
Abstract
Chikungunya virus (CHIKV) is a mosquito-borne alphavirus. The outbreak of CHIKV infection has been seen in many tropical and subtropical regions of the biosphere. Current reports evidenced that after outbreaks in 2005–06, the fitness of this virus propagating in Aedes albopictus enhanced due to the epistatic mutational changes in its envelope protein. In our study, we evaluated the prevalence of intrinsically disordered proteins (IDPs) and IDP regions (IDPRs) in CHIKV proteome. IDPs/IDPRs are known as members of a ‘Dark Proteome’ that defined as a set of polypeptide segments or whole protein without unique three-dimensional structure within the cellular milieu but with significant biological functions, such as cell cycle regulation, control of signaling pathways, and maintenance of viral proteomes. However, the intrinsically disordered aspects of CHIKV proteome and roles of IDPs/IDPRs in the pathogenic mechanism of this important virus have not been evaluated as of yet. There are no existing reports on the analysis of intrinsic disorder status of CHIKV. To fulfil this goal, we have analyzed the abundance and functionality of IDPs/IDPRs in CHIKV proteins, involved in the replication and maturation. It is likely that these IDPs/IDPRs can serve as novel targets for disorder based drug design.
Similar content being viewed by others
Introduction
Chikungunya fever is triggered by an arthropod-borne virus (arbovirus) known as Chikungunya virus (CHIKV) that is transmitted by mosquitoes (Aedes aegypti and Aedes albopictus)1 and is disseminated at a higher rate in tropical regions. A Makonde word ‘Chikungunya’, which means: ‘The one which bends up’2, is taken from the Bantu language. The first epidemic of this disease was recognized in Tanzania in 1952. Since then, it had been considered as a tropical neglected disease3, despite the fact that in past 50 years, innumerable cases of re-emergence of CHIKV have been documented in the African and Asian continents4. However, after its recent outbreaks and the disease severity, it is listed now as a category C priority pathogen in US National Institute of Allergy and Infectious Diseases (NIAID)5. The main manifestations are flu-like symptoms, such as fever, headache, joint pain, and difficulties in movement2. The CHIKV infection has a rapid onset and it gets cleared in 5–7 days6. The fitness of this virus propagating in Aedes albopictus is enhanced due to the epistatic mutational changes in its envelope protein7,8. Dong et al., also explained the infection pattern of CHIKV in midguts and salivary glands of two different strains of Aedes aegypti, Higgs white eyes (HWE) and Orlando, Florida (ORL)9. Mutational pressure is generally high in the viral genome, and similar patterns are observed in CHIKV and Zika virus7,10.
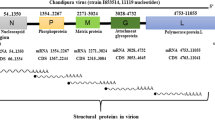
CHIKV is a small spherical (diameter of about 60–70 nm) shaped virus having a single-stranded positive-sense RNA genome (11,811 nucleotides)11 organises as 5′UTR-nsP1-nsP2-nsP3-nsP4-J-C-E3-E2-6K-E1-polyA-3′UTR12. It has two open reading frames (ORFs) placed between the 5′and 3′ UTRs. First ORF (7,422 nucleotides long) encodes four non-structural proteins, nsP1 (535 residues, involved in capping and GTPase activity), nsP2 (798 residues, shows 5′ RTPase, helicase and protease activity), nsP3 (530 residues, has replicase activity and involved in RNA synthesis), and nsP4 (611 residues, has RNA-dependent RNA polymerase activity)13 (Fig. 1). Second ORF (3,744 nucleotides long) encodes five structural proteins, such as capsid (261 residues, involved in growth and assembly) and envelope glycoproteins E1 (439 residues, facilitate membrane fusion), E2 (423 residues, helps in receptor binding), E3 (64 residues, protects the E2-E1 heterodimer from premature fusion with cellular membrane), and J (junction) region (used as the promoter for subgenomic RNA synthesis)13. Recently, the structure of CHIKV was determined by the cryo-electron microscopy (PDB ID: 3J2W)14.

Confinement of all proteins within CHIKV genome (Q8JUX6 and Q8JUX5). The CHIKV RNA (11811 nucleotides) translates into non-structural precursor proteins of 2474 residues and structural precursor protein of 1244 residues. On maturation, non-structural precursor protein cleaved into nsP1, nsP2, nsP3 and nsP4, and structural precursor protein cleaved into C, E1, E2, E3 and 6K proteins.
The prime focus of our article is to analyse the dark proteome of CHIKV. The dark proteome defined as proteins not amenable to structure determination by conventional methods, such as x-ray crystallography and electron microscopy15. Most of the dark proteome are IDPs of hybrid proteins containing ordered domains and IDPRs16, which show specific functions without being folded into unique 3D structure under physiological conditions17. It is known, hydrophobic interaction plays a driving force in protein folding18. Since IDPs/IDPRs are more hydrophilic than ordered proteins19, this could be one of the reasons for their disorderness20. These proteins exist as highly dynamic conformational ensembles and can attain highly diverse conformations, such as random coils, molten globules, pre-molten globules, flexible linkers, and much more21,22,23,24,25. A globular structure attained by the ordered proteins that typically prevent the formation of aggregate or misfolding because of the frustrations in amino acid sequence compositions26. IDPs/IDPRs are promiscuous binders that can be involved in numerous interactions with many partners, thereby acting as an important hub in protein-protein interaction networks to regulate multiple signaling pathways at a time27,28,29. Many IDPs/IDPRs show disorder to order transition after binding to their partners30,31,32. For example, transactivation domain of c-Myb is intrinsically disordered in its unbound form, but after interaction with KIX, this domain gains an α-helical conformation33. Some IDPs/IDPRs facilitating the interaction with partners are known as molecular recognition features (MoRFs). Most of the viral disordered proteins show MoRF regions. For example, structural and non-structural proteins of Zika virus has MoRF regions that regulate the functionality of this virus34.
Being disordered, these proteins increase the probability of binding to their partners by providing greater capture radii; i.e., utilizing a binding mode known as a fly-casting mechanism35. Despite this fly-casting mechanism, it was proposed that some disordered systems such as transactivation domain of c-Myb region, cannot speed up the recognition events between an IDP and its partner36. In recent studies, it is evidenced IDPs/IDPRs play a vital role in the establishment of several macromolecular complexes37 and also serve as assembly hubs38. Furthermore, it is recognized now that disordered regions represent new and attractive targets for drug design39,40,41,42. For example, IDPRs in c-Myc are the new targets for the development of drugs to cure cancer43,44. In our work, we have analyzed the dark proteome of CHIKV and evaluated disordered regions of viral proteins in terms of their functional significance.
Results and Discussion
Intrinsically disordered regions in CHIKV polyprotein
X-ray crystallography represents a unique technique for the determination of the 3D structure of proteins. It is based on the analysis of the scattering patterns of X-rays, which reads electron density maps to understand protein 3D structure45. One should keep in mind though, that the crystallization process typically requires various additives, such as PEG, high salt concentration, and many more, that might affect protein structure. IDPs/IDPRs are highly flexible and therefore characterized by the lack of specific electron densities. Since crystal structure of most of the CHIKV proteins are not reported at Protein Data Bank (PDB), we used limited crystallographic structural information that is currently available for a macro domain of nsP3 (PDB ID: 3GPG)46, nsP2 protease domain (PDB ID: 3TRK), and an envelope glycoprotein complex (E1, E2 and E3; PDB ID: 3N40)47. In light of the limited structural information, the use of computational analysis to observe disordered regions of the query protein may deliver great advantages23.
Analysis of disordered regions in non-structural proteins
Non-structural precursor polyprotein P1234. Genomic RNA of alphavirus translates into a short-lived non-structural precursor polyprotein P1234 that helps in the replication process. It is synthesized because of a fraction of ribosome unable to terminate the translation when reaching the opal stop codon48. There are three cleavage sites lie between the residue 535–536, 1333–1334, and 1863–1864 in the P1234 (Fig. 2). These three cleavage sites are also represented with respect to disordered regions predicted at the cleavage junction in Fig. 3a–c. At the initial stage of infection, P1234 is cleaved in trans by the nsP2 protease, forming P123 and nsP4 protein. Then, the P123 complex and nsP4 start replicating the viral genome into antigenome. Further, P123 is cleaved in cis into nsP1 and P23 by the nsP2 protease. At the N-terminus of P23, an ‘activator’ is exposed and induces the cleavage of P23 into nsP2 and nsP149. At the later stage of infection, P1234 is quickly cleaved by nsP2 protease into P12 and P34 and then into nsP1, nsP2, nsP3 and nsP449. We will discuss the results of disorder analysis of each protein in subsequent paragraphs.

Intrinsically disordered cleavage sites in CHIKV non-structural polyprotein (Q8JUX6). Prediction of intrinsically disordered regions of non-structural CHIKV polyprotein by PONDR® VSL2 (magenta line), PONDR® VL3 (violet line), PONDR® VLXT (red color), and PONDR-FIT (olive line). Mean disorder predisposition is shown by a dashed blue line. Localization of individual protein is represented by horizontal color bars at the top: nsP1 (black), nsP2 (yellow), nsP3 (orange), and nsP4 (light blue). Grey color vertical bar shows three cleavage sites. During the course of maturation, polyprotein cleaves into individual protein by nsP2 protease. These three cleavage sites (shown by grey vertical bars) lies at 535–536 (1st), 1333–1334 (2nd), and 1863–1864 (3rd) amino acid residues that show disorder regions. The amino acids and regions that have PONDR score ≥ 0.5 are considered as intrinsically disordered.

Contribution of intrinsically disordered regions in the maturation of specific proteins of CHIKV virus. The plots a-g represent the cleavage sites with respect to disordered regions present at the cleavage junction (grey vertical bar) of both CHIKV precursor proteins (cleavage of non-structural precursor protein by the nsP2 protease)96. Plot shows the cleavage site between (a) nsP1 (pink horizontal bar) and nsP2 (yellow horizontal bar) protein, (b) nsP2 (yellow horizontal bar) and nsP3 (black horizontal bar) protein, (c) nsP3 (black horizontal bar) and nsP4 (orange horizontal bar), (d) capsid (dark red horizontal bar) and E3 (cyan horizontal bar) protein, cleaved by capsid protease97, (e) E3 (cyan horizontal bar) and E2 (green horizontal bar) proteins, cleaved by furin cleavage, (f) E2 (green horizontal bar) and 6K (yellow horizontal bar) protein, cleaved by host signal peptidase, (g) 6K (yellow horizontal bar) and E1 (pink horizontal bar) protein, cleaved by host signal peptidase.
nsP1 protein. The nsP1 protein contains the methyltransferase (MTase) and guanylyltransferase (GTase) domains that have the role in 5′ capping, attachment of replication complex to the cytosolic membrane, induction of tapered pseudopodium-like structure, and in the synthesis of subgenomic RNA50. In the capping mechanism, methyl group from S-adenosylmethionine is transferred at the 7th position of GTP by the nsP1 enzyme, which forms a covalent complex with the m7GMP and releases pyrophosphate. The methylated residue is transferred to 5′ end of viral RNA to complete the capping process51. The amino acid residues Pro34 and His37 of nsP1 are involved in the capping process and serve as the binding site for m7GMP, whereas the amino acids Asp89, Arg92, and Tyr248 play an important role in the methyltransferase activity52. The average predicted percent of intrinsic disorder (PPID) based on the outputs of four predictors in nsP1 protein (Fig. 4a) is 15.14% (Fig. 5a), which defines this protein as moderately disordered. The disordered regions lie within both N and C-terminal domain of nsP1, where the N-terminal region is required for capping process and C-terminal IDPR helps in the regulation of downstream translation.

Diagrammatic representation of disordered aspect of CHIKV non-structural proteins. PONDRR VSL2, VL3, VL-XT and PONDR_FIT predictors used for disorder analysis. Plots represent disorder analysis of (a) nsP1 (residues 1–535, denoted as 1–535 at x-axis), shows disorder regions at its C-terminal region (Mean disorder propensity ≥ 0.5, from 394th to 535th residues) and black encircle represents binding site for m7GMP (bI) nsP2 (residues 536–1333, denoted as 1–798 at x-axis), black encircles represent domains (bII) the crystal structure of nsP2 protease where C1013 and H1083 serve as catalytic dyad, are represented as C478 and H548 respectively in this figure (cI) nsP3 (residues 1334–1863, denoted as 1–530 at x-axis), black encircles represent domains of nsP3 (cII) crystal structure of macro domain of nsP3 protein (d) nsP4 (residues 1864–2474, denoted as 1–611 at x-axis), shows the disorder region at its N-terminal domain, black encircle represents domain.

Results of predicted percentage of intrinsic disorder (PPID) in CHIKV proteins. PPID value predicted for various proteins of CHIKV is indicated at y-axis. Four softwares were used to predict the percentage disorder in proteins by VLXT, VSL2, VL3 and PONDER_FIT indicated by red, blue, violet and green colour bar. The mean PPID value of the results of these predictors is represented by a black bar. Plot (a) shows the disorder prediction for all CHIKV proteins and (b) represents disorder prediction for specific domains of non-structural and capsid proteins.
nsP2 protein. This is the largest CHIKV protein that plays an important role in the pathogenic mechanism and in the life cycle of the virus53. In the viral replication, nsP2 display three key roles as helicase, triphosphatase, and protease54. nsP2 has two domains, N-terminal and C-terminal. The N-terminal domain shows NTPase-dependent helicase activity having helicase sequence motifs, such as motif 1 (residues 715–733), motif 1A (residues 738–752), motif 2 (residues 782–791), motif 3 (residues 811–818), motif 4 (residues 839–848), motif 5 (residues 908–929), and motif 6 (residues 940–954)54. The N-terminal domain is also associated with the NTPase-dependent RTPase activity to remove phosphate from the 59th terminus of nascent RNA that acts as a substrate for capping reaction54. It also has nucleolar localization signal (NoLS region) and nuclear localization signal (NLS motif), positioned at residues 1005–1024 and 1182–1186, respectively55.
In other alphaviruses, nsP2 C-terminal domain (residues 1004–1327) is a papain-like protease domain, known as cysteine protease (thiol protease). However, a recent report revealed that in CHIKV, a cysteine residue can be catalytically replaced by serine. Hence, this study suggests that protease domain of CHIKV is not a papain-like protease56. It consists of two structural domains, such as a protease domain (residues 471–605) and a methyltransferase-like domain (residues 606–791). These two domains function as a single unit and are crucial for protease activity57. The functional mechanism of protease domain is related to deprotonation of a thiol group of cysteine residue from catalytic dyad at the active site (Cys1013 and His1083)58,59. The crystal structure of nsP2 protease domain of CHIKV have resolved (PDB ID: 3TRK) (Fig. 4bII). Although the analysis of the amino acid composition indicated that nsP2 has large net positive charge (+21), the disorder prediction revealed that the PPID value of the full-length nsP2 protein is only 0.50% (Fig. 5a). However, the helicase N-terminal ATP binding domain which plays a role in viral replication showing the PPID of 0.856% (Fig. 5b).
nsP3 protein. nsP3 protein constitutes the N-terminal macro domain, the Zn2+ binding domain, and the C-terminal variable tail domain. The crystal structure of macro domain (160 residues) has been resolved (PDB ID: 3GPG)46, where residues 30–37 represent a loop region and residues Thr111, Gly112, and Val113 form a binding site. The macro domain can interact with both mono and poly ADP-ribose and with RNA as well60. It has the ability to hydrolyze small substrate analog ADP-ribose-1-phosphate. A small Zn2+ binding domain follows macrodomain and contains four conserved cysteines (Cys263, Cys265, Cys288, and Cys306) where Cys263 and Cys265 are situated in the loop between the last two α-helices, crucial for replication61. Both macro domain and Zn2+ binding domain play a key role as regulators for the processing of non-structural polyprotein61. The third proline-rich C-tail domain is 205 residues long. It provides an insertion site for marker proteins. During the infection, components of stress granules G3BP1 and G3BP2 (host cells) directly bind to nsP3 protein to arrest RNA translation, and the interaction of G3BPs with nsP3 inhibits the stress granules formation62. Two short conserved repeat sequences near the C-terminal region of nsP3 interact with G3BPs. This interaction results in the depletion of G3BPs which leads to a reduction in CHIKV replication63. In disorder analysis, we found that the PPID value of this protein is 38.49% (Fig. 5a), and its C-terminal tail region shows the PPID value of 48.37% (Fig. 5b). Functional role of the C-terminal domain is not well known as of yet. However, it is assumed that it provides an insertion site for marker proteins. In other words, this analysis also showed the relevance of disordered regions of nsP3 for biological activities.
nsP4 protein. nsP4 acts as RNA-dependent RNA polymerase (RdRp) enzyme in CHIKV. It inhibits the phosphorylation at Ser51 residue of the α-subunit of elF2α, a eukaryotic translation initiation factor64. In addition to its polymerase activity, nsP4 also shows terminal adenylyltransferase activity, which is responsible for the synthesis of poly(A) tail in template-independent manner65. A recent report revealed that nsP4 is required for RNA synthesis assisted by other non-structural protein and possibly play a role in proper folding65. The N-terminal region (100 residues long) of nsP4 forms a partly unstructured domain, is necessary for the proper functioning of nsP4. This partly unstructured domain is followed by the catalytic domain with the established polymerase fold. A catalytic triad of GDD polymerase motif (residues Gly464, Asp465, and Asp466) is also important for polymerase activity66. The disorder analysis of this whole protein (Fig. 4d) gives the PPID value of 20.94% (Fig. 5a), but the N-terminal region (regulates polymerase activity) is characterized by the PPID of 76.00% (Fig. 5b). Therefore, one can target this region to stop its polymerase activity and thereby can inhibit virus replication within the host cell.
Analysis of the CHIKV structural proteins
Capsid protein. Capsid protein (CP) of alphavirus has an N-terminal RNA binding domain (residues 1–113) and the C-terminal protease domain (residues 114–261). The N-terminal domain constitutes two nuclear localization signal (NLS), residues 60–77 and 84–99. Although capsid protein is a cytoplasmic protein, it is assumed that its NLS may be involved in translocation of CP to the host nucleus67. The highly disordered N-terminal domain enriched with positively charged amino acid residues (Arg, and Lys) with high proline content. It has ribosome binding region (91–100 amino acid residues) that involved in protein-protein interaction. This domain also binds to the genomic RNA via 18 amino acid long coiled-coil α-helix68 and is required for the dimerization of capsid protein that negatively regulates host transcription67. This N-terminal domain is also involved in the interaction with RNA bound capsid proteins to form nucleocapsid core assembly in the cytoplasm of infected cells67. The nucleocapsids are then secreted out to the plasma membrane to interact with the C-terminal region of E2 for the initiation of virion budding. Residues 81–105 at the N- terminal domain of CP interact with RNA to form Capsid-RNA complex69. The C-terminal domain possesses a serine protease activity and is characterized by a chymotrypsin-like fold. This domain autoproteolytically cleaves CP from the nascent structural polyprotein. The report suggests that the C-terminal domain has numerous conserved amino acids, including a catalytic triad His139, Asp161, and Ser213, involved in the autoproteolytic activity that occurs at Trp261. Close to the substrate binding site of protease, a hydrophobic pocket is present, where binding of capsid to endodomain of the E2 glycoprotein occurs70. The predicted disordered analysis at the cleavage junction for CP is shown in Fig. 3d. IDP analysis of whole capsid protein (Fig. 6aI) shows PPID score of 45.59% (Fig. 5a). However, the PPID score of the N-terminal region comes out to be 100% (Fig. 5b). This N-terminal domain of CP is required for nucleocapsid core assembly that helps in virus budding.

Diagrammatic representation of IDPs analysis of structural proteins of CHIKV. Plot represents the disorder analysis of (aI) capsid protein (residues 1–261, denoted as 1–261 at x-axis), shows complete disorderness in the N-terminal domain, black encircles represent domains (aII) the homology model of the C-terminal domain (blue color) of capsid protein (b) E3 protein (residues 262–325, denoted as 1–64 at x-axis), black encircle represents furin loop (c) E2 protein (residues 326–748, denoted as 1–423 at x-axis), black encircles represent domain A, B, and C (d) E1 protein (residues 810–1248, denoted as 1–439 at x-axis), black encircle represents fusion loop (e) 6K (residues 749–809, denoted as 1–61 at x-axis) protein (f) ribbon diagram of the crystal structure of p62-E1 heterodimer (PDB ID: 3N40); E1 domains I, II, and III are in red, yellow and blue colors respectively and fusion loop is in orange color; E2 domains A, B and C are in cyan, dark green and pink colors respectively; E3 protein is in grey color having furin loop (grey color) at the junction of E2 and E3 proteins.
p62 (pE2) protein. p62 precursor protein is a premature complex of E3-E2 protein. Processing and maturation of the glycosylated pE2 and E1 proteins takes place at the ER of infected host cells, where the polysaccharide chains are embedded on the surface of the envelope proteins to regulate cell receptor recognition, cell attachment, and entry into host membrane after viral fusion71. The envelope polyprotein encoded by CHIKV has four proteins arranged as E3-E2-6K-E1. Crystal structure of immature glycoprotein complex has been resolved (PDB ID: 3N40) (Fig. 6f). It forms a stable heterodimer with E1 protein to prevent premature cleavage of envelope polyprotein. After maturation, p62 is processed by host furin to E3 and E2-E1 heterodimer72 where we found disordered regions at E3-E2, E2-6K, 6K-E1 cleavage junction of envelope protein complex (Fig. 3e–g).
E3 protein. E3 is an α/β glycoprotein where β-hairpin is packed with three α-helices that forms a horseshoe-shaped structure47. E3 protein helps in the structural stability of E2 glycoprotein through modification of A and B domains in such a way that it can create a groove to accommodate fusion loop47. Another important function of E3 is to protect and stabilize the immature trimer (p62-E1) complex from acidic environment73. It also helps in low pH-mediated endocytosis of spikes (three E2-E1 heterodimers). Numerous polar residues are present at the N-terminal region (serve as E3 signal), required for the translocation and processing of p62 into ER lumen74. This signal plays a major role in trimer formation among E3-E2-E175. The disorder analysis of E3 protein (Fig. 6b) gives PPID value of 65.62% (Fig. 5a), the highest score among all full-length CHIKV proteins. E3 glycoprotein has many disordered regions involved in stabilization of the E2-E3 complex structure and in the pH-mediated endocytosis of spikes. A furin loop present at the junction of E3 and E2 in the p62 precursor protein prevents premature cleavage of E1-p62 heterodimer47. This furin loop becomes disordered after the cleavage of the E1-p62 heterodimer.
E1 protein. Voss et al. reported the crystal structures of mature envelope glycoprotein complex (complex of E1, E2, and E3; PDB ID: 3N41, 3N42, 3N42, 3N43 and 3N44)47. The E1 protein is N-glycosylated at position N14173. It consists of three domains I, II, and III. The Domain II has a fusion loop (residues 893–910) at its tip, essential for the association of E1 and E2 glycoproteins. This fusion loop forms two H-bonds, first, between Ala92 of E1 and His226 of E2 (Domain B) and second, between Phe87 of E1 and His29 of (Domain A)47. The structure of CHIKV envelope is made up of spikes (three E2-E1 heterodimers). Eighty such spikes are present in a CHIKV virion, and those spikes mediate membrane fusion to deliver the viral genome into the host cell. The viral fusion is triggered by low pH (~5.5 to 6.0) dependent secretory pathway of endocytosis73. This is the reason why E1 fusion protein is expressed with E2 protein. However, disorder analysis of E1 protein (Fig. 6d) shows lower PPID value of 0.45% (Fig. 5a).
E2 protein. E2, a β-protein is crucial for cell receptor recognition, belongs to immunoglobulin superfamily consist of three domains (A, B, and C)74. Domain A and B of E2 protein forms a groove and this domain with unfolded fusion loop of E1 protein insert a β-hairpin into the groove to form a heterodimer complex. Further, these heterodimer forms spike in the envelope protein47. Domain B makes a contact with E3 protein via a long connector β ribbon. N-linked glycosylation of this protein occurs at Asn263 and Asn273 residues73. An acid sensitive region (ASR) (at 234th residue) has been identified in E2 protein that triggers the initiation of conformational changes in the E1-E2 complex47. The disorder analysis of E2 protein (Fig. 6c) shows the PPID value of 12.29% (Fig. 5a) only. Though, these proteins do not show more disordered regions, however, domain B shows some disordered regions that are important for its functionality.
6K protein. 6K is a hydrophobic acylated protein involved in the envelope development and membrane permeabilization. The C-terminal domain of this protein mediates ER translocation of the E1 protein. 6K protein has the ability to increase membrane permeability by flippase of lipids from one side of the membrane to the other side that helps in virus budding57. The disorder analysis of this protein (Fig. 6e) gives the PPID score of 14.75% (Fig. 5a). Although the overall biological significance of IDPRs in this protein is not very clear, the disordered region of this protein plays an important role in virus budding.
CHIKV proteins play important role in the regulation of viral life cycle, pathogenic mechanisms, and immune evasion1. Our study revealed the abundant presence of disordered regions in structural and non-structural CHIKV proteins. A recent study evidenced the mutational changes happen in the E1 and E2 proteins that are identified as K211E and A226V (E1) and V264A (E2) polymorphism7.
Concluding Remarks
It has been found that mutational changes occur in the disordered loop regions of E1 and E2 protein. These mutational changes trigger the conformational change in E1 protein to enhance the fitness of CHIKV propagating in Indian Aedes agepty & Aedes albopictus7. Fusion loop is disordered in nature that helps in dimer (E1 and E2 protein) formation. A recent study evidenced the mutational changes happen in the E1 and E2 proteins that are identified as K211E and A226V (E1) and V264A (E2) polymorphism7. These mutational changes are confined to the loop region of envelope protein, which is disordered in nature. They enhance the fitness of CHIKV residing in Indian Aedes agepty & Aedes albopictus. These mutations direct the conformational changes in the E1 protein. The current study suggests that IDPRs play a vital role in the structural flexibility and functional diversity of the CHIKV proteome and play diverse biological roles, such as cell cycle regulation, signaling, and protein stability of viral proteome. Therefore, we concluded that the disordered side of CHIKV proteome may provide a new angle to consider the pathogenic characteristic and virus-host interaction mechanism. In our study, we found that the N-terminal domain of capsid protein is completely disordered. This domain is required for the nucleocapsid assembly. Furthermore, the C-terminal domains of nsP3, E2 and E3 proteins have functional disordered regions. Since the role of IDPRs is well characterized in the regulation of life cycle of viruses, research has moved towards the development of the disorder based drug discovery strategies. The abundant disordered regions in CHIKV proteome could be an attractive target for drug designing. Since there is no vaccine/drug against chikungunya virus, a detailed analysis of CHIKV proteome is necessary to develop new drug by targeting the disordered regions.
Methods
For disorder analysis, we used a reviewed polyprotein sequence of the S27-African CHIKV prototype, because this strain is a most causative strain of Chikungunya disease [UniProt ID: Q8JUX6 and Q8JUX5]. Previously, for disorder analysis, several specialized predictors have been developed, for example, PONDR® pool [PONDR® FIT76, PONDR® VLS277, PONDR® VLXT78], as well as IUPred79, DisoPred80, DisEMBL81, GlobPlot82, SPRITZ83, and much more84. To evaluate the precision of disorder predictors, several aforementioned tools were compared within the frames of the Critical Assessment of Protein Structure Prediction (CASP)85. It was also indicated that because different predictors consider the occurrence of the intrinsic disorder in proteins from diverse perspectives, it is sagacious to use several computational tools to catch on the profusion of intrinsic disordered17. Typically, disorder predictors consider residues and regions as intrinsically disordered if their disorder score is above the 0.5 thresholds. The peculiarities of all predictors were considered while calculating the mean of all predictors.
PONDR® VSL286 gives one of the more accurate evaluations of disordered regions in a query protein, whereas PONDR® VLXT is known as the most sensitive predictor for finding disordered based interaction sites87, and a meta-predictor PONDR-FIT as is more accurate than its individual component predictors, such as PONDR® VLS277, PONDR® VLXT78, IUPred, FoldIndex, TopIDP. In our study, we have used PONDR® FIT76, PONDR® VSL277, and PONDR® VLXT78 for disordered analysis in polyprotein of CHIKV. Several reports suggested that IDPs/IDPRs play a central role in various molecular recognition events and in protein-protein interaction networks30,88,89,90,91,92. Some IDPs/IDPRs can undergo at least partial disorder-to-order transitions, when they get involved in interactions with specific binding partners that are needed for recognition, signaling, control, and regulation93,94,95.
References
Thiberville, S. D. et al. Chikungunya fever: Epidemiology, clinical syndrome, pathogenesis and therapy. Antiviral Research, https://doi.org/10.1016/j.antiviral.2013.06.009 (2013).
Mavalankar, D., Shastri, P., Bandyopadhyay, T., Parmar, J. & Ramani, K. V. Increased Mortality Rate Associated with Chikungunya Epidemic, Ahmedabad, India. Emerg. Infect. Dis. 14, 412–415 (2008).
ROBINSON, M. C. An epidemic of virus disease in Southern Province, Tanganyika Territory, in 1952–53. I. Clinical features. Trans. R. Soc. Trop. Med. Hyg. 49, 28–32 (1955).
Powers, A. M. & Logue, C. H. Changing patterns of chikungunya virus: re-emergence of a zoonotic arbovirus. J. Gen. Virol. 88, 2363–2377 (2007).
NIAID Emerging Infectious Diseases/Pathogens | NIH: National Institute of Allergy and Infectious Diseases. Available at: https://www.niaid.nih.gov/research/emerging-infectious-diseases-pathogens (Accessed: 31st October 2017).
Schwartz, O. & Albert, M. L. Biology and pathogenesis of chikungunya virus. Nat. Rev. Microbiol., https://doi.org/10.1038/nrmicro2368 (2010).
Agarwal, A., Sharma, A. K., Sukumaran, D., Parida, M. & Dash, P. K. Two novel epistatic mutations (E1:K211E and E2:V264A) in structural proteins of Chikungunya virus enhance fitness in Aedes aegypti. Virology, https://doi.org/10.1016/j.virol.2016.06.025 (2016).
Tsetsarkin, K. A., Vanlandingham, D. L., McGee, C. E. & Higgs, S. A Single Mutation in Chikungunya Virus Affects Vector Specificity and Epidemic Potential. PLoS Pathog. 3, e201 (2007).
Dong, S. et al. Infection pattern and transmission potential of chikungunya virus in two New World laboratory-adapted Aedes aegypti strains. Sci. Rep. 6, 1–13 (2016).
Khrustalev, V. V., Khrustaleva, T. A., Sharma, N. & Giri, R. Mutational Pressure in Zika. Virus: Local ADAR-Editing Areas Associated with Pauses in Translation and Replication. 7, 1–17 (2017).
Khan, A. H. et al. Printed in Great Britain Complete nucleotide sequence of chikungunya virus and evidence for an internal polyadenylation site. J. Gen. Virol. 83, 3075–3084 (2002).
Jose, J., Snyder, J. E. & Kuhn, R. J. A structural and functional perspective of alphavirus replication and assembly. Future Microbiol. 4, 837–56 (2009).
Li, X.-F. et al. Complete Genome Sequence of a Chikungunya Virus Isolated in Guangdong, China. J. Virol. 86, 8904–8905 (2012).
Sun, S. et al. Structural analyses at pseudo atomic resolution of Chikungunya virus and antibodies show mechanisms of neutralization. elife.elifesciences.org Sun al. eLife 2 (2013).
Bhowmick, A. et al. Finding Our Way in the Dark Proteome. J. Am. Chem. Soc. 138, 9730–9742 (2016).
Perdigão, N. et al. Unexpected features of the dark proteome. Proc. Natl. Acad. Sci. 112, 15898–15903 (2015).
Uversky, V. N. Intrinsically disordered proteins from A to Z. International Journal of Biochemistry and Cell Biology, https://doi.org/10.1016/j.biocel.2011.04.001 (2011).
Camilloni, C. et al. Towards a structural biology of the hydrophobic effect in protein folding. Sci. Rep. 6, 28285 (2016).
Uversky, V. N., Gillespie, J. R. & Fink, A. L. Why are "natively unfolded" proteins unstructured under physiologic conditions? Proteins 41, 415–27 (2000).
van der Lee, R. et al. Classification of Intrinsically Disordered Regions and Proteins. Chem. Rev. 114, 6589–6631 (2014).
Dunker, A. K. & Obradovic, Z. The protein trinity–linking function and disorder. Nat. Biotechnol. 19, 805–806 (2001).
Uversky, V. N. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 11, 739–756 (2002).
Uversky, V. N. A decade and a half of protein intrinsic disorder: Biology still waits for physics. Protein Sci. 22, 693–724 (2013).
Uversky, V. N. Unusual biophysics of intrinsically disordered proteins. Biochim. Biophys. Acta - Proteins Proteomics 1834, 932–951 (2013).
Wright, P. E. & Dyson, H. J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 16, 18–29 (2015).
Gianni, S. et al. Understanding the frustration arising from the competition between function, misfolding, and aggregation in a globular protein. Proc. Natl. Acad. Sci. 111, 14141–14146 (2014).
Gianni, S., Dogan, J. & Jemth, P. Deciphering the mechanisms of binding induced folding at nearly atomic resolution: The Φ value analysis applied to IDPs. Intrinsically Disord. proteins 2, e970900 (2014).
Uversky, V. N. Intrinsically disordered proteins and their (disordered) proteomes in neurodegenerative disorders. Front. Aging Neurosci. 7, 18 (2015).
Babu, M. M. The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochem. Soc. Trans. 44, 1185–1200 (2016).
Oldfield, C. J. et al. Coupled Folding and Binding with alpha-Helix-Forming Molecular Recognition Elements. Biochemistry 44, 12454–12470 (2005).
Cheng, Y. et al. Mining alpha-Helix-Forming Molecular Recognition Features with Cross Species Sequence Alignments. Biochemistry 46, 13468–13477 (2007).
Oldfield, C. J. et al. Flexible nets: disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics 9(Suppl 1), S1 (2008).
Toto, A. et al. Molecular Recognition by Templated Folding of an Intrinsically Disordered Protein 1–9, https://doi.org/10.1038/srep21994 (2016).
Mishra, P. M., Uversky, V. N. & Giri, R. Molecular Recognition Features in Zika Virus Proteome. J. Mol. Biol. https://doi.org/10.1016/j.jmb.2017.10.018 (2017).
Shoemaker, B. A., Portman, J. J. & Wolynes, P. G. Speeding molecular recognition by using the folding funnel: The fly-casting mechanism. Proc. Natl. Acad. Sci. 97, 8868–8873 (2000).
Giri, R., Morrone, A., Toto, A., Brunori, M. & Gianni, S. Structure of the transition state for the binding of c-Myb and KIX highlights an unexpected order for a disordered system. Proc. Natl. Acad. Sci. 110, 14942–14947 (2013).
Fuxreiter, M. et al. Disordered Proteinaceous Machines. Chem. Rev. 114, 6806–6843 (2014).
Kim, D. Y. et al. New World and Old World Alphaviruses Have Evolved to Exploit Different Components of Stress Granules, FXR and G3BP Proteins, for Assembly of Viral Replication Complexes. 1–18, https://doi.org/10.1371/journal.ppat.1005810 (2017).
Cheng, Y. et al. Rational drug design via intrinsically disordered protein. Trends Biotechnol. 24, 435–442 (2006).
Dunker, A. K. & Uversky, V. N. Drugs for ‘protein clouds’: targeting intrinsically disordered transcription factors. Curr. Opin. Pharmacol. 10, 782–788 (2010).
Uversky, V. N. Intrinsically disordered proteins and novel strategies for drug discovery. Expert Opin. Drug Discov. 7, 475–488 (2012).
Hu, G., Wu, Z., Wang, K., Uversky, V. N. & Kurgan, L. Untapped Potential of Disordered Proteins in Current Druggable Human Proteome. Curr. Drug Targets 17, 1198–205 (2016).
Yu, C. et al. Structure-based Inhibitor Design for the Intrinsically Disordered Protein c-Myc. Sci. Rep. 6, 22298 (2016).
Kumar, D., Sharma, N. & Giri, R. Therapeutic Interventions of Cancers Using Intrinsically Disordered Proteins as Drug Targets: c-Myc as Model System, https://doi.org/10.1177/1176935117699408 (2017).
Ringe, D. & Petsko, G. A. Study of protein dynamics by X-ray diffraction. Methods Enzymol. 131, 389–433 (1986).
Malet, H. et al. The Crystal Structures of Chikungunya and Venezuelan Equine Encephalitis Virus nsP3 Macro Domains Define a Conserved Adenosine Binding Pocket. J. Virol. https://doi.org/10.1128/JVI.00189-09 (2009).
Voss, J. E. et al. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 468, 709–712 (2010).
Utt, A. et al. Versatile Trans-Replication Systems for Chikungunya Virus Allow Functional Analysis and Tagging of Every Replicase Protein. PLoS One 11, e0151616 (2016).
Rausalu, K. et al. Chikungunya virus infectivity, RNA replication and non-structural polyprotein processing depend on the nsP2 protease’s active site cysteine residue. Sci. Rep 6, 37124 (2016).
Mounce, B. C. et al. Interferon-Induced Spermidine-Spermine Acetyltransferase and Polyamine Depletion Restrict Zika and Chikungunya Viruses. Cell Host Microbe 20, 167–177 (2016).
Li, C. et al. mRNA Capping by Venezuelan Equine Encephalitis Virus nsP1: Functional Characterization and Implications for Antiviral Research. J. Virol. 89, 8292–8303 (2015).
Delang, L. et al. The viral capping enzyme nsP1: a novel target for the inhibition of chikungunya virus infection. Sci. Rep. 6, 31819 (2016).
Leung, J. Y.-S., Ng, M. M.-L. & Chu, J. J. H. Replication of Alphaviruses: A Review on the Entry Process of Alphaviruses into Cells. Adv. Virol. 2011, 1–9 (2011).
Karpe, Y. A., Aher, P. P. & Lole, K. S. NTPase and 5′-RNA triphosphatase activities of chikungunya virus nsP2 protein. PLoS One 6 (2011).
Nguyen, P. T. V., Yu, H. & Keller, P. A. Journal of Molecular Graphics and Modelling Identification of chikungunya virus nsP2 protease inhibitors using structure-base approaches. 57, 1–8 (2015).
Saisawang, C. et al. Chikungunya nsP2 protease is not a papain-like cysteine protease and the catalytic dyad cysteine is interchangeable with a proximal serine. Sci. Rep. 5, 17125 (2015).
Okeoma, C. M. Chikungunya Virus. https://doi.org/10.1007/978-3-319-42958-8 (2016).
Owen, K. E. & Kuhn, R. J. Identification of a Region in the Sindbis Virus Nucleocapsid Protein That Is Involved in Specificity of RNA Encapsidation. J. Virol. 70, 2757–2763 (1996).
Ten Dam, E., Flint, M. & Ryan, M. D. Printed in Great Britain Virus-encoded proteinases of the Togaviridae. J. Gen. Virol. 80, 1879–1888 (1999).
Rosenthal, F. et al. Macrodomain-containing proteins are new mono-ADP-ribosylhydrolases. Nat. Struct. Mol. Biol. 20, 502–507 (2013).
Shin, G. et al. Structural and functional insights into alphavirus polyprotein processing and pathogenesis. https://doi.org/10.1073/pnas.1210418109.
Gorchakov, R., Garmashova, N., Frolova, E. & Frolov, I. Different Types of nsP3-Containing Protein Complexes in Sindbis Virus-Infected Cells. J. Virol. 82, 10088–10101 (2008).
Panas, M. D., Ahola, T. & McInerney, G. M. The C-terminal repeat domains of nsP3 from the Old World alphaviruses bind directly to G3BP. J. Virol. 88, 5888–93 (2014).
Novoa, I. et al. Stress-induced gene expression requires programmed recovery from translational repression. EMBO J. 22, 1180–1187 (2003).
Tomar, S., Hardy, R. W., Smith, J. L. & Kuhn, R. J. Catalytic Core of Alphavirus Nonstructural Protein nsP4 Possesses Terminal Adenylyltransferase Activity. J. Virol. 80, 9962–9969 (2006).
Koonin, E. V. The phylogeny of RNA-dependent RNA polymerases of positive-strand RNA viruses. J. Gen. Virol. 72, 2197–2206 (1991).
Thomas, S. et al. Chikungunya virus capsid protein contains nuclear import and export signals. Virol. J. https://doi.org/10.1186/1743-422X-10-269 (2013).
Goh, L. Y. H. et al. The chikungunya virus capsid protein contains linear B cell epitopes in the N- and C-terminal regions that are dependent on an intact C-terminus for antibody recognition. Viruses, https://doi.org/10.3390/v7062754 (2015).
Perera, R., Owen, K. E., Tellinghuisen, T. L., Gorbalenya, A. E. & Kuhn, R. J. Alphavirus Nucleocapsid Protein Contains a Putative Coiled Coil -Helix Important for Core Assembly. J. Virol. 75, 1–10 (2001).
Thomas, S. et al. Chikungunya virus capsid protein contains nuclear import and export signals. 1–13 (2013).
Simizu, B., Yamamoto, K., Hashimoto, K. & Ogata, T. Structural Proteins of Chikungunya Virus. J. Virol. 51, 254–258 (1984).
Firth, A. E., Chung, B. Y., Fleeton, M. N. & Atkins, J. F. Discovery of frameshifting in Alphavirus 6K resolves a 20-year enigma. Virol. J. 5, 108 (2008).
Uchime, O., Fields, W. & Kielian, M. The role of E3 in pH protection during alphavirus assembly and exit. J. Virol. 87, 10255–62 (2013).
Metz, S. W. et al. Functional processing and secretion of Chikungunya virus E1 and E2 glycoproteins in insect cells. Virol. J. 8, 353 (2011).
Metz, S. W. et al. Effective Chikungunya Virus-like Particle Vaccine Produced in Insect Cells. PLoS Negl. Trop. Dis. 7, e2124 (2013).
Xue, B., Dunbrack, R. L., Williams, R. W., Dunker, A. K. & Uversky, V. N. PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 1804, 996–1010 (2010).
Obradovic, Z., Peng, K., Vucetic, S., Radivojac, P. & Dunker, A. K. Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins Struct. Funct. Bioinforma. 61, 176–182 (2005).
Romero, P. et al. Sequence complexity of disordered protein. Proteins 42, 38–48 (2001).
Dosztanyi, Z., Csizmok, V., Tompa, P. & Simon, I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 21, 3433–3434 (2005).
Ward, J. J., Sodhi, J. S., McGuffin, L. J., Buxton, B. F. & Jones, D. T. Prediction and Functional Analysis of Native Disorder in Proteins from the Three Kingdoms of Life. J. Mol. Biol. 337, 635–645 (2004).
Linding, R. et al. Protein disorder prediction: implications for structural proteomics. Structure 11, 1453–9 (2003).
Linding, R., Russell, R. B., Neduva, V. & Gibson, T. J. GlobPlot: Exploring protein sequences for globularity and disorder. Nucleic Acids Res. 31, 3701–8 (2003).
Vullo, A., Bortolami, O., Pollastri, G. & Tosatto, S. C. E. Spritz: a server for the prediction of intrinsically disordered regions in protein sequences using kernel machines. Nucleic Acids Res. 34, W164–W168 (2006).
Ferron, F. et al. A practical overview of protein disorder prediction methods. Proteins Struct. Funct. Bioinforma. 65, 1–14 (2006).
Deng, X., Eickholt, J. & Cheng, J. A comprehensive overview of computational protein disorder prediction methods. Mol. Biosyst. 8, 114–21 (2012).
Peng, Z.-L. & Kurgan, L. Comprehensive comparative assessment of in-silico predictors of disordered regions. Curr. Protein Pept. Sci. 13, 6–18 (2012).
Dunker, A. K. et al. Intrinsically disordered protein. J. Mol. Graph. Model. 19, 26–59 (2001).
Dunker, A. K., Brown, C. J. & Obradovic, Z. Identification and functions of usefully disordered proteins. Adv. Protein Chem. 62, 25–49 (2002).
Dunker, A. K., Silman, I., Uversky, V. N. & Sussman, J. L. Function and structure of inherently disordered proteins. Current Opinion in Structural Biology, https://doi.org/10.1016/j.sbi.2008.10.002 (2008).
Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 27, 527–33 (2002).
Uversky, V. N. & Dunker, A. K. Understanding protein non-folding. Biochimica et Biophysica Acta - Proteins and Proteomics, https://doi.org/10.1016/j.bbapap.2010.01.017 (2010).
Uversky, V. N. Intrinsic disorder-based protein interactions and their modulators. Curr. Pharm. Des. 19, 4191–213 (2013).
Iakoucheva, L. M., Brown, C. J., Lawson, J. D., Obradović, Z. & Dunker, A. K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 323, 573–84 (2002).
Dunker, A. K., Brown, C. J., Lawson, J. D., Iakoucheva, L. M. & Obradović, Z. Intrinsic disorder and protein function. Biochemistry 41, 6573–82 (2002).
Toto, A., Giri, R., Brunori, M. & Gianni, S. The mechanism of binding of the KIX domain to the mixed lineage leukemia protein and its allosteric role in the recognition of c-Myb. Protein Sci. 23, 962–969 (2014).
Vasiljeva, L., Valmu, L., Kääriäinen, L. & Merits, A. Site-specific protease activity of the carboxyl-terminal domain of Semliki Forest virus replicase protein nsP2. J. Biol. Chem. 276, 30786–93 (2001).
Thomas, S. et al. Functional dissection of the alphavirus capsid protease: sequence requirements for activity. Virol. J. 7, 327 (2010).
Acknowledgements
Authors acknowledge the partial support of funds from Department of Science and Technology (DST), India (grant number YSS/2015/000613 to RG). RY is supported by the same grant. AS and AK is supported by MHRD fellowship in India.
Author information
Authors and Affiliations
Contributions
R.G. Conception and design, analysis and interpretation of data, writing, and review of the manuscript and study supervision. A.S., A.K. and R.Y generated the data. A.S. and A.K. re-analyzed the data. A.S., R.G. and V.U. writing and manuscript preparation.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Singh, A., Kumar, A., Yadav, R. et al. Deciphering the dark proteome of Chikungunya virus. Sci Rep 8, 5822 (2018). https://doi.org/10.1038/s41598-018-23969-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-23969-0
This article is cited by
-
Identifying and profiling structural similarities between Spike of SARS-CoV-2 and other viral or host proteins with Machaon
Communications Biology (2023)
-
Role of ORF4 in Hepatitis E virus regulation: analysis of intrinsically disordered regions
Journal of Proteins and Proteomics (2021)
-
Understanding COVID-19 via comparative analysis of dark proteomes of SARS-CoV-2, human SARS and bat SARS-like coronaviruses
Cellular and Molecular Life Sciences (2021)
-
The dark side of Alzheimer’s disease: unstructured biology of proteins from the amyloid cascade signaling pathway
Cellular and Molecular Life Sciences (2020)
-
In silico study of chikungunya polymerase, a potential target for inhibitors
VirusDisease (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
