
Abstract
The aim of this study was to extensively characterise natal dental pulp stem cells (nDPSC) and assess their efficiency to generate human induced pluripotent stem cells (hiPSC). A number of distinguishing features prompted us to choose nDPSC over normal adult DPSC, in that they differed in cell surface marker expression and initial doubling time. In addition, nDPSC expressed 17 out of 52 pluripotency genes we analysed, and the level of expression was comparable to human embryonic stem cells (hESC). Ours is the first group to report comprehensive characterization of nDPSC followed by directed reprogramming to a pluripotent stem cell state. nDPSC yielded hiPSC colonies upon transduction with Sendai virus expressing the pluripotency transcription factors POU5F1, SOX2, c-MYC and KLF4. nDPSC had higher reprogramming efficiency compared to human fibroblasts. nDPSC derived hiPSCs closely resembled hESC in terms of their morphology, expression of pluripotency markers and gene expression profiles. Furthermore, nDPSC derived hiPSCs differentiated into the three germ layers when cultured as embryoid bodies (EB) and by directed differentiation. Based on our findings, nDPSC present a unique marker expression profile compared with adult DPSC and possess higher reprogramming efficiency as compared with dermal fibroblasts thus proving to be more amenable for reprogramming.
Similar content being viewed by others

Introduction
Adult stem cells have an edge over somatic cells in terms of being an efficient starting cell source for reprogramming1. This is because they have relatively fast cell cycle kinetics, plasticity and endogenously express a subset of pluripotency factors, albeit at lower levels compared to embryonic stem cells. As opposed to somatic cells, adult stem cells are relatively undifferentiated with pronounced plasticity2,3. Various studies have pointed out that adult stem cells are more amenable and efficient in reprogramming than somatic or mature cells4,5,6,7,8,9. In sharp contrast to differentiated cells, adult stem cells have more reprogramming enhancers than barriers. Expression of stemness related genes, permissible chromatin state, a decreased level of barriers (e.g. TGF-β and MAP kinase pathways) and increased levels of genetic and epigenetic facilitators (e.g. KDM2B)10 are some of the intrinsic features that act as reprogramming enhancer. In summary, expression of pluripotency factors, lack of lineage-specific gene expression and permissible chromatin state makes adult stem cells more efficient to reprogramming over differentiated cells.
Examples of stem cells that were successfully reprogrammed are as follows. CD133+ stem cells were efficiently reprogrammed to pluripotent stem cells state using just two factors namely POU5F1 and SOX25. While, ectopic expression of POU5F1 was sufficient to reprogram neural stem cells as they endogenously expressed high levels of SOX2, c-MYC and KLF46,11. Examples of few other stem cells lines, which were successfully reprogrammed are adipose stem cells12, dental pulp stem cells (DPSC)13, hematopoietic stem cells and endometrial stem cells4,14. All these stem cells were more amenable to reprogramming than fibroblasts.
There are a number of distinctive features of DPSC that make them an attractive candidate for dedifferentiation or reprogramming. Primarily, they are multipotent stem cell in nature and express a myriad of markers including STRO-1, vimentin, CD29, CD44, CD73, CD90, CD166, as well as the stem cell markers nestin and nucleostemin15. It is advantageous to use stem cells as a starting cell point for reprogramming because the efficiency of reprogramming is higher as compared to dermal fibroblasts or other terminally differentiated tissue types16. In addition, the risk of exomic mutations due to aging is minimal in DPSC17 and the acquisition of DPSC is a relatively less invasive procedure.
Natal teeth are teeth that are present at birth, the incidence of natal and neonatal teeth ranges from 1:2,000 to 1:3,50018. Natal teeth are extracted at the earliest time-point possible because they may induce deformity or mutilation of the tongue leading to dehydration, inadequate nutrient uptake by the infant followed by growth retardation19. The extracted teeth are normally considered as biological waste. However, dental pulp present within the tooth is a rich source of stem cells and this repertoire of stem cells can potentially be used for regenerative medicine20.
The recently described Sendai virus platform for human induced pluripotent stem cell (hiPSC) generation is an ideal vector system for delivering reprogramming factors because of the following reasons: (i) the virus replicates in the form of negative-sense single-stranded RNA in the cytoplasm of infected cells and it does not integrate into the host genome21; (ii) it efficiently delivers exogenous genes in a wide spectrum of host cell species and tissues22; (iii) it shows low cytotoxicity upon infection23,24; and (iv) allows high-level expression of the delivered exogenous genes25.
In the current study, we sought to isolate, characterise and reprogramme natal dental pulp stem cells (nDPSC). nDPSC were initially characterized by detecting the expression of cell surface markers using flow cytometry. Then the nDPSC derived hiPSC clones were characterised to confirm complete reprogramming to pluripotency by performing immunostaining, gene expression analysis and embryoid body based spontaneous differentiation and directed differentiation to all three germ lineages.
Results
nDPSC express mesenchymal stem cell markers
Natal DPSCs (nDPSC) were isolated from a natal tooth extracted from an 11-day-old baby girl using our previously described procedure26. The derived nDPSC cells exhibited a fibroblastic morphology when observed under phase contrast microscopy (See Fig. 1a). Doubling time of nDPSC during initial passaging (i.e. from 2 to 6) ranged from 21 to 25 hr while that of adult DPSC cell lines ranged from 22 to 58 hr (See Fig. 2a). Compared to adult DPSC cell lines, nDPSC had a faster doubling time from passage 2 to 6 (See Fig. 2a). Comparative gene expression analysis between human embryonic stem cells (hESC) and nDPSC revealed that while the expression pattern of 17 genes associated with pluripotency were similar (See Fig. 1c), however the core pluripotency factors such as POU5F1, SOX2 and NANOG were not highly upregulated in nDPSC (See Fig. 1c). Expression of above mentioned 20 pluripotency genes of nDPSC were also compared to human dermal fibroblasts (HF) and WI38 human embryonic lung fibroblasts (See Fig. 1b). IL6ST, KLF4, COMMD3, NR5A2, RUNX2 and TBX3 genes were highly expressed in nDPSC as compared to other genes. 2−∆∆CT formula was used to calculate the fold change and hESC was used as calibrator.

Morphology and gene expression profile of nDPSC. (a) Morphology of nDPSC under phase contrast microscope. (b) Comparison of expression of 20 pluripotency genes between nDPSC and two cell lines of human fibroblasts. Values represent fold change. 2−∆∆CT formula was used to calculate the fold change and hESC was used as calibrator sample. (c) RT-qPCR expression profiling of pluripotency genes in hESC and nDPSC. The heat map was generated by presenting −∆Ct (CT gene − CT ACTB) values of each gene. Red colour and lower value indicates higher expression. Scale bar = 200 µm.

Growth pattern and flow cytometry data. (a) Comparison of doubling time between nDPSC and adult DPSC. Doubling time of nDPSC is compared with three adult DPSC cell lines during initial passages. Data are presented as the average +/− standard deviation; n = 3. (b) Flow cytometry histograms representing expression of markers characteristic to nDPSC; these markers are not expressed or are expressed at low levels in adult DPSC. nDPSC exhibited high expression of CD34, CD45, CD271, CD71, HLA-DR, CD146 and CXCR4 markers.
Flow cytometry (FC) results confirmed expression of cell surface markers indicative of mesenchymal stem cells (MSC) such as CD44, CD73, CD271, CD90, CD105, CD166, CD45 and CD10. Apart from MSC markers, nDPSC also expressed markers related to hematopoietic stem cells (HSC) such as CD34, CXCR4, CD71, CD45 and CD10. Other markers expressed were CD222 and HLA-DR (See Table 1 and Fig. 2b). This indicates that nDPSC are multipotent and we predicted highly amenable to reprogramming towards pluripotency27.
nDPSC expressed various markers characteristic of MSC and HSC which might indicate that these cells are relatively immature progenitor cell population and that the expression profile of nDPSC is unique to this cell type, as the majority of markers excluding the MSC specific markers, are not normally expressed by adult DPSC (See Table 1)27,28,29,30,31.
Generation of hiPSC from nDPSC
hiPSC colonies began to emerge on 13th day post transduction. On 7th day post transduction, 75000 cells from each cell line were plated on recombinant human vitronectin (rhVTN) coated plates to determine reprogramming efficiency using alkaline phosphatase (AP) staining. nDPSC cells were stained for AP on 18th day post transduction (See Fig. 3a), human fibroblasts (HF) on 25th day (See Fig. 3b) and WI38 on 27th day (See Fig. 3c). In nDPSC, 966 colonies stained positive, 550 in HF and 14 in WI38. Hence, reprogramming efficiency of nDPSC was the highest with 1.3% followed by HF with 0.73% and finally WI38 with 0.018%. In conclusion, nDPSC were more efficient and reprogrammed earlier than the fibroblasts.

Alkaline phosphatase (AP) staining to calculate reprogramming efficiency. Reprogramming efficiency was calculated by counting number of AP positive colonies after emergence of iPSC colonies following transduction of Sendai reprogramming vectors. Each 6 well plate was seeded with 75000 cells on 7th day post transduction. nDPSC (a) showed 966 AP positive colonies, while dermal fibroblasts (b) showed 550 colonies and WI38 lung fibroblasts (c) showed 14 colonies.
hiPSC colonies were manually picked once they attained an appropriate size, which was around day 18. hiPSC clones were expanded on recombinant human rhVTN coated plates and maintained in Essential 8 medium (E8). In total 35 individual clones were isolated. hiPSC clones formed tightly packed, flat colonies (See Fig. 4a). hiPSC clones showed significant up-regulation of the critical pluripotent cell markers such as alkaline phosphatase and stage-specific embryonic antigen (SSEA-4) and core pluripotency factors POU5F1, SOX2 and NANOG (See Fig. 4).

(a) nDPSC derived hiPSC. Image of nDPSC derived hiPSC with typical hES like morphology. (b) Colorimetric detection of alkaline phosphatase. (c–f) Immunocytochemistry against (c) SSEA-4, (d) POU5F1, (e) SOX2, and (f) NANOG. Nuclei were counterstained with DAPI. Images are shown as overlap of the two channels (c–f). Scale bar = 200 µm.
Comparative gene expression analysis between nDPSC derived hiPSC, fibroblast derived hiPSC and hESC
For gene expression analysis, a critical set of 83 genes was assessed. These genes were broadly classified into three groups as follows: pluripotency markers comprising of 52 genes; early differentiation markers with 18 genes; and somatic cell markers with 13 genes (See Table S1). For constructing heat map −∆CT (CT gene − CT ACTB)32 values of six samples i.e. hESC (CCTL 4), DP/iP/C3, DP/iP/C28, DP/iP/C4, HF/iP/C8 and WI38/iP/C5 were used. DP/iP/C3, DP/iP/C28 and DP/iP/C4 are hiPSC clones derived from nDPSC, HF/iP/C8 from human dermal fibroblasts and WI38/iP/C5 from human embryonic lung fibroblasts.
The heat map (See Fig. 5) was sub-divided into three subgroups based on gene expression: high expression, medium expression and low expression. The top showing 32 genes were highly expressed i.e. up to OTX2; the next 26 genes in the map had medial expression i.e. from GRB7 up to RUNX2 and last 21 genes had low expression. In the high expression subgroup, except for five genes (CDH1, NES, FN1, TUBB3, and COL1A1) all belonged to the pluripotency marker group. Out of these five genes, CDH1 and NES are from the somatic cell markers group while FN1, TUBB3, and COL1A1 are from the early differentiation markers group. Upregulation of CDH1 in iPSC clones is very critical because, CDH1 plays crucial role in development33 and in mesenchymal to epithelial transition (MET) during reprogramming34. In medium expression subgroup, all genes are from the pluripotency marker group with the exception of FABP7, which is from the somatic cell markers group. The low expression subgroup is mainly comprised of early differentiation markers and markers of somatic cells, with the following exceptions, TBX3, which is from the pluripotency marker group.

RT-qPCR expression profiling of pluripotency genes in hESC, nDPSC hiPSC clones (DP/iP/C3, DP/iP/C28 and DP/iP/C4), HF hiPSC clone (HF/iP/C8) and WI38 hiPSC clone (WI38/iP/C5). The heat map was generated by presenting −∆Ct (CT gene - CT ACTB) values of each gene. Red colour and lower value indicates higher expression. Parameters for generating the heat map are as follows: No scaling was applied to rows and rows were clustered using Euclidean distance and complete linkage. Data are expressed as means of technical duplicates.
In summary, all the pluripotency marker genes are highly expressed in nDPSC derived hiPSC clones, HF hiPSC clones, WI38 hiPSC clones and hESC, with the abovementioned exceptions, while the early differentiation and somatic cell marker genes have low expression levels as expected for bona fide hPSCs.
From a glance at the heat map, we can say that the overall gene expression patterns between nDPSC derived hiPSC clones (DP/iP/C3, DP/iP/C28 and DP/iP/C4), HF hiPSC clone (HF/iP/C8) embryonic lung fibroblasts hiPSC clone (WI38/iP/C5) and hESC are very similar with exception of LEFTY1 and FOXD3 genes where the pattern is dissimilar. The pattern of expression is exactly opposite for LEFTY1 and FOXD3. LEFTY1 is highly expressed in hESC while in all hiPSC clones it is expressed at low to medium level. Expression level of FOXD3 is lower in hESC as compared to all hiPSC clones, where it is expressed at medium level.
The overall expression patterns between nDPSC hiPSC clones, hESC and HF hiPSC clones is very similar (See Fig. 5). Linear regression plot shows that there is strong correlation between hESC and one of the clones of nDPSC iPSC (See Fig. 6a). While there is moderate correlation between nDPSC and hESC; from these results we can speculate that, nDPSC have pluripotent stem cell like characteristics but have not attained pluripotency to the level of hESC (See Fig. 6b)27. nDPSC derived hiPSC clones are highly similar to hESC with the correlation coefficient greater than 0.88. Correlation of nDPSC derived iPSC clones is maximal within themselves and with HF hiPSC clones followed by hESC (See Fig. 6c).

Gene expression patterns were compared between DP/iP/C4 vs hESC and DP/Parental vs hESC using linear regression plot (a & b). −∆Ct (CT gene − CT ACTB) values were used for constructing the plot. The line indicates the trend of the graph. Data are expressed as means of technical duplicates. Statistical correlation analysis shows that there is statistically significant correlation in gene expression pattern between DP/iP/C4 and hESC (P < 0.0001) with coefficient of correlation 0.8854. This correlation is regarded as a strong correlation. There is also a statistically significant co-relation between hESC and DP Parental (P = 0.0001) with coefficient of correlation 0.4571 and this is categorized as moderate correlation. (c) Correlation analysis between hESC and hiPSC clones. Correlation coefficient values are represented in form of heat map in pairwise matrix. Light red colour indicates lowest value while light green indicates highest value.
Differentiation of nDPSC derived hiPSC by embryoid body formation and directed differentiation
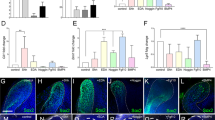
The differentiation potential of nDPSC derived hiPSCs was verified by generating embryoid bodies (EB) in order to induce spontaneous differentiation. EB were cultured for 21 days after which they were harvested and processed via paraffin embedding, followed by sectioning for analysis. Haematoxylin and eosin staining of EB sections revealed various structures characteristic of the three germ layers (See Fig. 7). Blood islands (mesoderm) (See Fig. 7b), neural rosettes (ectoderm) (See Fig. 7c), gut like epithelium (endoderm) (See Fig. 8d), covering epithelium (ectoderm) (See Fig. 7e) and optic plate (ectoderm) (See Fig. 7f) like structures were a few representative structures visible after staining the sections.

Embryoid body-mediated differentiation of nDPSC derived hiPSC. (a) EB derived from nDPSC derived hiPSC at day 5. (b-f) Haematoxylin and eosin stained embryoid bodies revealing various structures formed within them including (b) blood islands, (c) neural rosettes, (d) gut-like epithelium, (e) external covering epithelium, and (f) optic cup-like structure; arrows show the location of the optic cup primordium. Scale bar = 200 µm.

Relative telomere analysis was performed using qPCR. The graph represents log2 fold change of relative telomere length in parental cell DP/Parental and nDPSC-derived hiPSC (DP/iP/C4, DP/iP/C28 and DP/iP/C3). Data is presented as the average +/− standard deviation; n = 3.
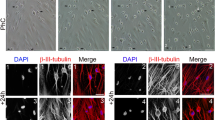
Immunohistochemistry of sectioned EB showed strong positivity for β-III tubulin (a marker of ectoderm), MAP2 (ectoderm), nestin (ectoderm), vimentin (mesoderm marker) and pan cytokeratin (endoderm marker) (See Supplementary Fig. S1). These results show that nDPSC-derived hiPSC have the potential to differentiate into the three germ layers, in vitro, during spontaneous differentiation in EB.
In addition, iPSC clones obtained from nDPSC, HF and WI38 cell lines were individually differentiated using directed differentiation approaches into neuroepithelium (ectoderm), cardiomyocytes (mesoderm) and hepatic endoderm (endoderm). Presence of neuroepithelium was confirmed by observing characteristic morphology of the differentiated cells and by RT-qPCR analysis (See Fig. S3). Beating cell clusters confirmed existence of cardiomyocytes (See supplementary videos). Cobblestone morphology together with immunostaining and qPCR results confirmed strong expression of hepatic markers, thus assuring presence of hepatic endoderm (See Fig. S2).
Morphological and immunohistochemistry results highlight that nDPSC derived hiPSC had attained pluripotency as they were capable of differentiation to all three lineages.
Telomere length and karyotype analysis
Telomere length analysis was carried out by qPCR according to the method described by Cawthon35. When compared to the parental nDPSC line, the relative telomere length increased by three-fold in DP/iP/C4 and DP/iP/C28 clones respectively (See Fig. 8) and by four fold in the DP/iP/C3 clone. DP/iP/C4 and DP/iP/C28 were established clones (passage 12) while DP/iP/C3 clone was at a relatively low passage (passage 7); this might have contributed to the observed differences in telomere lengths36,37,38.
Karyotype analysis of nDPSC derived hiPSC at passage 12 demonstrated that they had a normal karyotype of 46, XX without aneuploidy or polyploidy (See Supplementary Fig. S4).
Reprogramming of nDPSC led to a threefold increase in telomere lengths as compared to the starting parental cell line with a normal karyotype.
Discussion
Dental pulp harbours a stem cell population commonly referred to as DPSC, which possess properties similar to generic MSC39. Both adult DPSC and nDPSC had a similar expression profile for CD10, CD13, CD29, CD44, CD73, CD105, CD90 and CD166 but not for CD146, CXCR4, CD271, CD71, CD34, CD45 and HLA-DR. Adult DPSC expressed CD146moderate, CXCR4weak, CD271weak, CD71weak, CD34−, CD45− and HLA-DR− while nDPSC showed high positivity for all the markers27,28,29,30,31. It has been previously reported that nDPSC have an absence of CD34, CD45, CD71 and HLA-DR markers40,41. Surprisingly, our nDPSC were highly positive for CD34, CD45, CD71 and HLA-DR markers. Our finding demonstrates that the isolated population of nDPSC expressed HSC markers and HLA class II molecules in addition to MSC markers, which is a unique expression profile as compared to both adult DPSC and counter to the report of Akpinar and colleagues40.
Gene expression analysis revealed that nDPSC expressed 17 out of 52 pluripotency genes at levels equivalent to hESC. Overall gene expression analysis showed a moderate correlation between nDPSC and hESC. Previous studies have shown that adult DPSC express a number of pluripotency markers including POU5F1, NANOG, c-MYC, SOX2, stage specific embryonic antigens (SSEA-3, SSEA-4), and tumour recognition antigens (TRA-1–60 and TRA-1–81)40,42. However, the expression levels are significantly lower compared with the values observed in hESC43. From these studies and our data, we could speculate that promoter regions of pluripotency genes in nDPSC may have a low methylation status and favourable transcriptional as well as epigenetic state for reprogramming.
NR5A2 is medially expressed in nDPSC and the level of expression is higher as compared to hESC. Jian et al., had previously shown that, NR5A2 enhances kinetics of OSKM mediated reprogramming and NR5A2 functions synergistically with SOX2 and KLF4 to replace exogenous POU5F1 to mediate the successful reprogramming of murine somatic cells to pluripotent cells44. In addition, increase in NR5A2 expression directly causes an increase in NANOG expression44. An important point to be noted here is that, KLF4 is highly expressed in nDPSC and this same factor is used for reprogramming. For future experiments, it would be interesting to reprogramme nDPSC by eliminating KLF4 from the cocktail of transcription factor used for reprogramming. Another gene that grabs attention is IL6ST, which is an interleukin 6 signal transducer. This receptor activates Ras-ERK, JAK1-STAT3, and PI3K pathways45. Activation of STAT3 has been shown to enhance proliferation and stemness in glioma-associated-human MSCs46. Moreover, Hingwei et al., showed that STAT3 activity can reprogramme conventional (primed) hPSC to naive-like hPSC47. In case of nDPSC, they show high positivity for IL6 (IFN-beta) (flow cytometric data) as well as high expression of its corresponding receptor IL6ST. In summary, we can say that nDPSC are immaturely differentiated and could be classified as naïve multipotent stem cells.
Large and numerous reprogrammed colonies appeared on the 18th day post transduction, which indicates that nDPSC underwent reprogramming both rapidly and efficiently48. Reprogramming efficiency of nDPSC (1.3%) reported in this article is significantly higher than the efficiencies 0.1%, 0.09% and 0.24%, which are reported in previously published articles on DPSC13,49,50.
Based on the time of upregulation during reprogramming, pluripotency gene markers are classified into early upregulated group and late upregulated group51. LEFTY1 is expressed during the early phase of reprogramming while FOXD3 is upregulated at later stage51. FOXD3 and LEFTY1 have been reported to be crucial in maintaining pluripotency51. During reprogramming, the early upregulated group is activated between 10 to 20 days and the expression of these genes lasts beyond day 3051. However, late upregulated group is expressed only in a few of the colonies at day 3051. Colonies expressing late upregulated group can be used for isolating colonies because such colonies have high probability of becoming fully reprogrammed hiPSC51. This discussion explains the results observed in nDPSC derived hiPSC and hiPSC clones derived from HF and WI38 cell lines. Further comparative studies need to be performed in order to reveal the overall reprogramming competency of DPSC isolated from donors of different age groups and gender. The results presented here demonstrate that nDPSC are an efficient starting cell type for reprogramming.
To our knowledge, this is the first report for the complete characterization and reprogramming of nDPSC. In addition, nDPSC present a unique and beneficial gene and surface marker expression as well as superior proliferation capacity when compared to their adult counterparts. These characteristics make them ideal starting material for the generation of hiPSC.
Materials and Methods
Cell isolation, culture and reprogramming
A natal tooth was extracted from an 11 day old baby girl. Isolation and expansion of nDPSC was performed as previously described26,28. During the initial phase, nDPSC were cultured in media supplemented with human platelet rich plasma26.
For studying doubling time (DT), natal DPSC and adult DPSC were seeded at 5000 cells/cm2. After reaching 70% confluence, cells were harvested and were counted using Vi-CELL Cell Counter. The experiment was performed in triplicates for each time point. Doubling time was calculated using following formula:
For reprogramming, we employed the CytoTune-iPS 2.0 Sendai Reprogramming Kit (Life Technologies). nDPSC were maintained as previously described by Karbanova and colleagues28 prior to reprogramming as well as three days post transduction. The medium in which nDPSC were cultured, will henceforth be referred as DPSC medium. After this period, cells were cultured in DPSC medium but without growth factors for next five days. On seventh day, cells were detached using TrypLE select cell dissociation reagent (Life Technologies). Cells were counted and were plated at 5 × 105 and 1 × 105 densities on rh-VTN (Life Technologies) coated 100 mm plates. Following trypsinization, cells were cultured in DPSC medium without growth factors for 24 hrs. From eighth day onwards, cells were cultured in E8 (Life Technologies) medium and the spent medium was replaced every day. hiPSC clones were manually picked and subsequently expanded on rh-VTN coated plates with Essential 8 medium. For passaging, hiPSC clones were washed twice with DPBS (Life Technologies; Cat no. 14190169) without calcium and magnesium and then incubated with 0.5 mM EDTA at 37 °C for approximately 3 min52. The cells were detached from the well by gentle pipetting, ensuring that the colonies remained in chunks. The split ratio was routinely 1:3.
In addition to nDPSC, we used adult human fibroblasts and embryonic lung fibroblasts WI-38 (ATCC) for reprogramming. WI-38 fibroblasts were cultured in MEM (Life Technologies) media supplemented with 10% FBS (Sigma) while adult human fibroblasts were cultured in DMEM (Life Technologies) media supplemented with 10% FBS (Sigma). Adult human fibroblasts were isolated from skin biopsy of female patient undergoing cosmetic surgery, using a previously described protocol53.
Flow cytometry
nDPSC were harvested by trypsin/EDTA treatment for 5 min at 37 °C. After inactivation of trypsin and centrifugation (5 min at 300 g), 100 µl of the cell suspension (105 cells) was incubated for 30 min at room temperature in the dark with the following fluorochrome-conjugated antibodies: anti-CD10 (eBiosciences; Clone: CB-CALLA), anti-CD13 (eBiosciences; Clone: WM-15), anti-CD29 (Chemicon, Clone: TDM29), anti-CD34 (Biolegend; Clone: 4H11), anti-CD44 (Caltag; Clone: MEM-85), anti-CD45 (Caltag; Clone: HI30), anti-CD71 (Biolegend; Clone: MEM-75), anti-CD73 (BD Pharming; Clone: AD2), anti-CD90 (Chemicon; Clone: F15-42-1), anti-CD105 (Caltag; Clone: SN6), anti-CD146 (Millipore; Clone: P1H12), anti-CD166 (Beckman; Clone: 3A6), anti-CD222 (eBiosciences; Clone: MEM-238), anti-CD271 (Biolegend; Clone: ME20.4), anti-CXCR4 (Caltag; Clone: 12G5), anti-INF-beta (INFsource; Clone: MMHB-3) and anti-HLA-DR (Caltag; Clone: Tu36). After washing with PBS, at least 10,000 events were acquired using a Beckman Coulter FC500 flow cytometer. Instrument settings and gating strategies were established using isotype controls, and data were analysed using Kaluza software (Beckman Coulter). All experiments were carried out in duplicates.
Alkaline phosphatase staining and immunocytochemistry
Alkaline phosphatase staining was performed using a colorimetric method. hiPSC were incubated with NBT and BCIP substrates (nitro-blue tetrazolium and 5-bromo-4-chloro-3′-indolyphosphate respectively), which yielded an intense, insoluble black-purple precipitate on reaction with alkaline phosphatase. For immunofluorescence staining, hiPSC were grown on rh-VTN coated 12-well plates. After reaching the desired confluency, the medium was removed by aspiration and the cells were washed with DPBS-T (DPBS (Life Technologies; cat. no. 14190169) containing 0.1% Tween 20), followed by fixation with 4% paraformaldehyde for 10 min at room temperature. After washing with DPBS-T, the cells were permeabilized using 0.5% Triton X-100 for 10 min at room temperature. The cells were washed again with DPBS-T, after which the cells were then blocked with DPBS supplemented with 5% normal goat (Jackson ImmunoResearch) or donkey serum (Jackson ImmunoResearch) and incubated at room temperature for 30 min. Without washing, cells were incubated with primary antibodies overnight at 4 °C. The following primary antibodies were used: anti-SSEA4 (1:20, MC-813-70 (SSEA-4); Developmental Studies Hybridoma Bank), anti-POU5F1 (OCT-4) (1:100, sc-8629; Santa Cruz), anti-SOX2 (1:100, ab92494; Abcam) and anti-NANOG (1:100, PA1-097X; Life Technologies). The primary antibody was removed and the cells were washed with DPBS-T, followed by incubation with secondary antibodies for 45 min at room temperature, in the dark. The following secondary antibodies were used: cyanine 3 (Cy3) (Jackson ImmunoResearch) conjugated and Alexa488 (Jackson ImmunoResearch) conjugated IgG. The nuclei were counterstained with DAPI (Sigma) for 5 min.
Fluorescent images were captured using a Nikon Eclipse Ti-E inverted fluorescence microscope equipped with Nikon DS-Qi2 camera. Images were analysed and processed using NIS-Elements for advanced research (NIS-Elements AR) software.
RNA isolation and reverse transcription for SYBR RT-qPCR
Total RNA was purified using Direct-zol RNA Kit (Zymo Research). Isolated total RNA was quantified and reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (Life Technologies, Cat no.4368814). The primers were arrayed in 96-well plate and were subsequently lyophilized. cDNA was prepared from 2 µg of RNA in a 20 µl reaction. For a single well, RT-qPCR was performed using 10 ng of cDNA, along with 200 nM of each primer and 7.5 µl of SYBR Select Master Mix (Life Technologies, Cat no. 4472908) in a total reaction volume of 15 µl. Cycling condition for the analysis were: 50 °C for 2 min (Hold); 95 °C for 2 min (Hold); 95 °C for 15 sec; 60 °C for 40 sec, for 40 cycles. All results were normalized against ACTB as the endogenous control gene. qRT-PCR was performed using QuantStudio 6 Flex Real-Time PCR System (Applied Biosystems).
For generating the heat maps, ClustVis online software was used54. The statistical correlation analysis function was performed using NCSS software. Pairwise matrix of correlation values was generated using heatmapper software55. All experiments were carried out in duplicates, and the results are presented as mean.
Sequences of the primers are proprietary. Array plate designing was done in accordance with the objectives of grant project TACR GAMA TG1010108. The primers were synthesized by Generi Biotech bearing the reference number UKLFHK-160518.
In vitro differentiation (EB formation) and immunoperoxidase staining
For in vitro differentiation, hiPSC were cultured as floating EBs using a standard protocol56. These EBs were cultured for 21 days, and after this period they were fixed using 10% formalin (Lachner; Czech Republic) and embedded in paraffin. Serial sections (5–6 µm) were cut, mounted on glass slides pretreated with chrome alum-gelatin, and dried at room temperature overnight. Paraffin-embedded sections were deparaffinized by xylol (Lachner; Czech Republic) treatment, hydrated with decreasing concentrations of ethanol (Penta; Czech Republic) (96, 80 and 70%), and then rinsed twice with distilled water.
Haematoxylin and eosin staining was performed to identify various structures formed within embryoid bodies, dehydrated and mounted in DPX (Fluka).
For antigen retrieval, deparaffinized sections were exposed to microwaves (700 W) in sodium citrate solution (Lachner; Czech Republic) (pH = 6.0) twice, for 5 min each. After extensive washing with distilled water, endogenous peroxidase activity was quenched by incubating samples in 3% H2O2 (Penta; Czech Republic) (3 × 10 min). Sections were then incubated in blocking buffer (5% normal goat serum (Jackson ImmunoResearch) in PBS (Life Technologies; Cat no. 20012019)) for 20 min and exposed to primary antibodies (anti-β-III Tubulin (Exbio; Clone no: TU20), anti-MAP2 (Sigma; Clone no: HM2), anti-nestin (Life technologies; Clone no: SP103), anti-vimentin (Dako; Clone V9) and anti-pan cytokeratin (Dako; Clone AE1/AE3). Primary antibodies were diluted in Antibody Diluent with Background Reducing Components (Dako) and incubated at 4 °C overnight. After washing with PBS, sections were incubated with the following reagents: either an anti-mouse or an anti-rabbit EnVision peroxidase kit according to the manufacturer’s protocol. Colour reactions were performed using the chromogen DAB (3,3′-diaminobenzidine tetrahydrochloride, 2 µg/ml; Fluka, Darmstadt, Germany). After washing with distilled water, DAB precipitate was intensified with 3% CuSO4 (Penta; Czech Republic) solution for 5 min. Sections were then counterstained with Mayer’s haematoxylin (Merck) dehydrated and mounted in DPX (Fluka).
Images were captured using Olympus BX51 fluorescence microscope equipped with Olympus DP71 camera. Images were analysed and processed using Quick Photo 3.0 software.
Directed differentiation of hiPSC to 3 lineages
Endodermal Lineage
hiPSC were differentiated to the endodermal lineage as described previously57. Briefly, the nDPSC-hiPSC, HF Eye-hiPSC and W138-hiPSC were seeded as single cells using accutase (Life Technologies) at a density of 47,500 cells/cm2 into Geltrex (Life Technologies) coated tissue culture plates in E8 Medium (homemade) supplemented with Rock Inhibitor Y-27632 (BOC Sciences) as described previously58,59. After being allowed to adhere overnight, the differentiation was initiated in order to screen for the optimal differentiation conditions for definitive endoderm (DE) formation59,60. Briefly, on day 0, the E8 medium was removed and the cells were washed once with DPBS without Calcium and Magnesium (DPBS−/−). After the wash, the cells were treated with 4 different medium combinations as follows: RPMI1640 supplemented with B27 with Insulin (Life Technologies–RB+) and additionally supplemented with 3 μM CHIR99021 (BOC Sciences); RB+ supplemented with 4 μM CHIR99021; RPMI1640 supplemented with B27 without Insulin (Life Technologies—RB−) supplemented with 3 μM CHIR99021; and RB− supplemented with 4 μM CHIR99021. On day 1, after 24 hours culture in RB with CHIR, the spent medium was changed to RB medium without any additional small molecules, and the Insulin regime was maintained the same as for day 0. On day 2, the cells were assessed morphologically for the presence of DE cells via microscopy analysis. The optimal conditions were then used for subsequent experiments to further differentiate the cells to hepatic endoderm (HE). For HE differentiation, the hiPSC lines were seeded as described above and the differentiation was induced using the optimal conditions determined by the first experiment (in all cases the optimal condition was RB− supplemented with 4 μM CHIR99021). After DE differentiation, the cells were treated with hepatic endoderm specification medium (SRDMSO) that was prepared and formulated as described previously57,58,59,60,61,62. At the HE stage, (day 7 of differentiation) the cells were collected for gene expression analysis or fixed and stained for the presence of key HE markers alpha-fetoprotein (AFP) and hepatocyte nuclear factor 4 alpha (HNF4A). Details of the primary used for immunostaining are as follows. Anti-AFP (1:500; Sigma; Cat no. A8452) and anti-HNF4A (1:100; Santa Cruz; Cat no. sc8987). Details of the secondary antibodies are as follows. Alexafluor 488 (1:400; Life Technologies; Cat no. A21206) and Alexafluor 594 (1:400; Life Technologies; Cat no. A11005).
Mesodermal Lineage
hiPSC were differentiated to cardiomyocytes (representing the mesodermal lineage) as described previously63. Briefly, cells were seeded out as single cells as described above but at different cell densities (25,000/cm2, 35,000/cm2, and 45,000/cm2). After the cells had adhered overnight, the cells were washed with DPBS−/− and fresh E8 medium was added to the wells. The cells were cultured for an additional 2 days with medium exchanged every 24 hours. At this time point, the cells had reached between 60–90% confluence depending on initial seeding density and the differentiation commenced as described by Lian and colleagues: the concentration of CHIR99021 was tested in a range including 6, 8, 10 and 12 μM. The differentiation proceeded exactly as described by Lian and colleagues until the cells began to spontaneously contract.
Ectodermal Lineage
For differentiation to the ectodermal lineage, the hiPSC were differentiated to neuroepithelium using a modified version of Maroof et al.64. When hiPSC had reached 80% confluency, they were passaged at a 1:3 ratio using 0.5 mM EDTA and seeded into Geltrex coated tissue culture plates. After adhering overnight, the neural specification was commenced according to Maroof and colleagues64 using dual SMAD and WNT inhibition as follows. Briefly, the cells were approximately 30% confluent when starting the differentiation. The spent medium was removed and the cells were washed with DPBS−/−. After washing, the cells were treated with Neural Differentiation Phase I Medium consisting of Advanced DMEM/F12, 1 × Glutamax, 1 × Penicillin/Streptomycin, 1 × N2 Supplement (all from Life Technologies), and further supplemented with 10 μM SB431542 (Tocris), 100 nM LDN 193189 (Selleckchem), and 2 μM XAV-939 (Tocris). The cells were fed with this medium every 48 hours (days 0, 2, and 4). From day 5 until day 7, the medium was exchanged every 24 hours. After 7 days of differentiation, the cells were collected for gene expression analysis.
RT-qPCR (TaqMan)
All purchased from Life Technologies. RNA was isolated using Trizol according to the manufacturer’s instructions. cDNA synthesis was performed using the High Capacity cDNA Reverse Transcription kit (Thermo Fischer). 5ng of cDNA was used in each RT-qPCR reaction. Mastermix was the SSOAdvanced Universal Probes Supermix from Bio-rad. A 15 μl reaction volume was used. Details of Taqman probes are as follows.
AFP - Hs00173490_m1; BACT - Hs99999903_m1; FOXG1 - Hs01850784_s1; GATA4 - Hs00171403_m1; HNF4A - Hs00230853_m1; NES - Hs04187831_g1; PAX6 - Hs00240871_m1; PROX1 - Hs0086294_m1; SOX1 - Hs01057642_s1
Telomere length analysis
Genomic DNA was extracted from the cells using the DNeasy Tissue Kit (Qiagen, Germany). Telomere length measurement was performed by qPCR according to the method described by Cawthon35. Relative telomere length was calculated by normalizing the telomere repeat copy number to single-gene copy number and the formula used was 2−ΔCt (where ΔCt = Cttelomere − Ctsingle-copy gene). 36B4, encoding ribosomal protein lateral stalk subunit P0 (RPLP0), was used as the single copy gene. Telomere and 36B4 gene PCRs were performed in separate 96-well plates with each sample run in triplicate in the same well position on an ABI 7500 HT Detection System (Applied Biosystems, USA). Each 20 μL reaction consisted of 20 ng genomic DNA, 1 × SYBR Green master mix (Applied Biosystems), and 200 nM telomere forward primer, 200 nM telomere reverse primer. Primer pair sequences used for telomere length analysis and 36B4 gene are given in Supplementary Table S2.
Cycling conditions (for both telomere and 36B4) were 10 min at 95 °C, followed by 50 cycles of 95 °C for 15 sec and 60 °C for 1 min. Following amplification, a dissociation curve was performed to confirm the specificity of the reaction. All experiments were carried out in triplicates, and the results were presented as means.
Karyotyping
Chromosomal G-band and R-band analyses were performed at the Department of Medical Genetics, Faculty Hospital, Hradec Kralove, Czech Republic.
Biosafety Level 2
As per biosafety guidelines, all viral transduction related experiments were performed in Biosafety Level 2 (BL-2) containment using biological safety cabinets and laminar flow hoods, and with appropriate personal safety equipment to prevent mucosal exposure/splash.
Ethical guidelines statement
This study is based on biological material obtained for medical reasons and with the subjects’ legal (representatives) informed consent, after they have been given all the research relevant information. The extraction of the natal tooth was indicated for medical reasons. The local Ethical Committee of University Hospital Hradec Kralove ref. 200712 S01P approved the use of the dental pulp stem cells for research purposes while the usage of human platelet rich plasma was approved by Ethical Committee of the University Hospital Hradec Kralove, ref. 201011 S14P.
Human skin biopsies were obtained, with both informed donor consent and Ethical Committee approval, from donors undergoing cosmetic surgery.
Natal tooth extraction and human skin biopsies were performed in accordance with the relevant guidelines and regulations.
References
Ebrahimi, B. Reprogramming of adult stem/progenitor cells into iPSCs without reprogramming factors. J. Med. Hypotheses Ideas 9, 99–103 (2015).
Korbling, M., Estrov, Z. & Champlin, R. Adult stem cells and tissue repair. Bone Marrow Transplant. 32, S23–S24 (2003).
Ebrahimi, B., Yaghoobi, M., Kamal-abadi, A. & Raoof, M. Human dental pulp stem cells express many pluripotency regulators and differentiate into neuronal cells. Neural Regen. Res. 6, 2666–2672 (2011).
Eminli, S. et al. Differentiation stage determines reprogramming potential of hematopoietic cells into iPS cells. Nat. Genet. 41, 968–976 (2009).
Giorgetti, A. et al. Generation of induced pluripotent stem cells from human cord blood using OCT4 and SOX2. Cell Stem Cell. 5, 353–357 (2009).
Kim, J. B. et al. Oct4-induced pluripotency in adult neural stem cells. Cell 136, 411–419 (2009).
Niibe, K. et al. Purified mesenchymal stem cells are an efficient source for iPS cell induction. PLoS One 6, https://doi.org/10.1371/journal.pone.0017610 (2011).
Wang, J. et al. Generation of induced pluripotent stem cells with high efficiency from human umbilical cord blood mononuclear cells. Genomics Proteomics Bioinformatics 11, 304–311 (2013).
Yulin, X. et al. Efficient generation of induced pluripotent stem cells from human bone marrow mesenchymal stem cells. Folia Biol. (Praha) 58, 221–230 (2012).
Vidal, S., Amlani, B., Chen, T., Tsirigos, A. & Stadtfeld, M. Combinatorial modulation of signaling pathways reveals cell-type-specific requirements for highly efficient and synchronous iPSC reprogramming. Stem Cell Reports 3, 574–584 (2014).
Kim, J. B. et al. Direct reprogramming of human neural stem cells by OCT4. Nature 461, 649–643 (2009).
Sun, N. et al. Feeder-free derivation of induced pluripotent stem cells from adult human adipose stem cells. Proc. Natl. Acad. Sci. USA 106, 15720–15725 (2009).
Yan, X. et al. iPS cells reprogrammed from human mesenchymal-like stem/progenitor cells of dental tissue origin. Stem Cells Dev. 19, 469–480 (2010).
Park, J. H., Daheron, L., Kantarci, S., Lee, B. S. & Teixeira, J. M. Human endometrial cells express elevated levels of pluripotent factors and are more amenable to reprogramming into induced pluripotent stem cells. Endocrinology 152, 1080–1089 (2011).
Mokry, J. et al. Telomere attrition occurs during ex vivo expansion of human dental pulp stem cells. J. Biomed. Biotechnol. 2010, https://doi.org/10.1155/2010/673513 (2010).
Beltrao-Braga, P. C. et al. Feeder-free derivation of induced pluripotent stem cells from human immature dental pulp stem cells. Cell Transplant. 20, 1707–1719 (2011).
Lo Sardo, V. et al. Influence of donor age on induced pluripotent stem cells. Nat. Biotechnol. 35, 69–74 (2017).
Dyment, H., Anderson, R., Humphrey, J. & Chase, I. Residual neonatal teeth: a case report. J. Can. Dent. Assoc. 71, 394–397 (2005).
Mhaske, S. et al. Natal and neonatal teeth: an overview of the literature. ISRN Pediatr. 2013, https://doi.org/10.1155/2013/956269 (2013).
Verma, K. et al. Therapeutic potential of dental pulp stem cells in regenerative medicine: an overview. Dent. Res. J. (Isfahan) 11, 302–308 (2014).
Lamb, R. A. & Kolakofsky D. in Fields Virology (eds Fields, N., Knipe, M. & Howley, M.) (Lippincott-Raven Press, 1996).
Tokusumi, T. et al. Recombinant Sendai viruses expressing different levels of a foreign reporter gene. Virus Res. 86, 33–38 (2002).
Li, H. O. et al. A cytoplasmic RNA vector derived from nontransmissible Sendai virus with efficient gene transfer and expression. J. Virol. 74, 6564–6569 (2000).
Inoue, M. et al. Nontransmissible virus-like particle formation by F-deficient sendai virus is temperature sensitive and reduced by mutations in M and HN proteins. J. Virol. 77, 3238–3246 (2003).
Fusaki, N., Ban, H., Nishiyama, A., Saeki, K. & Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B. Phys. Biol. Sci. 85, 348–362 (2009).
Suchanek, J., Suchankova- Kleplova, T., Rehacek, V., Browne, K. Z. & Soukup, T. Proliferative capacity and phenotypical alteration of multipotent ecto-mesenchymal stem cells from human exfoliated deciduous teeth cultured in xenogeneic and allogeneic media. Folia Biol. (Praha) 62, 1–14 (2016).
Atari, M. et al. Dental pulp of the third molar: a new source of pluripotent-like stem cells. J. Cell. Sci. 125, 3343–3356 (2012).
Karbanova, J. et al. Characterization of dental pulp stem cells from impacted third molars cultured in low serum-containing medium. Cells Tissues Organs 193, 344–365 (2011).
Ferro, F., Spelat, R., Beltrami, A. P., Cesselli, D. & Curcio, F. Isolation and characterization of human dental pulp derived stem cells by using media containing low human serum percentage as clinical grade substitutes for bovine serum. PLoS One 7, https://doi.org/10.1371/journal.pone.0048945 (2012).
Alvarez, R., Lee, H. L., Hong, C. & Wang, C. Y. Single CD271 marker isolates mesenchymal stem cells from human dental pulp. Int. J. Oral Sci. 7, 205–212 (2015).
Suchanek, J. et al. Dental pulp stem cells and their characterization. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech. Repub. 153, 31–35 (2009).
Schmittgen, T. D. & Livak, K. J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108 (2008).
Larue, L. et al. A role for cadherins in tissue formation. Development 122, 3185–3194 (1996).
Chen, T. et al. E-cadherin-mediated cell-cell contact is critical for induced pluripotent stem cell generation. Stem Cells 28, 1315–1325 (2010).
Cawthon, R. M. Telomere measurement by quantitative PCR. Nucleic Acids Res. 30, https://doi.org/10.1093/nar/30.10.e47 (2002).
Wang, F. et al. Molecular insights into the heterogeneity of telomere reprogramming in induced pluripotent stem cells. Cell Res. 22, 757–768 (2012).
Suhr, S. T. et al. Telomere dynamics in human cells reprogrammed to pluripotency. PLoS One 4, https://doi.org/10.1371/journal.pone.0008124 (2009).
Vaziri, H. et al. Spontaneous reversal of the developmental aging of normal human cells following transcriptional reprogramming. Regen. Med. 5, 345–363 (2010).
Laino, G. et al. A new population of human adult dental pulp stem cells: a useful source of living autologous fibrous bone tissue (LAB). J. Bone Miner. Res. 20, 1394–1402 (2005).
Akpinar, G. et al. Phenotypic and proteomic characteristics of human dental pulp derived mesenchymal stem cells from a natal, an exfoliated deciduous, and an impacted third molar tooth. Stem Cells Int. 2014, https://doi.org/10.1155/2014/457059 (2014).
Karaoz, E. et al. Isolation and in vitro characterisation of dental pulp stem cells from natal teeth. Histochem. Cell Biol. 133, 95–112 (2010).
Kerkis, I. et al. Isolation and characterization of a population of immature dental pulp stem cells expressing OCT-4 and other embryonic stem cell markers. Cells Tissues Organs 184, 105–116 (2006).
Lengner, C. J. et al. Oct4 expression is not required for mouse somatic stem cell self-renewal. Cell. Stem Cell. 1, 403–415 (2007).
Heng, J. C. et al. The nuclear receptor Nr5a2 can replace Oct4 in the reprogramming of murine somatic cells to pluripotent cells. Cell Stem Cell. 6, 167–174 (2010).
Kishimoto, T. Interleukin-6: from basic science to medicine - 40 years in immunology. Annu. Rev. Immunol. 23, 1–21 (2005).
Hossain, A. et al. Mesenchymal stem cells isolated from human gliomas increase proliferation and maintain stemness of glioma stem cells through the IL-6/gp130/STAT3 Pathway. Stem Cells 33, 2400–2415 (2015).
Chen, H. et al. Reinforcement of STAT3 activity reprogrammes human embryonic stem cells to naive-like pluripotency. Nat. Commun. 6, https://doi.org/10.1038/ncomms8095 (2015).
Illich, D. J. et al. Concise review: induced pluripotent stem cells and lineage reprogramming: prospects for bone regeneration. Stem Cells 29, 555–563 (2011).
Takeda-Kawaguchi, T. et al. Derivation of iPSCs after culture of human dental pulp cells under defined conditions. PLoS One 9, https://doi.org/10.1371/journal.pone.0115392 (2014).
Tamaoki, N. et al. Dental pulp cells for induced pluripotent stem cell banking. J. Dent. Res. 89, 773–778 (2010).
Pomeroy, J. E. et al. Stem cell surface marker expression defines late stages of reprogramming to pluripotency in human fibroblasts. Stem Cells Transl. Med. 5, 870–882 (2016).
Beers, J. et al. Passaging and colony expansion of human pluripotent stem cells by enzyme-free dissociation in chemically defined culture conditions. Nat. Protoc. 7, 2029–2040 (2012).
Rittie L & Fisher, G. J. in Fibrosis Research: Methods and Protocols (eds Varga, J., Brenner, D. A. & Phan, S. H.) 83–98 (Humana Press, Totowa, NJ, 2005).
Metsalu, T. & Vilo, J. ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 43, https://doi.org/10.1093/nar/gkv468 (2015).
Babicki, S. et al. Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res. 44, https://doi.org/10.1093/nar/gkw419 (2016).
Lin, Y. & Chen, G. Embryoid body formation from human pluripotent stem cells in chemically defined E8 media. https://www.stembook.org/node/6632 (2014).
Siller, R., Greenhough, S., Naumovska, E. & Sullivan, G. J. Small-molecule-driven hepatocyte differentiation of human pluripotent stem cells. Stem Cell Rep. 4, 939–952 (2015).
Mathapati, S. et al. Small-molecule-directed hepatocyte-like cell differentiation of human pluripotent stem cells. Curr. Protoc. Stem Cell Biol. 38, https://doi.org/10.1002/cpsc.13/full (2016).
Siller, R. & Sullivan, G. J. Rapid screening of the endodermal differentiation potential of human pluripotent stem cells. Curr. Protoc. Stem Cell Biol. 43, https://doi.org/10.1002/cpsc.36/full (2017).
Siller, R. et al. Development of a rapid screen for the endodermal differentiation potential of human pluripotent stem cell lines. Sci. Rep. 6, https://doi.org/10.1038/srep37178 (2016).
Hay, D. C. et al. Highly efficient differentiation of hESCs to functional hepatic endoderm requires ActivinA and Wnt3a signaling. Proc. Natl. Acad. Sci. USA 105, 12301–12306 (2008).
Sullivan, G. J. et al. Generation of functional human hepatic endoderm from human induced pluripotent stem cells. Hepatology 51, 329–335 (2010).
Lian, X. et al. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/beta-catenin signaling under fully defined conditions. Nat. Protoc. 8, 162–175 (2013).
Maroof, A. M. et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell. 12, 559–572 (2013).
Acknowledgements
We thank, Dr. Ales Tichy (Biomedical Research Center, University Hospital in Hradec Kralove, Hradec Kralove, Czech Republic) for providing WI38 human fibroblasts, Prof. Dr. Ales Hampl, Ph.D. (Department of Embryology and Histology, Masaryk University, Brno, Czech Republic) for providing total RNA of hESC, Dr. Lenka Bydzovska (Department of Medical Genetics, Faculty Hospital, Hradec Kralove, Czech Republic) for performing karyotyping analysis on DP hiPSC clones, Dr. Petra Matoušková (Department of Biochemical Sciences, Faculty of Pharmacy, Hradec Kralove, Czech Republic) for allowing to use her lab and instruments to perform qPCR analysis and Dr. Doris Vokurkova (Department of Clinical Immunology, Faculty Hospital, Hradec Kralove, Czech Republic) for performing flow cytometry analysis on nDPSC. This study was supported by project PROGRES Q40/06.
Author information
Authors and Affiliations
Contributions
R.P. conceived and designed the experiments; R.P., J.S., R.S., H.H., performed experiments; A.B., performed statistical analysis; S.M., performed telomere length analysis; T.S., contributed analytic tools; D.K., gene selection, primer selection, plate designing, primer validation; R.P., T.S., S.F., R.S., G.S., J.M., analysed data and R.P., G.S., R.S., J.M. wrote the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pisal, R.V., Suchanek, J., Siller, R. et al. Directed reprogramming of comprehensively characterized dental pulp stem cells extracted from natal tooth. Sci Rep 8, 6168 (2018). https://doi.org/10.1038/s41598-018-24421-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-24421-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
