. Visible-light-driven BiOI-based Janus micromotor in pure water. J. Am. Chem. Soc. 139, 1722–1725 (2017).
Zhang, Q. et al. Light-driven Au-WO3@C Janus micromotors for rapid photodegradation of dye pollutants. ACS Appl. Mater. Interfaces 9, 4674–4683 (2017).
Wu, Z. et al. Near-infrared light-triggered ‘On/Off’ motion of polymer multilayer rockets. ACS Nano 8, 6097–6105 (2014).
Wu, Z. et al. Superfast near-infrared light-driven polymer multilayer rockets. Small 12, 577–582 (2015).
Xuan, M. et al. Near infrared light-powered Janus mesoporous silica nanoparticle motors. J. Am. Chem. Soc. 138, 6492–6497 (2016).
Suzuki, K. & Sugawara, T. Phototaxis of oil droplets comprising a caged fatty acid tightly linked to internal convection. ChemPhysChem 17, 2300–2303 (2016).
Xu, L., Mou, F., Gong, H., Luo, M. & Guan, J. Light-driven micro/nanomotors: from fundamentals to applications. Chem. Soc. Rev. 46, 6905–6926 (2017).
Jang, B. et al. Multiwavelength light-responsive Au/B-TiO2 Janus micromotors. ACS Nano 11, 6146–6154 (2017).
Palacci, J., Sacanna, S., Vatchinsky, A., Chaikin, P. M. & Pine, D. J. Photoactivated colloidal dockers for cargo transportation. J. Am. Chem. Soc. 135, 15978–15981 (2013).
Mou, F. et al. Single-component TiO2 tubular microengines with motion controlled by light-induced bubbles. Small 11, 2564–2570 (2015).
Palagi, S. et al. Structured light enables biomimetic swimming and versatile locomotion of photoresponsive soft microrobots. Nat. Mater. 15, 647–653 (2016).
Kaneko, S., Asakura, K. & Banno, T. Phototactic behavior of self-propelled micrometer-sized oil droplets in a surfactant solution. Chem. Commun. 53, 2237–2240 (2017).
Uchida, E., Azumi, R. & Norikane, Y. Light-induced crawling of crystals on a glass surface. Nat. Commun. 6, 7310 (2015).
Zhou, D. et al. Visible-light controlled catalytic Cu2O–Au micromotors. Nanoscale 9, 75–78 (2017).
Li, Y. et al. Light-controlled bubble propulsion of amorphous TiO2/Au Janus micromotors. RSC Adv. 6, 10697–10703 (2016).
Dai, B. et al. Programmable artificial phototactic microswimmer. Nat. Nanotechnol. 11, 1087–1092 (2016).
Enachi, M. et al. Light-induced motion of microengines based on microarrays of TiO2 nanotubes. Small 12, 5497–5505 (2016).
Mou, F. et al. Light-controlled propulsion, aggregation and separation of water-fuelled TiO2/Pt Janus submicromotors and their “on-the-fly” photocatalytic activities. Nanoscale 8, 4976–4983 (2016).
Upadhyaya, A. & van Oudenaarden, A. Biomimetic systems for studying actin-based motility. Curr. Biol. 13, R734–R744 (2003).
Upadhyaya, A., Chabot, J. R., Andreeva, A., Samadani, A. & van Oudenaarden, A. Probing polymerization forces by using actin-propelled lipid vesicles. Proc. Natl. Acad. Sci. USA 100, 4521–4526 (2003).
Cameron, L. A., Footer, M. J., van Oudenaarden, A. & Theriot, J. A. Motility of ActA protein-coated microspheres driven by actin polymerization. Proc. Natl. Acad. Sci. USA 96, 4908–4913 (1999).
Bosques, C. J. & Imperiali, B. Photolytic control of peptide self-assembly. J. Am. Chem. Soc. 125, 7530–7531 (2003).
Haines, L. A. et al. Light-activated hydrogel formation via the triggered folding and self-assembly of a designed peptide. J. Am. Chem. Soc. 127, 17025–17029 (2005).
Matsuzawa, Y. et al. Assembly and photoinduced organization of mono- and oligopeptide molecules containing an azobenzene moiety. Adv. Funct. Mater. 17, 1507–1514 (2007).
Qiu, Z., Yu, H., Li, J., Wang, Y. & Zhang, Y. Spiropyran-linked dipeptide forms supramolecular hydrogel with dual responses to light and to ligand-receptor interaction. Chem. Commun. 3342–3344 (2009).
Furutani, M. et al. A photoinduced growth system of peptide nanofibres addressed by DNA hybridization. Chem. Commun. 51, 8020–8022 (2015).
Shigenaga, A., Yamamoto, J., Hirakawa, H., Yamaguchi, K. & Otaka, A. FRET-based assay of the processing reaction kinetics of stimulus-responsive peptides: influence of amino acid sequence on reaction kinetics. Tetrahedron 65, 2212–2216 (2009).
Umezawa, N., Noro, Y., Ukai, K., Kato, N. & Higuchi, T. Photocontrol of peptide function: backbone cyclization strategy with photocleavable amino acid. ChemBioChem 12, 1694–1698 (2011).
Toebes, M. et al. Design and use of conditional MHC class I ligands. Nat. Med. 12, 246–251 (2006).
Jiménez-Balsa, A., Pazos, E., Martínez-Albardonedo, B., Mascareñas, J. L. & Vázquez, M. E. Temporary electrostatic impairment of DNA recognition: light-driven DNA binding of peptide dimers. Angew. Chem. Int. Ed. 51, 8825–8829 (2012).
Veatch, S. L. & Keller, S. L. Separation of liquid phases in giant vesicles of ternary mixtures of phospholipids and cholesterol. Biophys. J. 85, 3074–3083 (2003).
Beales, P. A. & Vanderlick, T. K. Partitioning of membrane-anchored DNA between coexisting lipid phases. J. Phys. Chem. B 113, 13678–13686 (2009).
McGrath, J. L. et al. The force-velocity relationship for the actin-based motility of Listeria monocytogenes. Curr. Biol. 13, 329–332 (2003).
Tsumoto, K., Matsuo, H., Tomita, M. & Yoshimura, T. Efficient formation of giant liposomes through the gentle hydration of phosphatidylcholine films doped with sugar. Colloid Surf. B 68, 98–105 (2009).
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas ‘Molecular Robotics’ (No. 24104004) of The Ministry of Education, Culture, Sports, Science, and Technology, Japan.
Author information
Authors and Affiliations
Contributions
H.I., A.U. and K. Morishita prepared conjugates 1 and 2 and analysed the light-induced fibre formation. They also prepared 1- and 2-conjugated liposomes and analysed the tracking. T.K., A.S. and A.O. synthesized the Fmoc-X derivative. H.I. and K. Matsuura supervised the study and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Inaba, H., Uemura, A., Morishita, K. et al. Light-induced propulsion of a giant liposome driven by peptide nanofibre growth. Sci Rep 8, 6243 (2018). https://doi.org/10.1038/s41598-018-24675-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-24675-7
This article is cited by
-
Direct light-induced propulsion of vessels filled with a suspension of graphene particles and methanol
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
