
Abstract
The Chinese white wax scale insect (Ericerus pela) is an economically valuable species with an important role in wax production. Recently, in a greenhouse in Kunming, we identified a genus of fungus that infects and kills E. pela females. This study sought to perform the molecular detection of entomopathogens and analyze the changes in the host microbiota after entomopathogen infection. We used library construction, high-throughput sequencing and real-time quantitative polymerase chain reaction (RT-qPCR) to identify the fungi infecting adult E. pela, to understand the changes in the host organism, and to determine the distribution of the entomopathogens. Cladosporium langeronii and C. sphaerospermum were the main pathogenic species that infected the E. pela females, and they were most prevalent in the dorsal cuticle. In vivo, after infection, the proportion of Cladosporium clearly increased. The infection had little influence on the fungal community but had a strong influence on the bacterial community. After infection, Arsenophonus was dominant, and numerous bacterial genera disappeared. However, Rickettsia, instead of Arsenophonus, became dominant in the Cladosporium-infected individuals that had also been infected with Rickettsia. We identified the species that infected E. pela females and determined the influence of infection on the host microorganisms.
Similar content being viewed by others
Introduction
The scale insects are usually immobile on the host plants. They encounter adverse environmental conditions, predators, bacteria, and fungi. The wax secretion and other scale covers are produced by the scale insects to serve the protective function1. For the wax secretions, surface hydrophobic properties and antimicrobial activity come naturally. Therefore, the secretions produced by some scale insects have economic value and are used in industrial field2. The Chinese white wax scale insect, Ericerus pela Chavannes (Hemiptera: Coccoidae), is an economically valuable insect with an important role in wax production. The white wax secreted by the male second-instar nymph of E. pela is composed of natural esters and has wide applications in the medicine, food, chemical and other industries3,4,5,6,7,8,9. The white wax layer protects E. pela against attack by natural enemies and pathogens. It has evolved associations with microorganisms2. In a previous study, we analyzed the diversity of microorganisms in the white wax layer secreted by E. pela, and found the changes of bacterial and fungal communities in the wax layer with different thickness2. However, the studies of microorganisms in female secretion are lacking. Such studies are needed to better understand the influence of microorganisms in secretions on the host.
In traditional white wax production, the female and male E. pela are both reared to produce the eggs and white wax, respectively. In this process, the growth and development of females are influenced by several negative factors, which may result in decreased oviposition and hatchability. Because of the sedentary life on higher plant, females are susceptible to various pathogens. Phloem-feeding females excreted sugar-reach honeydew during adult stage. The excreted sugar increase sharply, and adhere to the immobile female adults with the development of females. The excreted sugar is prone to be contaminated by pathogens. Contamination of the excreted sugar is a serious threat to the survival of females.
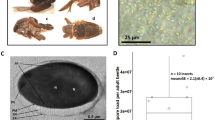
Over the past two years, we found that many females were infected with a green microorganism and died in the greenhouse in Kunming. Through detailed observations of the infected insects, we found that the microorganism parasitized initially on the sugar excreted from the female adult vent during the oviposition period, on the cuticle of E. pela (Fig. 1A). After the excreted sugar dries up, the microorganism surrounds the cuticle (Fig. 1B). Ultimately, the infected insects shrivel and die (Fig. 1C). We also observed that the color of the microorganism changes from olive green to dark green during the drying of the excreted sugar and the degeneration of the female E. pela in the infection process. We observed that the pathogen had spread and infected many E. pela in the greenhouse within one month.

E. pela females at different infection stages. (A) Initial infection stage. The excreted sugar was contaminated and part of the sugar showed brown; however, the color of the female didn’t change. (B) Middle infection stage. The excreted sugar dries up and become olive green or more dark, the color of the female become brown. (C) Last infection stage. The female shrivel and die.
Because the microorganism that we observed harmed the females of E. pela, we inferred that it might be a new type of pathogenic microorganism that kills the host by constantly expanding the regions that it parasitizes from the outside in. In this study, we detected the major microorganism parasitizing on the sugar secreted by female E. pela by 16S and ITS (internal transcribed spacer) library construction and sequencing; then, we used the relative RT-qPCR technique to identify the infected tissues. The microbial communities of the infected females were analyzed by high-throughput sequencing. This study lays a foundation for further investigations of the microorganisms parasitizing E. pela and should provide a scientific basis for the management of microorganism infection during E. pela breeding. In addition, the study may be helpful in the management of other scale insects through use of this microorganism as a biological control.
Results
Library analysis and sequencing
The 16S library
No 16S rDNA genes were amplified from bacterial DNA. The results demonstrated that there were no bacteria in the infected samples of excreted sugar (Figure S1).
The ITS library
The titers of the three ITS libraries were 5.39 × 106, 1.79 × 106 and 2.26 × 106, and the recombination rates were 95.91%, 97.46% and 94.46%. Therefore, the three libraries were suitable for identifying the fungi found on the excreted sugar and the outer cuticle of the females. The BLAST results showed that the 30 sequences were from 6 species of fungi: C. langeronii (10 sequences), C. sphaerospermum (16 sequences), C. cladosporioides (1 sequence), C. sp. MPI.04 (1 sequence), C. sp. NJP05 (1 sequence), and Cryptococcus sp. SJ8L03 (2 sequence).
The results of 16S and ITS MiSeq high-throughput sequencing
Statistical summaries
The read numbers and OTUs of bacteria and fungi in the control checks (CK) and the infection groups (IN) are shown in Table 1. The rarefaction curve and Shannon-Wiener curve showed the number of read samples reached represented saturated conditions (Figure S2).
The average number of bacterial OTUs of IN was 308 less than in CK. The average number of fungal OTUs of IN was 19 more than in CK (Table 1).
Bacterial diversity and community structure
After the female E. pela were infected, the bacterial community richness was clearly reduced, as shown by the significant reduction of the Chao index and Ace index in IN compared with CK. The bacterial community diversity was also markedly reduced, as shown by the increasing Simpson index and significant reduction of the Shannon index in IN compared with CK. The Simpson index was 0.2736 in CK and 0.8648 in IN, which indicated the proportion of dominant species was not high in CK but was high in IN (Table 2).
In total, 25 phyla, and 253 genera were identified. The bacterial community in CK was dominated by the Proteobacteria phyla (68.15%), followed by Firmicutes (7.18%). It was clearly different in IN. The proportion of Proteobacteria was dominant (93.86%) after infection. However, the proportions of six phyla were reduced significantly after infection, and ten other phyla disappeared after infection (Table S1).
A total of 236 genera were identified in CK, whereas only 89 genera were identified in IN. A total of 164 genera disappeared after infection, including Desulfobulbus, Nitrosococcus, and Sh765B-TzT-29. However, 17 genera appeared after infection, the main genus of which was Arsenophonus (Table S1).
One CK sample (CK3) and one IN sample (IN3) was found to have been infected with Rickettsia, which accounted for the vast majority of bacteria (90.22% in CK and 88.06% in IN). To avoid the influence of the Rickettsia bacteria on the analysis, the IN3 and CK3 pair was selected for comparison alone.
In the other two CK groups (CK1 and CK2), we did not find Rickettsia was dominant, as expected, but after infection (IN1 and IN2), Arsenophonus was dominant and several bacterial genera disappeared. The details are as follows:
In the two CK groups, a total of 226 genera were identified, with the highest proportion represented by Rhodospirillaceae_uncultured (5.66%). In the two IN groups, the community was entirely different. There were 181 bacterium genera that disappeared after infection, and only 57 genera were identified, of which Arsenophonus was dominant (95.20%), followed by Bacillus (1.64%), Paenibacillus (1.12%), and others (2.04%).
However, findings in IN3 (has been infected with Rickettsia), were different. The proportion of Arsenophonus was only 0.37% in IN3. A total of 69 genera were identified, and Rickettsia was dominant (88.06%), followed by Bacillus (4.12%) and Paenibacillus (2.27%). In CK3 (has been infected with Rickettsia), only 51 genera were identified, of which Rickettsia was dominant (90.22%), followed by Bacillus (4.10%) and Paenibacillus (2.58%). Aside from the reduced proportion of Rickettsia in IN3, we did not find additional differences between IN3 and CK3.
Fungal diversity and community structure
The fungal community richness increased after the female E. pela were infected, as shown by the increased Chao index and Ace index after infection (IN vs. CK). The fungal community diversity also increased, as shown by the reduction of the Simpson index and the increased Shannon index (Table 2).
A total of 4 phyla, and 69 genera were identified. At the phylum level, we did not find significant differences between CK and IN. However, Zygomycota was absent in CK but present in IN (0.02%). The phylum Ascomycota (93.92%) was dominant in CK. This composition was similar in IN (Table S1).
At the genus level, in CK, Cladosporium was dominant (64.09%), followed by Ophiocordycipitaceae (24.31%), Basidiomycota (5.74%) and Davidiellaceae (2.78%). However, after infection, the proportions of Cladosporium increased to 75.68%, followed by Ophiocordycipitaceae (11.39%), Basidiomycota (5.54%) and Cryptococcus (2.78%). In addition, Davidiellaceae (2.78%) was found in CK but was absent in IN, and Cryptococcus (2.78%) was found in IN but was absent in CK (Table S1).
Because Cladosporium was identified in both the excreted sugar and normal female individuals, we further analyzed the sequence of Cladosporium in detail. The sequences of C. langeronii and C. sphaerospermum from the ITS library were aligned with the sequences of OTUs, which were classified into the Cladosporium genus. The ITS I fragment of C. langeronii had 100% similarity with OTU 73, and the ITS I fragment of C. sphaerospermum had 100% similarity with OTU 23 (Figure S3). The proportion of OTU 23 and OTU 73 in each sample is shown in Fig. 2. However, the proportion of OTU 73 was very low in each sample (Fig. 2).

The proportion of OTU 23 and OTU 73 in the three control checks (CK1, CK2, and CK3) and three infection groups (IN1, IN2, and IN3). The OUT 23 was shown in red color, and the OUT 73 was shown in green color.
C. langeronii and C. sphaerospermum distribution in the female body after infection
From the results of relative RT-qPCR, we found that C. langeronii and C. sphaerospermum were expressed in all the tissues we dissected. However, in different individuals, different tissues had different expression levels. We found that the expression levels of the two genes were highest in the dorsal cuticle, followed by fat body. The lowest expression level was in the ovaries (Figure S4).
Discussion
In this study, we identified the fungi that infected E. pela. ITS library analysis showed that Cladosporium was the major genus that infected E. pela. There were two species: C. langeronii and C. sphaerospermum. Cladosporium belongs to the anamorphic fungi, the Cladosporiaceae. Cladosporium has a worldwide distribution and often parasitizes plants, humans and animals, causing sickness. The spore can be allergenic and can cause subcutaneous phaeohyphomycosis10,11. Many researchers have found that Cladosporium is an effective entomopathogenic fungus against many insect pests12,13. However, Cladosporium had not previously been found to infect scale insects. This finding might provide a new means for controlling scale insect pests.
C. langeronii and C. sphaerospermum showed the highest expression levels in the dorsal cuticle of the adults, a result consistent with the infection process from outside to inside. However, Cladosporium was also found in normal female individuals. It is thought that the pathogen may be suppressed by the insect’s resistance measures, such as cellular immunity and cellular immunity, antimicrobial peptides (AMPs) and symbiotic bacteria14. It can be inferred that, the low content of C. langeronii and C. sphaerospermum cause no harm to the normal individuals (Figure S5). C. langeronii and C. sphaerospermum in the normal female individuals will dormant forever until they meet favorable conditions for expansion. It is similar to the Wolbachia, which is a symbiont normally, can cause early death of Drosophila15. We also identified Cladosporium in the wax layer secreted by male E. pela. Their content decreased from 17.01 to 0.49% with the wax layer thickening2. It seemed that Cladosporium in the wax layer has no influence on the survival of the male E. pela. More specific primers that facilitate more accurate identification will be needed to analyze the distribution of Cladosporium in the female and male E. pela16,17.
After infection, the proportion of OTU 23 increased significantly but the proportion of OTU 73 did not substantially change. The variation in OTU 23 proportion among the three infection samples maybe results from the individual differences during female defense to fungal infection.
On the basis of the observation of infected female E. pela, most of the dead individuals were infected from the outside in. Hence, we speculate that one of the female E. pela died may also because of fungal mechanical force or fungal toxins. In a study of Beauveria bassiana, researchers have also found that the fungus can damage the host Carposina sasakii through mechanical force18. Other researchers have found that fungal toxins can lead to host death19,20. Hence, the death of E. pela females may be caused by these factors.
Arsenophonus is considered to be a secondary endosymbiont in E. pela21. Researchers have found that symbiotic bacteria are helpful in protecting the host against pathogens and play an important role in host physiology and metabolism22,23. In this study, we found that after C. langeronii and C. sphaerospermum infection, female E. pela were more susceptible to infection with Arsenophonus (Fig. 3). C. langeronii and C. sphaerospermum infection may promote Arsenophonus transmission horizontally or infection in E. pela females (Fig. 4). A similar finding has also been reported in Glycaspis brimblecombei larvae24 and Ixodiphagus hookeri25. Thus, we speculate that Arsenophonus infection might be a form of countermeasure against C. langeronii and C. sphaerospermum.

The sketch map that showed the distribution of Cladosporium, Arsenophonus and Rickettsia in the six samples (CK1, CK2, CK3, IN1, IN2, and IN3). The sketch map showed that, Arsenophonus increased after infection (IN1, IN2); however, if the individuals have been infected with Rickettsia, Arsenophonus colonies may be inhibited by Rickettsia.

The relationship among microorganisms in E. pela females and the interaction between females and endosymbiont. The figure showed that, C. langeronii and C. sphaerospermum infection may promote Arsenophonus colonies in E. pela; however, Rickettsia infection can shield against this effects.
Interestingly, we found that Arsenophonus colonies may be inhibited by Rickettsia. The proportion of Arsenophonus was only 0.37% in IN3 (Fig. 3). Thus, after the host is infected with Rickettsia, the promotion effect of C. langeronii and C. sphaerospermum on Arsenophonus infection is negligible. In addition, Rickettsia infection can shield against the effects of C. langeronii and C. sphaerospermum infection of the host (Fig. 4). Rickettsia is also considered to be a secondary endosymbiont in E. pela26. Many researchers have found that Rickettsia can aid in preventing pathogen invasion27,28,29.
Secondary endosymbionts have been shown to help protect the host against parasitoids and fungal entomopathogens and to potentially influence host physiology, evolution and distribution24,30,31. In this study, we identified the lethal fungus infects E. pela and inferred the microorganism interactions. It is useful to identify the insect-endosymbiont interactions and may provide a reference for future studies investigating entomopathogens in E. pela.
Methods
Materials
Insects. E. pela females were collected from Chinese privet (Ligustrum sinense Lour) in the greenhouse of the Research Institute of Resource Insects in Kunming (102.73°E, 25.04°N).
Infected samples of excreted sugar used for microorganism isolation were collected from the infected females’ bodies during the middle infection stage (Fig. 1B) because the biomass of microorganisms from the middle stage is higher than that in the initial stage, and the females in this stage of infection are still alive, as shown in Fig. 1.
Library construction of 16S and ITS
Isolation of microorganism genomic DNA
The microorganism samples (which were dark green and surrounded by the dry excreted sugar, as shown in Fig. 1) were extracted from six different females, three of which were used to isolate fungal DNA (F1, F2, F3) and the other three of which were used to isolate bacterial DNA (B1, B2, B3). Then, the bacterial DNA of the three microorganism samples was isolated separately by using a Bacterial DNA Miniprep Kit (Omega, Atlanta, GA, USA), and the fungal DNA of the other three microorganism samples was isolated separately using the Fungal DNA Miniprep Kit (Omega, Atlanta, GA, USA).
16S and ITS amplification, cloning, and library construction
Three 16S rDNA genes were amplified using the bacterial DNA as a template and the universal primers (27F and 1492R)32. However, when the production was separated on a 1% agarose gel, no amplification bands were visible (Figure S1).
The ITS genes containing the ITS I and ITS II regions were amplified (ITS1 and ITS4; the primer sequences were according to Bokulich et al., 2013)33 by using the fungal DNA as a template. Three biological repeats were used. Then, three ITS rDNA libraries were constructed.
Library analysis and sequencing
After blue-white selection and positive clone identification by using bacterial PCR, the colony-forming units (cfu) and recombination rate were calculated according to Sambrook et al., 201434.
Ten positive recombinant monoclones were selected at random from every library and then sequenced by BioSune (Kunming, China). Then, the sequences were annotated with the NCBI basic local alignment search tool (BLAST) (https://blast.ncbi.nlm.nih.gov/Blast.cgi) under the default parameter values, and the results were selected on the basis of identity thresholds ≥95%.
E. pela female microbial diversity analysis based on 16S and ITS MiSeq high-throughput sequencing
Isolation of the genomic DNA of E. pela females
A total of six females were collected for high-throughput analysis: three individuals were infected with the microorganism (IN1, IN2, IN3), and the other three individuals were control checks (CK1, CK2, CK3). The six samples were crushed into powder separately, as previously described for DNA isolation, with a DNeasy Blood and Tissue Kit (QIAGEN, Hilton, Germany). Then, the DNA was stored at −20 °C for MiSeq sequencing of 16S and ITS.
MiSeq sequencing of 16S and ITS
The genomic DNA from the six females was used separately as a template to amplify 16S rRNA genes or the ITS regions by using primers (338F and 806R for 16S rRNA genes; ITS1F and ITS2R for ITS regions; the primer sequences were according to Xu et al. and Bokulich et al.)21,33 with different barcodes. The 100 μL PCR reaction system contained 500 ng DNA template, 0.2 mM primer, 0.2 mM dNTP mix and 2.5 U of Q5® High-Fidelity DNA Polymerase (NEB, Ipswich, MA, USA). The PCR conditions were 94 °C for 5 minutes, then 25 cycles at 94 °C for 30 seconds, 55 °C for 30 seconds, and 72 °C for 1 minute. The PCR was purified with a Gel Extraction Kit (Thermo, Waltham, MA, USA) and then stored at −20 °C used.
The products were detected and quantified with QuantiFluorTM-ST (Promega, Madison, WI, USA). The loading quantities were calculated to determine the sequencing requirements.
The 16S and ITS MiSeq PE (paired-end) libraries were constructed as follows: First, the products were linked with “Y”-type adapters. Then, the filtered double-stranded DNA segments were enriched via PCR amplification. The products were denatured by sodium hydroxide to produce single-stranded DNA segments. Finally, the single-stranded DNA segments were sequenced by using Illumina MiSeq (Illumina, San Diego, California, USA).
Data analysis
According to the overlap nexus, the PE reads were assembled using FLASH software (Version 3+). The quality of reads and the effect of assembly were simultaneously detected. The detected reads were quality filtered to remove the adapters, unreplicated single sequences and chimeric reads, and effective reads were obtained. Because the six samples resulted in different numbers of reads, 12,476 reads in bacteria 16S and 24,227 reads in fungi ITS were selected at random for each sample to provide the same amount of data for further analysis.
Then, the reads were analyzed by using Usearch software (Version 7.1 http://drive5.com/uparse/) to generate operational taxonomic units (OTUs) at the 97% identity level. On the basis of the OTUs data, the taxonomy and the alpha-diversity were analyzed and calculated.
In the taxonomy analysis, the OTUs were analyzed by using the RDP Classifier (Version 2.2 http://sourceforge.net/projects/rdp-classifier/), and the community composition was analyzed at different taxonomic levels, including domain, kingdom, phylum, class, order, family, genus and species.
In the alpha-diversity analysis, the Chao, Ace, Shannon, Simpson and Coverage indexes were calculated by using Mothur software (Version v.1.30.1 http://www.mothur.org/wiki/Schloss_SOP#Alpha_diversity) on the basis of the OTUs.
The rarefaction curve and Shannon-Wiener curve were drawn by using Mothur software (Version v.1.30.1 http://www.mothur.org/wiki) and R software.
Relative RT-qPCR
DNA isolation in different tissues
The fat body, enteron, cuticle, and ovary were dissected from 3 infected females by using sterile microforceps under an anatomical lens. Each tissue was separately washed for 2 minutes in sterile water, and the tissue was immersed in lysis buffer containing proteinase K (Roche, Mannheim, Germany) and then incubated for 30 minutes in hot water. After a brief centrifugation, the same volume of sterile water was added to the supernatant, mixed gently, and stored at −20 °C for relative RT-qPCR analysis. Each tissue was subjected to 3 replications and used for DNA isolation.
Relative RT-qPCR
According to the results from the ITS library sequencing, specific primers (Figure S6) for C. langeronii and C. sphaerospermum (the fungus infects E. pela identified by the method stated above) were designed. The 10 µL PCR solution with SsoFast EvaGreen Supermix (Bio-Rad, Hercules, CA, USA) was used. The RT-PCR reactions were performed with a CFX96TM real-time system (Bio-Rad, Hercules, CA, USA). The cycling conditions were 98 °C for 3 minutes, then 39 cycles at 98 °C for 10 seconds and 62 °C for 20 seconds. The solubility curves were generated by increasing the annealing temperature at 5 °C/5 seconds from 65 °C to 95 °C. The succinate dehydrogenase subunit A (SdhA) gene and myosin (Myo) gene were selected as two reference genes for relative RT-qPCR. Each PCR run was repeated 3 times.
The results were analyzed using the LSD (least significant difference) method at P < 0.01 level in the Data Processing System (DPS) software35.
References
Gullan, P. J. & Kosztarab, M. Adaptations in Scale Insects. Annu. Rev. Entomol. 42(1), 23 (1997).
Wang, X. Q. et al. Analysis of the diversity of microoraganisma in the wax secreted by the Chinese white wax scale insect, Ericerus pela (Chanvannes) (Homoptera: Coccidae). Acta. Entomol. Sin. 59(10), 1086–1092 (2016).
Yang, P. et al. Transcriptome analysis of the Chinese white wax scale Ericerus pela with focus on genes involved in wax biosynthesis. PLoS. ONE. 7(4), e35719 (2012).
Zhao, J. J., Wang, Z. L., Chen, X. M. & Chen, Y. Sex differences in piercing-sucking sites on leaves of Ligustrum lucidum (Oleaceae) infested by the Chinese white wax scale insect, Ericerus pela (Chavannes) (Hemiptera: Coccidae). Neotrop Entomol. 2, 158–163 (2013).
Liu, W. W., Yang, P., Chen, X. M., Xu, D. L. & Hu, Y. H. Cloning and expression analysis of four heat shock protein genes in Ericerus pela (Homoptera: Coccidae). J. Insect. Sci. 14, 1 (2014).
Yang, P. & Chen, X. M. Protein profiles of Chinese white wax scale, Ericerus pela, at the male pupal stage by high-throughput proteomics. Arch. Insect Biochem. 87, 214–233 (2014).
Yang, P., Chen, X. M., Liu, W. W., Feng, Y. & Sun, T. Transcriptome analysis of sexually dimorphic Chinese white wax scale insects reveals key differences in developmental programs and transcription factor expression. Sci. Rep. 5, 8141 (2015).
Yu, S. H. et al. Identification and evaluation of reference genes in the Chinese white wax scale insect Ericerus pela. Springerplus. 1, 791 (2016).
Yu, S. H. et al. Transcriptomic and proteomic analyses on the supercooling ability and mining of antifreeze proteins of the Chinese white wax scale insect. Insect Sci. 3, 430–437 (2016).
Grinn-Gofroń, A., Strzelczak, A., Stępalska, D. & Myszkowska, D. A 10-year study of Alternaria and Cladosporiumin two Polish cities (Szczecin and Cracow) and relationship with the meteorological parameters. Aerobiologia. 32, 83–94 (2016).
Zhou, Y. B., Chen, P., Sun, T., Wang, X. J. & Li, D. M. Acne-like subcutaneous phaeohyphomycosis caused by Cladosporium cladosporioides: a rare case report and review of published literatures. Mycopathologia. 181, 567–573 (2016).
Eken, C. & Hayat, R. Preliminary evaluation of Cladosporium cladosporioides (Fresen.) de Vries in laboratory conditions, as a potential candidate for biocontrol of tetranychus urticae koch. World J Microb Blot. 25, 489–492 (2009).
Bahar, M. H., Backhouse, D., Gregg, P. C. & Mensah, R. Efficacy of a Cladosporium sp. fungus against Helicoverpa armigera (Lepidoptera: Noctuidae), other insect pests and beneficial insects of cotton. Biocontrol. Sci. Technol. 21, 1387–1397 (2011).
Qu, S. & Wang, S. Interaction of entomopathogenic fungi with the host immune system. Dev. Comp. Immunol. pii: S0145-305X(17)30554-2 (2018).
Min, K. T. & Benzer, S. Wolbachia, normally a symbiont of Drosophila, can be virulent, causing degeneration and early death. Proc. Natl. Acad. Sci. USA 94(20), 10792–10796 (1997).
Sung, G. H. et al. Phylogenetic classification of Cordyceps and the clavicipitaceous fungi. Stud. Mycol. 57, 5–59 (2007).
Divakar, P. K. et al. Using a temporal phylogenetic method to harmonize family- and genus-level classification in the largest clade of lichen-forming fungi. Fungal Divers. 84(12), 1–17 (2017).
Xiong, Q. et al. Morphologicaland ultrastructural characterization of Carposina sasakii, larvae (Lepidoptera: CarposInidae) infected by Beauveria bassiana, (Ascomycota: Hypocreales: Clavicipitaceae). Micron. 44, 303–311 (2013).
Behar, A., Yuval, B. & Jurkevitch, E. Gut bacterial communities in the mediterranean fruit fly (Ceratitis capitata) and their impact on host longevity. J. Insect Physiol. 54, 1377–1383 (2008).
Khan, S. et al. Identification and characterization of an insect toxin protein, Bb70p, from the entomopathogenic fungus, Beauveria bassiana, using Galleria mellonella, as a model system. J. Invertebr. Pathol. 133, 87–94 (2016).
Xu, D. L., Liu, W. W., Hu, Y. H., Yang, P. & Chen, X. M. Analysis of the gut microbes of the Chinese White Wax Scale Ericerus pela and molecular detection of Rickettsia. Forest Research. 25, 780–783 (2012).
Clark, E. L., Karley, A. J. & Hubbard, S. F. Insect endosymbionts: manipulators of insect herbivore trophic interactions? Protoplasma. 244, 25–51 (2010).
Huang, X., Huang, Y., Zhang, J. & Jiang, M. Interactions of Various Microbes in Insects: A Review. Chinese J. Biol. Control. 31, 936–945 (2015).
Hansen, A. K., Jeong, G., Paine, T. D. & Stouthamer, R. Frequency of secondary symbiont infection in an invasive psyllid relates to parasitism pressure on a geographic scale in California. Appl Environ Microbiol. 73, 7531–7535 (2007).
Bohacsova, M., Mediannikov, O., Kazimirova, M., Raoult, D. & Sekeyova, Z. Arsenophonus nasoniae and Rickettsiae Infection of Ixodes ricinus Due to Parasitic Wasp Ixodiphagus hookeri. PLoS ONE. 11, e0149950 (2016).
Liu, W. et al. Molecular detection of symbiotic bacteria Arsenophonus from Ericerus pela Chavannes. Acta. Microbiol. Sinica. 52, 1002–1010 (2012).
Dillon, R. J. & Dillon, V. M. The gut bacteria of insects: nonpathogenic interactions. Annu. Rev. Entomol. 49, 71–92 (2004).
Dillon, R. J., Vennard, C. T. & Charnley, A. K. Diversity of locust gut bacteria protects against pathogen invasion. Ecol. Lett. 8, 1291–1298 (2010).
Hendry, T. A., Hunter, M. S. & Baltrus, D. A. The facultative symbiont Rickettsia protects an invasive whitefly against entomopathogenic Pseudomonas syringae strains. Appl. Environ. Microbiol. 80, 7161–7168 (2014).
Oliver, K. M., Moran, N. A. & Hunter, M. S. Variation in resistance to parasitism in aphids is due to symbionts not host genotype. P. Natl. Acad. Sci.USA 102, 12795–12800 (2005).
Jahan, S. M. H., Lee, K. Y., Howlader, M. I. A., Bashar, H. M. K. & Hasan, G. N. Molecular divergence of secondary endosymbiont, cardinium in Bemisia tabaci (gennadius) and associates. Bangladesh J. Agr. Res. 40, 1 (2015).
Sagaram, U. S. et al. Bacterial diversity analysis of huanglongbing pathogen-infected citrus using phylochips and 16s rdna gene clone library sequencing. Appl. Environ. Microbiol. 75, 1566–1574 (2009).
Bokulich, N. A. & Mills, D. A. Improved selection of internal transcribed spacer-specific primers enables quantitative, ultra-high-throughput profiling of fungal communities. Appl. Environ. Microbiol. 79, 2519–2526 (2013).
Sambrook, J. & Russell, D.W. Molecular cloning: a laboratory manual; VCH: Science Press 2014.
Tang, Q. Y. & Zhang, C. X. Data processing system (dps) software with experimental design, statistical analysis and data mining developed for use in entomological research. Insect Sci. 20, 254–260 (2013).
Acknowledgements
This work was financially supported by the Special Fund for Forestry Research in the Public Interest (201504302, 201304808, 201204602), the National Natural Science Foundation of China (Grant No. 31572337, 31000983), the National High Technology Research and Development Program (‘‘863” Program) of China (2014AA021801), the Applied Basic Research Foundation of Yunnan Province (Grant No. 2013FA052, 2010ZC235), and a RIRI-CAF National Nonprofit Institute Research Grant (Grant No. riricaf200904M-3, riricaf2011006M).
Author information
Authors and Affiliations
Contributions
P.Y. conceived and designed the research. T.S., X.-Q.W., Z.-L.Z. and S.-H.Y. performed the experiments, wrote the manuscript and prepared the supplementary material. X.-M.C. analyzed the data. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sun, T., Wang, XQ., Zhao, ZL. et al. A Lethal Fungus Infects the Chinese White Wax Scale Insect and Causes Dramatic Changes in the Host Microbiota. Sci Rep 8, 5324 (2018). https://doi.org/10.1038/s41598-018-23671-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-23671-1
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
