
Abstract
Individuals with depression differ substantially in their response to treatment with antidepressants. Specific predictors explain only a small proportion of these differences. To meaningfully predict who will respond to which antidepressant, it may be necessary to combine multiple biomarkers and clinical variables. Using statistical learning on common genetic variants and clinical information in a training sample of 280 individuals randomly allocated to 12-week treatment with antidepressants escitalopram or nortriptyline, we derived models to predict remission with each antidepressant drug. We tested the reproducibility of each prediction in a validation set of 150 participants not used in model derivation. An elastic net logistic model based on eleven genetic and six clinical variables predicted remission with escitalopram in the validation dataset with area under the curve 0.77 (95%CI; 0.66-0.88; p = 0.004), explaining approximately 30% of variance in who achieves remission. A model derived from 20 genetic variables predicted remission with nortriptyline in the validation dataset with an area under the curve 0.77 (95%CI; 0.65-0.90; p < 0.001), explaining approximately 36% of variance in who achieves remission. The predictive models were antidepressant drug-specific. Validated drug-specific predictions suggest that a relatively small number of genetic and clinical variables can help select treatment between escitalopram and nortriptyline.
Similar content being viewed by others


Introduction
The reasons why some patients respond well to antidepressant medications but others do not benefit sufficiently from treatment are still poorly understood. Investigations of biologically related individuals from family studies1, non-related individuals from candidate gene studies2 and large-scale genome-wide association studies3,4,5,6,7 identified genetic contributions to treatment outcome. However, few associations with specific genetic variants were replicated and genetic polymorphisms explained only a small fraction of individual differences in antidepressant response. Other factors affecting the response to antidepressant drugs include the severity and type of depressive symptoms, prior exposure to adverse environment, and demographic factors. However, none of these provided differential prediction of alternative treatments outcomes with a clinically meaningful accuracy8,9,10,11,12.
The modest contributions of multiple clinical and genetic predictors suggest that a multivariate approach that combines genetic variants and clinical variables could improve the prediction of antidepressant treatment outcome. An initial application of statistical learning suggested that a combination of multiple clinical variables can improve the prediction over any single factor12. However, it is unknown whether a combination of genetic and clinical variables can improve the prediction of treatment outcomes further. Here, for the first time, we aim to maximise prediction of outcomes of treatment with alternative antidepressants using a combination of genetic, demographic and clinical measurements in patients with major depressive disorder. We report on a statistical learning analysis using more than 500,000 common genetic variants and 139 demographic and clinical variables to optimize the prediction of remission during treatment with a serotonergic or noradrenergic antidepressant.
Results
Prediction of remission during treatment with escitalopram
In the training dataset of escitalopram-treated participants, 17 variables were selected including HRSD total score and item Somatic Symptoms - General, the symptom dimensions of loss of interest-activity and appetite, BDI item sleep, SCAN item fatigability and 11 genetic markers (Tables 1 and 2).
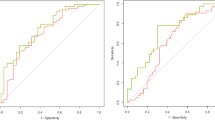
An elastic net logistic model based on these variables predicted remission in the training set with AUC 0.80 (95%CI [0.73–0.88]; p value < 0.001), sensitivity 0.71, specificity 0.77 and pseudo R2 0.37. In external validation, the same model predicted remission in the non-overlapping validation dataset with AUC 0.77 (95%CI [0.66–0.88]; p value = 0.004), sensitivity 0.69, specificity 0.71 and pseudo R2 0.30 (Fig. 1).

Remission prediction accuracy and specificity to antidepressant drug. AUC (Area Under the ROC curve) is shown for models trained and validated in samples treated with the same drug, and for models trained and validated in samples treated with different drug (cross-drug analysis). The horizontal dashed line marks the no discrimination level (AUC of 0.5). Vertical bars indicate a 95% confidence interval of the AUC estimate.
In cross-drug specificity analyses, the escitalopram-derived elastic net model predicted remission in nortriptyline-treated participants at chance level, with AUC 0.57 (95%CI [0.44–0.71]; p value = 0.29), sensitivity 0.46, specificity 0.67 and pseudo R2 0.03, suggesting that prediction is drug-specific (Fig. 1).
Prediction of remission during treatment with nortriptyline
In the training dataset of nortriptyline-treated participants, 20 variables were selected, all of them genetic variants (Tables 1 and 2). The elastic net logistic regression model derived from these 20 genetic variables predicted remission in the training set with AUC 0.83 (95%CI [0.76–0.91]; p value 0.003), sensitivity 0.7, specificity 0.83 and pseudo R2 0.36. The model predicted remission in the non-overlapping validation dataset of nortriptyline-treated participants with an AUC 0.77 (95%CI [0.65–0.90]; p value < 0.001), sensitivity 0.68, specificity 0.87 and a pseudo R2 0.36 (Fig. 1).
In cross-drug specificity analyses, the nortriptyline-derived elastic net model predicted remission in escitalopram-treated participants at chance level, with AUC 0.62 (95%CI [0.50–0.75]; p value = 0.062), sensitivity 0.29, specificity 0.52 and pseudo R2 0.04, suggesting that prediction is drug-specific (Fig. 1).
Discussion
The present results show that a combination of relatively few genetic and clinical variables can predict whether an individual with depression may reach remission with a specific antidepressant. The prediction models are parsimonious, based on only 17 and 20 variables, and the predictions are reproducible in non-overlapping validation datasets. These results demonstrate that a combination of genomic and clinical information in statistical learning framework has the potential to serve as a clinical decision support tool that may help select an antidepressant that an individual is more likely to benefit from.
The prediction was largely antidepressant-specific. The models predicted remission in validation sample treated with the same antidepressant, but not in samples treated with the other antidepressant. The drug-specificity makes the multivariate prediction more useful and applicable to clinical decision making. While the prediction of remission with escitalopram was driven by a combination of clinical and genetic variables, the achievement of remission with nortriptyline was predicted from genetic variants only. The clinical variables that contributed to the prediction of remission with escitalopram overlapped with previously reported predictors. Our model suggested that patients who had low levels of interest and activity, sleep problems, somatic symptoms and severe depression were less likely to reach remission, reflecting previously identified associations with symptom profiles10,12,13. For the prediction of response to nortriptyline, the procedure selected only genetic variables. The selection of only genetic variables in the nortriptyline-treated group suggests that the information predictive of nortriptyline response was better captured by genetic variables than the information predictive of response to escitalopram. The genetic variants selected into the prediction models were distinct from those identified in univariate genome-wide association studies3,4,5,6,7. For example, the genetic variants that predicted remission with nortriptyline in the multivariate model did not include the variant rs2500535 in UST that was previously identified as significantly associated with response to this antidepressant in the same dataset7. These results demonstrate that a statistical learning framework uses a multidimensional pool of predictors in a way that is partially distinct from traditional univariate approaches and has the potential to build novel prediction models that are relevant to clinical outcomes and robust in generalisation.
It is widely accepted that multiple genes/alleles are involved in determining response to antidepressants, some of which may not have been yet discovered. Interestingly, some of the genes containing variants that we reported as predictive of antidepressant treatment response have been recently identified as depression risk genes, as well as associated with bipolar disorder, schizophrenia and other brain diseases (Tables 1 and 2). For example, the SGCZ gene, part of the sarcoglycan complex, a group of six proteins which bridge the inner cytoskeleton and the extra-cellular matrix, has been recently reported to be associated with major depression, schizophrenia and bipolar disorder14, as well as with alcohol and nicotine co-dependence15, and Parkinson’s disease16. The consistent down-regulation in major depression patients in three independent samples suggested that SCL25A37 may be used as a potential biomarker for major depression diagnosis17. This gene was also associated with fatigue18. The acid sensing ion channel (ACCN1) has been associated with response to lithium treatment in bipolar disorder19 and also associated with risk of autism20. The gene encoding the transmembrane protein 229 b has been associated with risk for Parkinson disease21 and with childhood obesity22. The gene TMEM170A encoding the transmembrane protein 170 A and the CFDP1, the craniofacial development protein 1, have been both associated with coronary risk disease23. The latter has been also associated with lung function24. Another variant identified in this work was located in the transmembrane protein 2 gene TMEM2, which has an essential role in coordination of myocardial and endocardial morphogenesis25. None of the selected genetic variations were located in genes previously associated with pharmacogenetics in depression treatment. However, it is a common finding in genomics that most predictive genetic variants are in locations other than the predicted candidate genes. This is responsible for the general failure of the candidate gene approach and it opens new ways for understanding pathogenesis and pharmacology. Surprising findings from genomic research in other disorders have open new ways of understanding and treating the disorders (e.g. the involvement of complement in macular degeneration, schizophrenia was previously unsuspected). Further functional characterization may provide potential targets for future therapeutic antidepressants.
The prediction was accurate enough to be clinically meaningful. Remission was predicted in validation data with an AUC of 0.77 in the escitalopram group and 0.77 in the nortriptyline group. Following the classification proposed by Hosmer & Lemeshow26 our models had “acceptable discrimination” (values of AUC of 0.7 or higher). The utility of biomarkers and prediction models in practice does not depend solely on their prediction accuracy, as reflected by the AUC, but also on clinical context, gravity of the predicted outcomes, cost and burden of the test. For example, a comparison among breast cancer prediction algorithms reported good performance for models having AUC’s below 0.727. The fact that genetic and clinical variables used in the present model can be obtained with high accuracy and low-cost measurements that do not burden participants suggest that such models may be useful in practice.
Most of our previous work reporting on GENDEP applied analytical methods from the traditional inferential statistical framework, based on the assessment of association of a single clinical or genetic variant with treatment response in any given test. Association analysis aims to test the effects of specific factors on the response. This approach will highlight the predictive variable that has the strongest relationship with outcome on its own. In contrast, our current report aims to achieve an optimized prediction of outcome with the use of all available predictor variables, thus following a substantially different aim. Statistical learning can be used to build a model that will predict treatment outcome for new (unseen) cases, with clinical utility in practice. While explanatory power provides information about the strength of an underlying causal relationship, it does not imply its predictive power. By capturing underlying complex patterns and relationships, predictive modeling can suggest improvements to existing explanatory models28.
GENDEP has several strengths that make it suitable for prediction modeling. It is a randomised controlled trial that allows optimal comparison between treatments and the development of treatment-specific predictors29,30. The longitudinal study design of GENDEP allowed the follow-up of patients and the prospective assessment of symptom change, this being the most appropriate approach to establish cause-effect relations and avoid inconsistencies in data collection. The study was specifically designed to assess remission as the primary outcome, with patients being followed for 12 weeks. All patients had four or more depression severity measurements, with more than eighty percent of the sample having eight or more depression measurements, enough time to observe a clinical trend that could lead to clinical remission. However, interpretation of the present results has to take into account several limitations. First, while a wealth of information was available in the GENDEP dataset, not all relevant predictors were measured. For example, history of maltreatment in childhood has been shown to predict outcome of treatment with antidepressants31, but information on childhood maltreatment is not available in GENDEP. Second, since GENDEP only included individuals of white European ancestry without family history of bipolar disorder, the results may not generalize to individuals of other ethnicities or those with family history of bipolar disorder. Third, GENDEP only included two antidepressant drugs distinct in their mechanisms of action. Similar prediction of outcomes with other antidepressants, with neurostimulation and psychological treatments will require investigation in large and richly assessed samples of individuals treated with different modalities. Fourth, the GENDEP study was used as an exploratory dataset to build and test the predictive models. The clinical application of these models will require a comparison of outcomes between individuals whose treatment is selected according to a prediction model with those whose treatment is selected by chance or according to the judgement of the treating physician.
In conclusion, the present results demonstrate that a combination of a relatively small number of clinical and genetic variables can meaningfully and robustly predict remission with escitalopram and nortriptyline antidepressants among individuals with major depressive disorder. Statistical learning methods may be used to derive similar models for individuals treated with various antidepressants and other treatment modalities to map the opportunities for individualized indications for treatments.
The models are available online at https://gist.github.com/raqini/669c38a6329aa2231268770200519d64.
Methods
Participants
We investigated treatment outcomes in 430 adults with major depressive disorder who were randomly allocated to receive either escitalopram, a selective serotonin reuptake inhibitor (SRI), or nortriptyline, a second-generation tricyclic antidepressant (TCA) that acts primarily as a norepinephrine reuptake inhibitor, and completed at least 4 weeks of treatment with the allocated antidepressant as part of the Genome-based Therapeutic Drugs for Depression (GENDEP)7,32. The two antidepressants were selected as representatives of different classes of antidepressants (SRI and TCA) that differ in their pharmacodynamics (serotonergic vs. primarily noradrenergic reuptake inhibition) and pharmacokinetics (distinct primary metabolizing enzymes). Genetic data for GENDEP participants were obtained in two phases. Firstly, 706 individuals were genotyped7. In a second phase, 105 more individuals were genotyped building a total sample of 811 individuals that were partially randomized to escitalopram and nortriptyline. Since our hypotheses concerned differential prediction and participants non-randomly allocated differed on some clinical characteristics2, we restricted the present analyses to the randomly allocated participants (n = 430). Randomisation has been shown to be crucial to avoid systematic confounding effect that might prevent predictive models from properly generalizing to other samples33. The participants were recruited from nine European centers and diagnosed with ICD-10/DSM-IV current depressive episode of at least moderate severity with the Schedules for Clinical Assessment in Neuropsychiatry (SCAN) interview34. Because of the genetic character of the study, the recruitment was restricted to individuals of white European parentage. Patients with personal or family history of bipolar disorder or schizophrenia and those with current substance dependence were excluded. They were treated for 12 weeks according to a protocol that guided dose adjustments according to response and tolerability, with 10 to 30 mg of escitalopram or 50 to 200 mg of nortriptyline daily. We randomly separated the participants into a training sample (65% of participants, a total of 280 patients) and a validation sample (the remaining 35%, a total of 150 patients) (Fig. 2) according to optimal percentage of split recommended to minimise predictive error35. The research ethic boards in all nine centers approved the study protocol. The ethics committee/institutional review board that approved GENDEP study in the lead center, King’s College London, was the Joint South London and Maudsley and the Institute of Psychiatry NHS Research Ethics Committee formed by Dr M Philpot (Co-Chair), Dr T Eaton (Co-Chair), Dr J Bearn, Professor T Craig, Professor A Farmer, Dr N Fear, Mr R Maddox, Mrs J Bostock, Dr V Kumari, Dr M Leese, Dr V Mouratoglou, Professor Sir Michael Rutter, Mr G Smith, Dr D Taylor, Dr U Ettinger, Mr J Watkins, Dr V Ng, Dr D Freeman and Dr T Joyce. All participants signed a written informed consent. All experiments were performed in accordance with relevant guidelines and regulations. The GENDEP study was registered at ISRCTN03693000 (www.controlled-trials.com) on 27th September 2007. Participant characteristics are described in Supplementary Table S2.

Flow diagram of sample division and analytic procedure used in variable selection, model derivation and validation.
Outcome
The outcome was remission, defined as scoring 7 points or less on the 17-item Hamilton Rating Scale for Depression (HRSD)36 at the last available measurement after 4–12 weeks of treatment.
Demographic and clinical predictors
All predictors were obtained at baseline, before participants received any study medication. Severity of depressive symptoms was assessed using three scales: the clinician-rated Montgomery–Åsberg Depression Rating Scale (MADRS)37, HRSD36 and the Beck Depression Inventory (BDI)38. Study interviewers collected information on gender, age, age at depression onset, body mass index (BMI), smoking (yes or not), years of education, marital status, occupation, and children (yes or not). The number of stressful life events in the 6 months previous to the interview was reported with the Brief List of threatening Events questionnaire (BLEQ)39. Medication information was recorded including the use of antidepressant at the time of recruitment, number of prior antidepressant trials and the types of antidepressants tried (SRI, tricyclic, dual, monoamine oxidase inhibitor or other antidepressants). Missing data were imputed by a bagged tree nonparametric method that allows inclusion of all cases without causing bias under a broad range of assumptions about missing data mechanisms40. Categorical data were rounded to plausible values after imputation41. In total, we included 139 clinical and demographic predictors (see Supplementary Table S1).
Genotyping
DNA was extracted from blood samples collected in ethylenediaminetetraacetic acid42 and genotyped using the Illumina Human610-quad bead chip (Illumina, Inc., San Diego). This chip assays more than 610,000 single nucleotide polymorphisms (SNPs) and copy number variant markers selected to provide a comprehensive coverage across populations, and captures the majority of known common variation in the human genome, based on HapMap (release 23). Of the 550,337 SNPs with a minor allele frequency >0.01, a total of 539,391 (98%) were at least 99% complete and retained for analyses. The 430 participants presented no sex mismatches, no ambiguous genotypic sex and no outliers on heterozygosity. One individual in each of six pairs of related individuals (three first- and three second-degree pairs of relatives) was retained for further analyses. No population structure outliers were detected. The 430 individuals had a mean genotyping completeness of 99.82%. Using the IMPUTE v2 program43, we imputed missing SNPs data up to the 1000genomes (build 37). Quality control procedures and imputation are described in detail in Supplementary materials. Variants showing linkage disequilibrium (LD) over 0.8 were excluded from analysis. A total of 524871 common genetic variants were analysed.
Data modeling
We randomly split the participants into mutually exclusive training dataset (65% of participants) and validation dataset (the remaining 35%; Fig. 2), a ratio that is optimal to minimise prediction error across a plausible range of achievable full dataset accuracy between 60% and 99%35. Within the training data set we performed 5-fold cross-validations to select informative variables and derive a statistical learning model to predict remission separately for escitalopram and for nortriptyline. The two resulting models (one for escitalopram and one for nortriptyline) were then externally validated in the validation dataset, a set of participants treated with the same drug that was not used in any way in the model derivation (Fig. 2). In addition, we probed drug-specificity of prediction by testing each predictive model in the validation dataset treated with the other drug. An additional analysis of the whole dataset of patients treated either with escitalopram or nortriptyline is reported in Supplementary materials.
Variable selection in training data
In training data, we performed variable selection in 20 repetitions of a 5-fold cross-validation, 100 rounds in total. In each round, we left out one fifth of the training dataset and, in the remaining four-fifths of the training dataset, we estimated a Correlation-Adjusted T (CAT) score (i.e. a multivariate generalization of the standard univariate T-test statistic that takes the correlation among variables explicitly into account44,45 and the Local False Discovery Rate (LFDR) (i.e. the probability of a variable to be non-informative with regard to remission prediction given its CAT score) for each potential predictor. We retained predictors that had a LFDR smaller than 0.8 more times than not across the 100 rounds.
Models development in training data
We used this set of variables to develop an elastic net logistic regression model in the training data set46. Elastic net model is a modified regression that allows to build multivariate models efficiently incorporating the correlation structure into the predictive accuracy calculation, whilst preventing the models from overfitting47. Parameters for the elastic net model need to be empirically determined. Following a procedure that optimizes the stability of results48, we carried out a 5-fold cross-validation with 100 repetitions to derive the parameters of a final predictive model.
External validation of the models
For each antidepressant drug, we validated the final predictive model in the validation data set, an independent non-overlapping set of participants not used in any way in models derivation. We externally validated the prediction robustness and accuracy in the validation dataset of participants treated with the same drug. In addition, we evaluated drug-specificity of prediction by comparing same-drug (training and validation datasets treated with the same drug) with a cross-drug analysis (training and validation datasets treated with a different drug).
Quantification of prediction accuracy
We indexed the accuracy of prediction with the Area Under the Curve (AUC) of a Receiver Operating Curve (ROC), sensitivity, specificity and Nagelkerke pseudo R2 coefficient. AUC49 can be interpreted as the probability that a classifier can identify (discriminate) a remitter when a remitter and a non-remitter cases are selected at random. The maximum value for the AUC is 1.0, thereby indicating a (theoretically) perfect discrimination (i.e., 100% sensitive, and 100% specific). An AUC value of 0.5 indicates no discriminative value (i.e., 50% sensitive and 50% specific). The Nagelkerke pseudo R2 approximates the proportion of outcome variance explained by the model.
Statistical software used for analysis
We used caret50, sda44,45, glmnet51 and pROC52 libraries from R 3.2.3 statistical software53.
Data availability statement
The data that support the findings of this study are available from the corresponding author on reasonable request. Data were used under license for the current study, and so are not publicly available.
References
Franchini, L., Serretti, A., Gasperini, M. & Smeraldi, E. Familial concordance of fluvoxamine response as a tool for differentiating mood disorder pedigrees. Journal of psychiatric research 32, 255–259 (1998).
Uher, R. et al. Genetic predictors of response to antidepressants in the GENDEP project. Pharmacogenomics J 9, 225–233 (2009).
Biernacka, J. M. et al. The International SSRI Pharmacogenomics Consortium (ISPC): a genome-wide association study of antidepressant treatment response. Translational psychiatry 5, e553 (2015).
Garriock, H. A. et al. A genomewide association study of citalopram response in major depressive disorder. Biological psychiatry 67, 133–138 (2010).
Gendep Investigators, Mars. Investigators. & Investigators, S. D. Common genetic variation and antidepressant efficacy in major depressive disorder: a meta-analysis of three genome-wide pharmacogenetic studies. The American journal of psychiatry 170, 207–217 (2013).
Tansey, K. E. et al. Contribution of common genetic variants to antidepressant response. Biological psychiatry 73, 679–682 (2013).
Uher, R. et al. Genome-wide pharmacogenetics of antidepressant response in the GENDEP project. The American journal of psychiatry 167, 555–564 (2010).
Novick, D. et al. Predictors of remission in the treatment of major depressive disorder: real-world evidence from a 6-month prospective observational study. Neuropsychiatric disease and treatment 11, 197–205 (2015).
Rush, A. J. et al. Report by the ACNP Task Force on response and remission in major depressive disorder. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology 31, 1841–1853 (2006).
Uher, R. et al. Depression symptom dimensions as predictors of antidepressant treatment outcome: replicable evidence for interest-activity symptoms. Psychological medicine 42, 967–980 (2012).
Keers, R. et al. Stressful life events, cognitive symptoms of depression and response to antidepressants in GENDEP. Journal of affective disorders 127, 337–342 (2010).
Iniesta, R. et al. Combining clinical variables to optimize prediction of antidepressant treatment outcomes. Journal of psychiatric research 78, 94–102 (2016).
Madhoo, M. & Levine, S. Z. Initial Severity Effects on Residual Symptoms in Response and Remission: A STAR*D Study During and After Failed Citalopram Treatment. Journal of clinical psychopharmacology 35, 450–453 (2015).
Chen, X. et al. A Novel Relationship for Schizophrenia, Bipolar, and Major Depressive Disorder. Part 8: a Hint from Chromosome 8 High Density Association Screen. Mol Neurobiol (2016).
Zuo, L. et al. Genome-wide search for replicable risk gene regions in alcohol and nicotine co-dependence. American journal of medical genetics. Part B, Neuropsychiatric genetics: the official publication of the International Society of Psychiatric Genetics 159B, 437–444 (2012).
Liu, X. et al. Increased Rate of Sporadic and Recurrent Rare Genic Copy Number Variants in Parkinson’s Disease Among Ashkenazi Jews. Mol Genet Genomic Med 1, 142–154 (2013).
Huo, Y. X. et al. Identification of SLC25A37 as a major depressive disorder risk gene. Journal of psychiatric research 83, 168–175 (2016).
Hsiao, C. P., Wang, D., Kaushal, A., Chen, M. K. & Saligan, L. Differential expression of genes related to mitochondrial biogenesis and bioenergetics in fatigued prostate cancer men receiving external beam radiation therapy. Journal of pain and symptom management 48, 1080–1090 (2014).
Squassina, A. et al. Evidence for association of an ACCN1 gene variant with response to lithium treatment in Sardinian patients with bipolar disorder. Pharmacogenomics 12, 1559–1569 (2011).
Stone, J. L., Merriman, B., Cantor, R. M., Geschwind, D. H. & Nelson, S. F. High density SNP association study of a major autism linkage region on chromosome 17. Human molecular genetics 16, 704–715 (2007).
Nalls, M. A. et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nature genetics 46, 989–993 (2014).
Comuzzie, A. G. et al. Novel genetic loci identified for the pathophysiology of childhood obesity in the Hispanic population. PLoS One 7, e51954 (2012).
Gertow, K. et al. Identification of the BCAR1-CFDP1-TMEM170A locus as a determinant of carotid intima-media thickness and coronary artery disease risk. Circulation. Cardiovascular genetics 5, 656–665 (2012).
Soler Artigas, M. et al. Genome-wide association and large-scale follow up identifies 16 new loci influencing lung function. Nature genetics 43, 1082–1090 (2011).
Totong, R. et al. The novel transmembrane protein Tmem2 is essential for coordination of myocardial and endocardial morphogenesis. Development 138, 4199–4205 (2011).
Hosmer, D. W., Lemeshow, S. & Sturdivant, R. X. Applied logistic regression. 3rd ed. (Wiley, 2013).
Anothaisintawee, T., Teerawattananon, Y., Wiratkapun, C., Kasamesup, V. & Thakkinstian, A. Risk prediction models of breast cancer: a systematic review of model performances. Breast cancer research and treatment 133, 1–10 (2012).
Shmueli, G. To Explain or to Predict? Statist. Sci. 25, 289–310 (2010).
Pourhoseingholi, M. A., Baghestani, A. R. & Vahedi, M. How to control confounding effects by statistical analysis. Gastroenterology and hepatology from bed to bench 5, 79–83 (2012).
Matsui, S., Buyse, M. & Simon, R. Design and Analysis of Clinical Trials for Predictive Medicine (2015).
Nanni, V., Uher, R. & Danese, A. Childhood maltreatment predicts unfavorable course of illness and treatment outcome in depression: a meta-analysis. The American journal of psychiatry 169, 141–151 (2012).
Uher, R. et al. Differential efficacy of escitalopram and nortriptyline on dimensional measures of depression. Br J Psychiatry 194, 252–259 (2009).
Perlis, R. H. Use of large data sets and the future of personalized treatment. Depression and anxiety 31, 916–919 (2014).
Wing, J. K. et al. SCAN. Schedules for Clinical Assessment in Neuropsychiatry. Archives of general psychiatry 47, 589–593 (1990).
Dobbin, K. K. & Simon, R. M. Optimally splitting cases for training and testing high dimensional classifiers. BMC Med Genomics 4, 31 (2011).
Hamilton, M. Development of a rating scale for primary depressive illness. Br J Soc Clin Psychol 6, 278–296 (1967).
Montgomery, S. A. & Asberg, M. A new depression scale designed to be sensitive to change. Br J Psychiatry 134, 382–389 (1979).
Beck, A. T., Ward, C. H., Mendelson, M., Mock, J. & Erbaugh, J. An inventory for measuring depression. Archives of general psychiatry 4, 561–571 (1961).
Brugha, T., Bebbington, P., Tennant, C. & Hurry, J. The List of Threatening Experiences: a subset of 12 life event categories with considerable long-term contextual threat. Psychological medicine 15, 189–194 (1985).
Grzymala-Busse, J. W. & Hu, M. A Comparison of Several Approaches to Missing Attribute Values in Data Mining. In International conference on Rough Sets and Current Trends in Computing 375–385, (Springer-Verlag London, 2001).
Schafer, J. L. Analysis of incomplete multivariate data. (Chapman & Hall, 1997).
Freeman, B. et al. DNA from buccal swabs recruited by mail: evaluation of storage effects on long-term stability and suitability for multiplex polymerase chain reaction genotyping. Behavior genetics 33, 67–72 (2003).
Howie, B., Fuchsberger, C., Stephens, M., Marchini, J. & Abecasis, G. R. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nature genetics 44, 955–959 (2012).
Zuber, V. & Strimmer, K. Gene ranking and biomarker discovery under correlation. Bioinformatics 25, 2700–2707 (2009).
Zuber, V. & Strimmer, K. High-dimensional regression and variable selection using CAR scores. Statistical Applications in Genetics and Molecular Biology 10 (2011).
Zou, Ha. H. T. Regularization and Variable Selection via the Elastic Net. Journal of the Royal Statistics Society, Series B 67, 301–320 (2005).
Bühlmann, P. & Geer, S. A. v. d. Statistics for high-dimensional data. (Springer, 2011).
Kim, J. H. Estimating classification error rate: Repeated cross-validation, repeated hold-out and bootstrap. Computational Statistics & Data Analysis 53, 3735–3745 (2009).
Fan, J., Upadhye, S. & Worster, A. Understanding receiver operating characteristic (ROC) curves. Cjem 8, 19–20 (2006).
Kuhn, M. a. J., K. Applied Predictive Modeling. (Springer, 2013).
Friedman, J., Hastie, T. & Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J Stat Softw 33, 1–22 (2010).
Robin, X. et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC bioinformatics 12, 77 (2011).
R: A Language and Environment for Statistical Computing. (Vienna, Austria, 2008).
Acknowledgements
We acknowledge Lundbeck for providing the medications free of charge to the study. We acknowledge the contributions of Andrej Marusic and Jorge Perez, who were the lead investigators at Ljubljana, Slovenia and at Brescia, Italy, and who passed away during the conduct of the study.
Author information
Authors and Affiliations
Contributions
R.U. and P.M.G. conceived and designed the work. R.U., K.M., W.M., M.R., O.M., J.H., N.H., M.Z.D., D.S. and K.J.A. collected the data. R.I. and R.U. performed data analysis. R.I., K.H., D.S., K.M., W.M., M.R., O.M., J.H., N.H., M.Z.D., D.S., R.D., K.J.A., A.F., P.M.G., C.M.L. and R.U. interpreted results. R.I. and R.U. drafted the article and got critical revision from all authors. All authors read and approved the final manuscript to be published.
Corresponding author
Ethics declarations
Competing Interests
R.I., K.H., P.M.c.G., A.F., W.M., D.S., M.R., M.Z.D., D.S., J.H., O.M., R.D. and K.M. have no conflicts of interest. C.M.L., R.U. and K.J.A. report grants from European Commission, during the conduct of the study. N.H. reports grant from European Commission (through Institute of Psychiatry, King’s College, London), and participation in clinical trials sponsored by pharmaceutical companies, including Lundbeck outside the submitted work. K.J.A. was previously (more than 48 months ago) a member of various advisory boards, receiving consultancy fees and honoraria (including from Lundbeck), and has received research grants from various companies including Johnson and Johnson Pharmaceuticals Research and Development and Bristol-Myers Squibb Pharmaceuticals Limited. She has also received consultancy fees and research support from Roche Diagnostics and Roche Molecular Systems She currently holds an Alberta Centennial Addiction and Mental Health Research Chair, funded by the Government of Alberta.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Iniesta, R., Hodgson, K., Stahl, D. et al. Antidepressant drug-specific prediction of depression treatment outcomes from genetic and clinical variables. Sci Rep 8, 5530 (2018). https://doi.org/10.1038/s41598-018-23584-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-23584-z
This article is cited by
-
The Burnout PRedictiOn Using Wearable aNd ArtIficial IntelligEnce (BROWNIE) study: a decentralized digital health protocol to predict burnout in registered nurses
BMC Nursing (2024)
-
The Precision in Psychiatry (PIP) study: Testing an internet-based methodology for accelerating research in treatment prediction and personalisation
BMC Psychiatry (2023)
-
Deep phenotyping towards precision psychiatry of first-episode depression — the Brain Drugs-Depression cohort
BMC Psychiatry (2023)
-
Predicting treatment outcome in depression: an introduction into current concepts and challenges
European Archives of Psychiatry and Clinical Neuroscience (2023)
-
Creating sparser prediction models of treatment outcome in depression: a proof-of-concept study using simultaneous feature selection and hyperparameter tuning
BMC Medical Informatics and Decision Making (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
