
Abstract
Transient receptor potential vanilloid 4 (TRPV4) cation channels are functional in all renal vascular segments and mediate endothelium-dependent vasorelaxation. Moreover, they are expressed in distinct parts of the tubular system and activated by cell swelling. Ischaemia/reperfusion injury (IRI) is characterized by tubular injury and endothelial dysfunction. Therefore, we hypothesised a putative organ protective role of TRPV4 in acute renal IRI. IRI was induced in TRPV4 deficient (Trpv4 KO) and wild–type (WT) control mice by clipping the left renal pedicle after right–sided nephrectomy. Serum creatinine level was higher in Trpv4 KO mice 6 and 24 hours after ischaemia compared to WT mice. Detailed histological analysis revealed that IRI caused aggravated renal tubular damage in Trpv4 KO mice, especially in the renal cortex. Immunohistological and functional assessment confirmed TRPV4 expression in proximal tubular cells. Furthermore, the tubular damage could be attributed to enhanced necrosis rather than apoptosis. Surprisingly, the percentage of infiltrating granulocytes and macrophages were comparable in IRI–damaged kidneys of Trpv4 KO and WT mice. The present results suggest a renoprotective role of TRPV4 during acute renal IRI. Further studies using cell–specific TRPV4 deficient mice are needed to clarify cellular mechanisms of TRPV4 in IRI.
Similar content being viewed by others

Introduction
Transient receptor potential (TRP) channels were initially identified in a mutant strain of Drosophila melanogaster as a novel component required in phototransduction1. Subsequently, they were identified in vertebrates and found to be ubiquitously expressed in many cells and tissues. Currently the mammalian TRP channel family comprises 28 members which share some structural similarity with each other2. Mutated TRP genes are causal for human hereditary diseases, the so–called ‘TRP channelopathies’3, for some TRP genes.
TRP vanilloid 4 (TRPV4) [initially named vanilloid receptor–related osmotically activated ion channel (VROAC), vanilloid receptor–like channel 2 (VRL–2), TRP protein 12 (TRP12), and osmosensory protein 9–like transient receptor potential channel, member 4 (OTRPC4)] represents the fourth member of the TRPV subfamily, and was originally identified as a mammalian homolog of the Caenorhabditis elegans OSM–93,4,5,6. TRPV4 is a polymodal, non–selective cation channel, which was initially demonstrated to be activated by hypotonicity–induced cell swelling7,8. Later studies revealed that it can also be activated by various other stimuli including mechanical stimulation6,9, moderate heat, endogenous chemicals such as anandamide, arachidonic acid and its epoxyeicosatrienoic acid metabolites, as well as by a number of exogenous chemical ligands10,11,12 or UVB13,14. Human TRPV4 mutations are known to cause skeletal dystrophic disorders of varying severity, and neurological conditions, spinal muscular atrophies and the predominantly motor-neuropathy, Charcot-Marie Tooth Disease, type 2C12.
Expression of TRPV4 in mammals is wide-spread. It is expressed in the central and peripheral nervous system15, noticeably in tissues involved in volume regulation and osmosensing such as the circumventricular organs, the vascular endothelium and the kidney12,16,17. In an earlier study expression of TRPV4 in the kidney has been reported to be restricted to the distal part of the renal nephron system, which is constitutively or conditionally water–impermeable18, thereby supporting its osmoregulatory role17. In other studies, TRPV4 channel has been shown to be activated by hypotonic cell swelling, which further suggested that TRPV4 functions in response to osmotic cues19. Indeed, osmotic and mechanical sensing are impaired in Trpv4-/- mice8,17. A later study showed that TRPV4 is also expressed in renal connecting tubule/collecting duct cells and is activated by fluid flow20. Flow-induced activation of TRPV4 in the thick ascending limb indicates that TRPV4 channels might be involved in salt homoeostasis21. Pharmacological and siRNA experiments suggested that hypotonicity-induced TRPV4 activation is a prerequisite for ATP release from epithelial cells in response to osmotic changes19. More recent data demonstrate a functional interaction between TRPV4 and aquaporin 2 in response to anisosmotic conditions22.
On the other hand, TRPV4 channels in endothelial and smooth muscle cells have been recognized to play an important role in the control of vascular tone23,24. Recently, we have shown that TRPV4 promotes endothelium-dependent relaxation in renal resistance arteries, renal conduit arteries and in medullary vasa recta25. TRPV4 has been shown to function as key endothelial ion channels for calcium influx to control vascular function26. Moreover, TRPV4 participates in hypoxic vascular relaxation which is critical for postischaemic ‘no–reflow’ tissue regions27. The outcome of acute kidney injury (AKI) has been primarily linked to renal tubular injury. In fact, most theories consider renal tubular cells as the main culprit, i.e, tubular obstruction, calcium overload, loss of cytoskeletal integrity, and loss of cell–matrix adhesion due to AKI28,29,30. However, more than forty years ago Flores and colleagues pointed out that ischaemic swelling of endothelial cells may occlude the flow in small blood vessels, which prevents reflow even if the initial cause of ischaemia is relieved. This hypothesis was supported by several other studies31,32,33 and these studies established the pathophysiological role of endothelial cell activation and dysfunction in AKI.
TRPV4 channels present a promising novel target to treat acute and chronic renal disease processes34. In line with the above described vasodilatory properties and the regional and functional distribution of TRPV4 in the renal vasculature and tubular system, we hypothesized that TRPV4 might function in a renoprotective manner in renal ischaemia/reperfusion injury (IRI). To elucidate the putative role of TRPV4 in AKI, we performed renal IRI studies using Trpv4 KO mice17 and wild–type (WT) control mice as littermates on the same genetic background. We show that Trpv4 deficiency deteriorates the immediate outcome of AKI, in that renal – and primarily cortical – tubular damage is aggravated in Trpv4 deficient mice after IRI which was attributable to increased necrosis rather than apoptosis.
Results
The Impact of TRPV4 Deficiency on Renal Damage after IRI
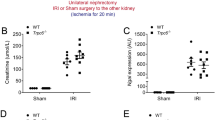
To gain insights into the (patho)physiological role of TRPV4 in renal IRI, we performed comparative in vivo studies using TRPV4 deficient (Trpv4 KO) mice and wild–type (WT) control mice. Although Trpv4 KO mice exhibited slightly higher serum concentrations of sodium and glucose, and blood haemoglobin and haematocrit levels at baseline, the kidney function markers creatinine and urea nitrogen were initially indistinguishable between Trpv4 KO and WT mice (Table 1). Already six hours after renal IRI, however, Trpv4 KO mice showed higher serum creatinine and urea nitrogen levels compared to WT mice; this difference in creatinine levels persisted over a period of twenty–four hours after ischaemia (Fig. 1A). Other lab parameters summarised in Supplementary Table 1 showed no significant differences at these time points after renal ischaemia. Sham–operated Trpv4 KO mice had similar creatinine levels compared to sham-operated WT mice (Supplemental Fig. 1). Kidney sections were haematoxylin and eosin (HE) stained to assess the degree of tubular damage after ischaemic AKI. Histological analyses and semi–quantitative scoring revealed aggravated cortical tubular damage in IRI–damaged kidneys of Trpv4 KO mice compared to IRI–damaged kidneys of WT mice (Fig. 1B,C). Of the five examined parameters tubular necrosis, tubular degeneration and brush border loss showed the most prominent differences between IRI–damaged kidneys of WT and Trpv4 KO mice, whereas sham-operated mice did not differ in any of the examined parameters (Fig. 1D). Extensive necrosis of tubular cells with brush border was verified by proximal tubular cell specific staining for aquaporin-1 (Supplemental Fig. 2). Additionally, we verified the existence of functional TRPV4 channels in the proximal tubular system. Proximal tubular cells (PTCs) were isolated from WT and Trpv4 KO mice and were stimulated in vitro with the TRPV4 agonist GSK1016790A at both 10 and 100 nM concentrations after loading with the calcium indicator fluo-4 AM. Calcium influx was detected in PTCs of WT mice after application of GSK1016790A but not in PTCs of Trpv4 KO mice (Supplemental Fig. 3). Histological analysis of the outer stripe of the outer medulla, a region characterized by a relative paucity of intertubular capillaries and therefore particularly prone to ischemic injury, showed less distinctive, but similar differences (Supplemental Fig. 3A–C).

Renal function and damage after renal ischaemia/reperfusion injury (IRI). (A) Serum creatinine levels of Trpv4 KO (▪) and WT (○) mice at baseline, 6 and 24 hours after renal IRI. Please note that at baseline all serum creatinine levels were below the measurement limit (18 µmol/L). n = 6 for both WT and Trpv4 KO mice, repeated measurement two-way ANOVA (PTime < 0.0001, PGenotype = 0.0007, PInteraction < 0.0001), Sidak’s multiple comparisons test, *P < 0.05, #P < 0.001. (B) Representative cortical images of haematoxylin and eosin stained sections of sham and I/R–injured kidneys of WT and Trpv4 KO mice (×200). * on the histological images represent necrosis and # represent tubular cast formation. (C) Semi–quantification of cortical tubular injury in sham and I/R–injured kidneys of WT and Trpv4 KO mice, two-way ANOVA (PTreatment < 0.0001, PGenotype = <0.0001, PInteraction < 0.0001), Sidak’s multiple comparisons test, #P < 0.001, n = 6 for both WT and Trpv4 KO mice. (D) Detailed analysis of cortical tubular damage, one-way ANOVA, Tukey post-hoc test, #P < 0.001, n = 6 for both WT and Trpv4 KO mice.
Surprisingly, renal mRNA levels of the AKI marker lipocalin 2 (Lcn2; also known as neutrophil gelatinase–associated lipocalin [Ngal]) and kidney injury molecule-1 (Kim1, also named Tim1 and Havcr1), as well as renal mRNA levels of the pro–inflammatory cytokines interleukin (Il)–1 beta, Il–6 and tumor necrosis factor (Tnf) did not differ between sham or IR-injured kidneys of Trpv4 KO and WT mice (Fig. 2A–E).

Expression of renal injury and pro-inflammatory markers. (A) Renal mRNA levels of lipocalin 2 (Lcn2; also known as neutrophil gelatinase–associated lipocalin [Ngal]), (B) kidney injury molecule 1 (Kim1) and pro–inflammatory cytokine (C) interleukin 1 beta (Il1b) and (D) Il6 and (E) tumor necrosis factor (Tnf) in sham and in Trpv4 KO and WT mice following IRI. Renal gene expression data are determined in n = 4 for sham-operated WT and Trpv4 KO mice and n = 6 for I/R–injured WT and Trpv4 KO mice, two-way ANOVA. In all cases PTreatment < 0.001, and PGenotype and PInteraction are not significant.
Expression of TRPV4 in the Renal Tubular System
We were somewhat surprised of the massive tubular necrosis and brush border loss seen in the renal cortex of Trpv4 KO mice, as it is believed that TRPV4 is not expressed in proximal tubular cells. Therefore, we used the advantage of our KO mice and tested several commercially available antibodies against TRPV4, however, only one of the tested commercially available antibodies worked in our hands for immunofluorescence detection of TRPV4 (Supplemental Fig. 4). A detailed analysis of renal tubular TRPV4 expression in WT kidneys revealed that TRPV4 is co-expressed in aquaporin-1 (proximal tubule), aquaporin-2 (collecting duct) and partly in sodium-chloride symporter (distal convoluted tubule) positive tubules but not in sodium-potassium-chloride co-transporter 2 (thick ascending limb) positive tubules (Fig. 3A–D).

Analysis of renal tubular TRPV4 expression in the kidney of WT mice by immunostaining. (A) The proximal tubule marker aquaporin-1 (Aqp1), (D) the collecting duct marker aquaporin-2 (Aqp2) and (C) partly the distal convoluted tubule marker sodium-chloride symporter (Slc12a3) co-localize with TRPV4. (B) The thick ascending limb marker sodium-potassium-chloride cotransporter 2 (Slc12a2) does not co-localize with TRPV4. White squares mark equivalent tubular sections represented on adjacent sections. White arrows point to the same tubular structures seen on two serial cuts stained by different antibodies. Yellow bar represents 50 μm.
The Impact of TRPV4 Deficiency on Renal Apoptosis after IRI
In renal ischaemia, there is mismatch of local tissue oxygen supply and demand and cells accumulate metabolic waste products. As a result of this imbalance, tubular epithelial cells undergo death by apoptosis or necrosis depending on injury severity. Moreover, renal tubular epithelial apoptosis can initiate reperfusion–induced inflammation and subsequent tissue injury35. To assess tubular epithelial apoptosis, TUNEL labeling was performed on paraffin sections of IRI–kidneys of WT and Trpv4 KO mice. Interestingly, injured kidneys of Trpv4 KO mice showed similar rates of apoptosis as injured kidneys of WT mice (Fig. 4A). Quantification by direct counting of TUNEL–positive tubular cells confirmed this observation (Fig. 4B). Nevertheless, the concentration of the tissue damage marker lactate dehydrogenase (LDH) was significantly higher in IRI-injured Trpv4 KO mice compared to WT mice, supporting the histological finding of more extensive renal necrosis in Trpv4 KO mice (Fig. 4C). Contralateral kidneys of Trpv4 KO and WT mice showed virtually no TUNEL–positive tubuli (Supplemental Fig. 5).

TRPV4 deficiency does not alter cortical tubular apoptosis upon acute ischaemic kidney injury. (A) Representative ×200 images of TUNEL labelling on WT and Trpv4 KO kidney sections 24 hours after renal ischaemia. Thin white arrows indicate TUNEL–positive tubular cells. Thick white arrows indicate TUNEL–positive interstitial cells. Yellow bars represent 100 μm. (B) Number of TUNEL–positive cortical tubular cells per ×200 field. Each dot represents a single animal and is the mean of 5 randomly selected cortical fields. n = 6 for both WT and Trpv4 KO mice, two-tailed unpaired t-test. G, glomerulus; N, necrosis. (C) Serum lactate dehydrogenase (LDH) levels in WT and Trpv4 KO mice 24 hours after renal ischaemia. n = 5 for both WT and Trpv4 KO mice, two-tailed unpaired t-test, *P < 0.05.
The Impact of TRPV4 Deficiency on Renal Cellular Infiltration after IRI
Renal IRI is associated with infiltration of granulocytes and monocytes/macrophages. These cells contribute to inflammation and subsequent repair of the injured kidney. Therefore, we characterized renal granulocyte and macrophage infiltration after IRI by flow cytometry in WT and Trpv4 KO. Whole kidney cell suspensions were immunolabeled for Ly6G and F4/80 as markers for granulocytes and macrophages, respectively. Among pre–gated singlet live cells (Fig. 5A), Ly6G–positive & F4/80–negative granulocytes as well as Ly6G–negative & F4/80–positive macrophages were detected (Fig. 5B). No significant differences in granulocyte or macrophage populations were detected between IRI–damaged kidneys of WT and Trpv4 KO mice at 24 hours after IRI (Fig. 5C,D). Renal cell populations were also similar in kidneys of Trpv4 KO and WT mice which underwent sham surgery (Fig. 5C,D). We also performed immunohistochemistry to detect IRI-induced granulocyte infiltration, and counted the average number of Ly6B-positive cells in the outer medulla after IRI. Similarly to our flow cytometry results, no differences in granulocyte counts were detected between WT and Trpv4 KO mice (Supplemental Fig. 6A,B).

Flow cytometric analysis of renal granulocyte infiltration. (A) Gating strategy: Pre–gating on live cells using Fixable Viability Dye eFluor 660 and further gating on single cells. (B) Representative flow cytometry data of infiltrating Ly6G–positive cells (granulocytes) and F4/80–positive cells (macrophages) in sham and I/R–injured kidneys of WT and Trpv4 KO mice. (C) Quantification of infiltrating Ly6G–positive cells and (D) F4/80-positive cells. n = 4 for sham-operated WT and Trpv4 KO mice and n = 6 for I/R–injured WT and Trpv4 KO mice, two-way ANOVA, Sidak’s multiple comparisons test. In both cases PTreatment < 0.0001, and PGenotype and PInteraction are not significant.
The Impact of TRPV4 Deficiency on Primary PTCs Treated with Cobalt Chloride (CoCl2)
The stabilization of hypoxia-inducible factor (HIF)1α protein is a hallmark of hypoxia. To mimic hypoxia in vitro, WT and Trpv4 KO PTCs were exposed to the HIF1α stabilizing agent CoCl2 in a low glucose medium for 24 hours. mRNA expression of the HIF1α target gene vascular endothelial growth factor (Vegfa) was upregulated by CoCl2 treatment in both WT and Trpv4 KO PTCs, indicating successful HIF1α-stabilization. We next measured mRNA levels of the stress cytokine interleukin 6 (Il6), of nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha (IkBa), a bona fide NF-κB target gene, and of the endoplasmic reticulum stress marker CCAAT/enhancer-binding protein homologous protein (Chop). In both WT and Trpv4 KO PTCs CoCl2 treatment caused a similar upregulation of these stress genes (Fig. 6A–D). We subsequently counted the average number of cells per view-field of untreated and CoCl2-treated WT and Trpv4 KO PTCs by microscopic analysis. Percentage of remaining PTCs were calculated and percentage of apoptotic PTCs were determined using Annexin V staining (Fig. 6E–G and Supplemental Fig. 8). To corroborate these data caspase 3/7 activity was determined using the Caspase-Glo 3/7 Assay (Fig. 6H) and number of viable cells were determined by the CellTiter-Glo Assay (Fig. 6I). In agreement with our in vivo data, we did not find any differences in the rate of apoptosis between WT and Trpv4 PTCs, whereas fewer Trpv4 PTCs could be detected after CoCl2 treatment when compared to WT PTCs suggesting an increased rate of necrotic cell death in Trpv4 KO PTCs. Low-glucose environment by itself - without CoCl2 - did not lead to increased cell death in Trpv4 PTCs compare to WT PTCs (Supplemental Fig. 9).

Treatment of primary proximal tubular cells (PTCs) of WT and Trpv4 KO mice with hypoxia-inducible factor 1-alpha (HIF1α)-stabilizing agent CoCl2. mRNA expression of (A) HIF1α target gene Vegfa, (B) stress cytokine interleukin 6 (Il6), bona fide NF-κB target gene nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor alpha (IkBa), and (D) endoplasmatic reticulum stress marker CCAAT/enhancer binding protein homologous protein (Chop). Each dot is a mean of 2 technical repeats and represents a pool of PTCs isolated from 2–3 animals of the same genotype. Data of two independent experiments. n = 4 for untreated and n = 5 for CoCl2-treated WT and Trpv4 KO PTCs, two-way ANOVA. In all cases PTreatment < 0.05, and PGenotype and PInteraction are not significant. (E) Representative ×200 images of Annexin V-stained (red) untreated WT and Trpv4 KO PTCs and of PTCs 24 hours after treatment of 300 μM hypoxia-mimetic agent CoCl2. Nucleus was stained with 4’,6-diamidino-2-phenylindole (DAPI) (blue). White arrows indicate Annexin V–positive tubular cells. Yellow bars represent 100 μm. (F) Quantification of Annexin V-positive PTCs in the respective groups. Each dot is a percentage of the mean Annexin V-positive cells/negative cells in 7–10 randomly selected high-power fields. Data of three independent experiments. Two-way ANOVA, PTreatment < 0.05, and PGenotype and PInteraction are not significant. (G) Quantification of DAPI-positive WT and Trpv4 KO PTCs after a 24-hour treatment with 300 μM CoCl2. Each dot represents the percentage of the remaining DAPI-positive PTCs. To calculate the mean DAPI-positive PTC number of untreated and treated PTCs, 7–10 randomly selected high-power fields were counted and percentage was calculated. Data of three independent experiments. (H) Caspase 3/7 activity measured by the Caspase-Glo 3/7 Assay. Each dot is a ratio of the relative fluorescence of CoCl2-treated PTC/untreated PTC (background). n = 5 for CoCl2-treated WT and Trpv4 KO PTCs. (I) Percentage of viable PTCs measured by CellTiter-Glo Luminescent Cell Viability Assay. Luminescence of WT and Trpv4 KO PTCs after a 24-hour treatment with 300 μM CoCl2 was measured and was divided by the luminescence of their respective untreated controls and multiplied by 100. Data of three independent experiments, each experiment is a mean of 3–5 samples.
Discussion
Acute kidney injury is a global public health problem in developed and developing countries. Recent data show that AKI is responsible for more than 1.5 million deaths yearly worldwide36. Thus, there is an urgent need for new therapeutic approaches. Recent pre–clinical data indicate that TRPV1 agonists can improve the outcome of ischaemic AKI37,38. Besides TRPV1, TRPV4 has been recently identified to serve as a major calcium influx pathway in endothelial cells to control vascular function26,39. Very recently, we were able to show that TRPV4 channels can promote endothelium-dependent relaxation in different vessels in the kidney, including vasa recta25. Others have demonstrated that TRPV4 plays an important role in the preservation of vasodilatation under hypoxic conditions27. Moreover, TRPV4 channels are highly expressed in renal tubular cells as shown in this study and by others40. We therefore hypothesized that loss-of-function of the osmotically-sensitive ion channel TRPV4, would impact the ability of tubular cells to recover from IRI.
In this study, we explored the pathophysiological role of TRPV4 in renal IRI. We uncovered massive necrosis of PTCs after renal IRI and provide evidence – contrasting to earlier reports – that PTCs express functional TRPV4. As early as 6 hours after renal IRI Trpv4 KO mice showed higher serum creatinine levels compared to WT mice, which was maintained over 24 hours after renal ischaemia. Surprisingly, renal expression of the AKI markers Ngal and Kim1, which are expressed by distal and proximal tubular cells, respectively, did not differ between WT and Trpv4 KO mice. In the kidney, NGAL and KIM1 have been reported to be expressed specifically in injured nephrons19,41. On the other hand, the expression profile of NGAL and KIM1 in the injured kidney has been described to be similar to TRPV4 expression in the intact kidney18,19,20,41. Therefore, one possible explanation for the lack of an increased Ngal and Kim1 expression Trpv4 KO mice after renal IRI could thus be the higher necrosis rate of TRPV4 deficient tubular cells, which will in parallel deplete the number of NGAL and KIM1 expressing cells making the evaluation of renal Ngal and Kim1 expression ambiguous in this study. Noteworthy, histological analysis of the IRI–damaged kidneys of Trpv4 KO mice revealed massively increased cortical tubular damage, in particular tubular necrosis and loss of aquaporin-1 positive proximal tubular cells, compared to the kidneys of WT mice which underwent the same surgical procedure. This finding surprised us for two reasons; first, proximal tubular cells were thus far unknown to express TRPV4 channels, and second, the renal cortex is known to be less prone to ischemic injury as compared to the outer stripe of the outer medulla18,20,41. Therefore, we performed diligent co-immunostainings which revealed that proximal tubular cells indeed highly express TRPV4. While our study was underway Janas and co-workers reported on the expression of TRPV4 in the mouse kidney by using qPCR on isolated nephron segments, immunohistochemistry, and a Trpv4 reporter mouse model40. Similarly to our findings they found TRPV4 to be expressed in the proximal tubule, in the distal convoluted tubule, and in the collecting duct, but not in the glomerulus and the thick ascending limb. These and our findings together revise earlier reports which could not demonstrate expression of TRPV4 in proximal tubular cells.
Tissue injury resulting from hypoxia can result in both necrosis and apoptosis. Apoptosis occurs in normal and disease states, whereas necrosis is induced only when cells or tissues are exposed to severe and acute injury42. Lack of TRPV4 has recently been linked to increased apoptosis and inhibited autophagy in a rat liver stellate cell line43. Therefore, we addressed the question whether a higher rate of apoptosis might be responsible for the extensive renal tubular damage in Trpv4 KO mice. Interestingly, injured kidneys of Trpv4 KO mice showed similar numbers of TUNEL–positive tubular cells as injured kidneys of WT mice. Moreover, in vitro treatment of WT and Trpv4 KO PTCs with the HIF1α-stabilizing agent CoCl2 caused similar levels of apoptosis as measured by either the caspase 3/7 assay or by counting the number of Annexin V positive PTCs. In addition, mRNA levels of stress genes like Il6, IkBa and Chop were similarly upregulated in CoCl2-treated WT and Trpv4 KO PTCs. However, CoCl2 treatment induced more cell death in Trpv4 KO PTCs compared to WT PTCs as assessed by direct counting and by luminescent cell viability assay. These data suggest that TRPV4 deficiency induces necrosis but does not interfere with cell apoptosis in a hypoxia model of HIF1α stabilization. To the best of our knowledge no HIF binding sites on the Trpv4 gene has been established. Nevertheless, it has recently been shown that HIF1α transcriptionally regulates Trpa1 expression presumably via binding to an unusual hypoxia-response element-like motif within the Trpa1 gene, which subsequently leads to suppressed secretion of interleukin-6 and interleukin-844. One might speculate that a similar mechanism could regulate Trpv4 expression in response to hypoxia. This in turn might be an indispensable mechanism for successful adaptation of PTCs to hypoxia.
Renal tubular epithelial apoptosis can initiate reperfusion–induced inflammation which is a hallmark of renal IRI35,45. Similar to the results for TUNEL–positive renal tubular cells, we found comparable degrees of infiltrating neutrophil granulocytes and F4/80–positive macrophages as well as mRNA expression levels of pro-inflammatory cytokines in IRI–kidneys of WT and Trpv4 KO mice. These finding in combination with our histological and immunofluorescence results strongly suggests that the lack of functional TRPV4 does not increase inflammation and apoptosis, but directly compromises the ability of the renal epithelium to recover from ischaemic injury leading to prompt necrosis. Higher serum LDH levels in Trpv4 KO mice compared to WT mice after renal IRI support this concept.
Calcium entry is a hallmark of the cellular reaction to ischemia in numerous cell types, including tubular epithelial cells. TRPV4 channels have been shown to be involved in ischemia-induced calcium entry in a number of cells, including reactive astrocytes and cardiac myocytes46,47. One may therefore expect that lack of TRPV4 channels should be rather beneficial. On the other hand, pharmacological inhibitors of TRPV4 channels are able to reduce cytokine production and preserve endothelial cell function, including Ca2+ signaling, and endothelium-dependent vasodilation, resulting in increased survival e.g. in septic mice48. Additionally, Trpv4 KO mice exhibit higher blood sodium concentrations and present impaired osmotic sensitivity upon challenge17. Sublethal tubular damage results in impaired proximal sodium reabsorption which is expected to activate the tubular glomerular feedback mechanism leading to increased afferent vascular resistance and a concomitant decrease in glomerular filtration rate49. The impaired tubular transport of sodium in Trpv4 KO mice might also contribute to the progression of IRI in a tubular glomerular feedback-dependent manner. In a recent study, the exogenous TPRV4 activator apigenin reduced hypertension-induced renal fibrosis through the AMP-activated protein kinase/sirtuin 1 pathway in a deoxycorticosterone acetate (DOCA)–salt rat model. Knockout of TRPV4 in mice abolished the beneficial effects of apigenin that were observed in the DOCA–salt hypertensive rats, suggesting a protective and anti-fibrotic role of TRPV4 in chronic renal disease50. Here, our data suggest that lack of TRPV4 impairs recovery after acute renal IRI and induces massive-scale necrosis, especially in the renal cortex.
We demonstrated earlier that activation of TRPV4 promotes endothelial relaxation in renal resistance arteries, renal conduit arteries and in the medullary vasa recta25. During renal IRI the most commonly injured epithelial cells are the proximal tubular cells. These cells are particularly susceptible to ischemic insult partly due to their high metabolic rate required for mediating ion transport and a limited capacity to undergo anaerobic glycolysis51. As Trpv4 KO mice exhibited significantly more necrosis of proximal tubular cells after renal IRI compared to WT mice, it is tempting to speculate that vascular mechanisms may also be responsible for the observed effect.
A limitation of our study is that we used Trpv4 pan-null mice. Global lack of TRPV4 can lead to a number of pathologies e.g. impaired osmotic and mechanosensing17 which become more prominent in stress situations. Although we carefully designed our study to circumvent possible confounding effects caused by the baseline phenotype of the Trpv4 KO mice, we cannot exclude that the above-mentioned limitation of global TRPV4 deficiency could have influenced the outcome of our study. This might apply particularly for haemodynamic changes after renal ischemia, since TRPV4 has an established role in the maintenance of vascular tone39. In addition, the increased sensitivity to ischaemia may not only be due directly to TRPV4 loss-of-function, but due to developmental-compensatory and other secondary mechanisms caused by genetically-engineered global absence on Trpv4. Indeed, TRPV4 have been recently described as a sensor of cell stress in the diseased fatty liver. These data lend solid support to the emerging concept of TRPV4 protecting a key epithelial cell – hepatocytes – from oxidative injury52.
In summary, genetically-encoded Trpv4 knockout (pan-null) aggravates acute renal tubular injury which is associated with enhanced renal epithelial necrosis. Although the results of our study suggest that this necrosis may be a direct effect of TRPV4 deficiency, additional studies are needed to clarify the exact molecular mechanism. Further studies using cell–specific TRPV4 mouse models are needed to clarify the precise pathophysiological sequence of events, and to clarify how the IRI–induced, TRPV4–dependent tubular damage influences long term renal recovery and survival.
Methods
Mice
Trpv4 KO mice have been generated and characterized previously.17 Wild–type (WT) control mice as littermates on the same genetic background were used. Mice were allowed free access to standard chow and water and were kept in a 12:12–h light–dark cycle. Animal studies were approved by the Berlin Animal Review Board in 2012 (No. G0082/12) and conducted in accordance with the standards of the American Physiological Society.
Renal IRI Model
Renal IRI was induced as described earlier53. Briefly, male mice (aged 14–17 weeks) were anaesthetized by isoflurane (2.3%) in air (350 mL/min). Mice were operated individually to ensure similar exposure to isoflurane (33.8 ± 3.0 min, mean ± SD)54. Body temperature was maintained at 37 °C and monitored during surgery using a temperature controller with a heating pad (TCAT–2, Physitemp Instruments). Ischaemia was induced after right–sided uni-nephrectomy by clipping the pedicle of the left kidney for 20 minutes with non–traumatic aneurysm clips (FE690K, Aesculap). Reperfusion was confirmed visually and the abdomen and the skin were sutured separately with a 5/0 braided-silk suture. After surgery mice had free access to water and chow. Body–warm sterile physiological saline solution (1 mL) and pre–emptive analgesia with tramadol (1 mg/kg) were applied subcutaneously to every mouse. Sham operation was performed in a similar manner, except for clamping the renal pedicle. Mice with bleeding during surgery, with incomplete renal reperfusion, with excessive exposure to isoflurane for any reason, with significant temperature fluctuation during surgery, or with signs of infection 24 h after IRI, were immediately euthanized and not used for further analysis. Twenty–four hours after reperfusion, mice were sacrificed, and kidney and blood samples were collected for further analysis. The kidneys were divided into two portions. Half of the kidney was immersed in 4% phosphate–buffered saline (PBS)–buffered formalin for histology, and the other half was snap–frozen in liquid nitrogen for RNA preparation.
Primary Proximal Tubular Cell Isolation and Calcium concentration (Ca2+) measurement
Cells were obtained and cultured as described previously53. Briefly, WT and Trpv4 KO mice aged 10–12 weeks were euthanized by an overdose of isoflurane and the kidneys were flushed with 10 ml ice-cold PBS (Sigma). Renal cortices were dissected visually and minced into small pieces which were transferred through two layers of stainless steel sieves (pore size 125 μm and 106 μm; Linker). Tubular fragments caught by the 106 μm sieve were flushed in the reverse direction with PBS and centrifuged for 5 minutes at 170 × g, washed, and then resuspended into the appropriate amount of culture medium: 1:1 DMEM/F12 (D 8437; Sigma) supplemented with fetal calf serum (FCS) 10%, penicillin 100 IU/ml and streptomycin 100 µg/ml buffered to pH 7.4. Plates were incubated in a standard humidified incubator at 5% CO2. Medium was changed two days later and maintained every other day until the monolayer of cells reached 90% confluence; at this time, over 90% of the isolated cells were megalin-positive (sheep-anti-LRP2 antibody (1:10000) was a gift from Prof. Thomas Willnow (Max–Delbrück Center for Molecular Medicine, Berlin); secondary antibody was donkey anti-sheep Alexa 555 (1:2000; Thermo Fisher Scientific)).
After 5–7 days cells were trypsinized and seeded on a 48-well plate at a density of 20,000 cells/well. Cells were let to adhere overnight. The next day cells were loaded with the Ca2+ indicator Fluo-4 AM (Invitrogen, F14201) at a final concentration of 10 µM at 37 °C in a standard humidified incubator at 5% CO2 in the presence of Pluronic F127 (0.005%; w/v, Calbiochem, 540025). After 1-hour incubation time, cells were washed twice with pre-warmed PBS and then stimulated with the TRPV4 agonist GSK1016790A (Sigma-Aldrich) at final concentrations of 10 and 100 nM. Unstimulated cells of each genotype served to measure background fluorescence. After stimulation fluorescence was measured in a SpectraMax M5 Multi-Mode Microplate Reader (Ex.: 488 nm, Em.: 535 nm) using the SoftMax Pro software package. Data are expressed as (F − Fo)/Fo, where F is the fluorescence intensity of GSK1016790A-treated cells and Fo is the fluorescence intensity of the unstimulated cells.
Quantitative Real–Time (qRT)–PCR
qRT–PCR was performed as described earlier53. Briefly, total RNA was isolated from snap–frozen kidneys after homogenization with a Precellys 24 homogenizer (Peqlab) using RNeasy RNA isolation kit (Qiagen). RNA quality and concentration were determined by a NanoDrop–1000 spectrophotometer (Thermo Fisher Scientific). Two micrograms of total renal RNA were transcribed to cDNA (Applied Biosystems). Quantitative analysis of target mRNA expression was calculated using the relative standard curve method. TaqMan and SYBR green analysis was conducted using an Applied Biosystems 7500 Sequence Detector (Applied Biosystems). The expression levels were normalized to the purinergic housekeeping gene hypoxanthine-guanine phosphoribosyltransferase. Primer sequences are provided in Supplementary Table 2.
Serum Measurements
To allow for repeated measurement of lab parameters within a short time interval in the same mouse, 90 μL blood was taken from the facial vein and parameters were measured using an i-STAT system with Chem8+ cartridges (Abbott). Serum creatinine levels of sham-operated mice were measured by Labor 28 GmbH (Berlin, Germany). Lactate dehydrogenase was measured at the Pathophysiological Platform of the Max–Delbrück Center for Molecular Medicine (Berlin, Germany) using a Fuji DRI-CHEM automated clinical chemistry analyser.
Microscopy
Histology
Formalin–fixed, paraffin–embedded, 5 µm thick kidney sections were stained with haematoxylin (Sigma) and 1% eosin (Sigma). Semi-quantitative scoring of tubular damage was performed in a blinded manner in that 10 to 18 images at 200× magnification per sample were scored on a 1 to 10 scale according to percentage (where 0 = 0% and 10 = 100%) of tubular dilation, brush border loss, tubular necrosis, tubule degeneration and tubular cast formation. The total score was calculated as sum of all five morphological parameters.
Immunofluorescence
Five μm thick cryosections were post-fixed in ice-cold acetone, air-dried, rehydrated and blocked with 10% normal donkey serum (Jackson ImmunoResearch) for 30 min. Next, sections were incubated overnight at 4 °C with the following primary antibodies: rat anti-Ly-6B.2 (Gr1) (1:300; MCA771G; AbD Serotec), rabbit anti-TRPV4 (1:50; acc-034, Alomond Labs), rabbit anti-aquaporin-1 (1:300; AB2219, Millipore), rabbit anti-sodium-chloride symporter (1:500; AB3553; Millipore), goat anti-aquaporin-2 (1:200, sc-9882; Santa Cruz Biotechnologies), rabbit anti-sodium-potassium-chloride cotransporter 2 (1:1000; a gift from Sebastian Bachmann, Charité Berlin). For co-localization experiments serial sections were stained. All incubations were performed in a humid chamber. For fluorescence visualization of bound primary antibodies, sections were further incubated with appropriate Cy3- or Alexa 488-conjugated secondary antibodies (1:500; Jackson ImmunoResearch) for 1 h in a humid chamber at room temperature.
CoCl2 and untreated primary proximal tubular cells isolated from WT and Trpv4 KO mice (see below) were stained with Annexin V (51-65875×; BD Pharmingen) and DAPI (final concentration 1 µg/mL). Annexin V positive cells and negative cells were counted in 7–10 randomly selected high-power fields (400×) using of a Zeiss Axio Imager.M2 fluorescence upright microscope.
TUNEL Assay
TMR In Situ Cell Death Detection Kit (Roche) was used according to the manufacturer’s instructions on formalin–fixed, paraffin–embedded 2 μm thick sections. In order to visualize cell nuclei slides were counterstained with the nuclear dye 4′,6-diamidino-2-phenylindole (DAPI, Sigma). The number of TUNEL–positive tubular cells was determined as mean of at least 5 non–overlapping fields in the cortex by use of a Zeiss Axio Imager.M2 fluorescence upright microscope using a 20× Plan–Apochromat objective.
Flow Cytometry
Granulocyte and macrophage infiltration was assessed in sham–operated and IRI–injured kidneys. GentleMacs C-tubes (Miltenyi Biotec) in the presence of 10 mg/mL collagenase IV (Sigma) and 200 U/mL DNase I (Roche) dissolved in Hank’s balanced salt solution were used to obtain single cell suspensions. Fixable Viability Dye eFluor 660 (eBioscience) was used for detection of dead cells. Granulocytes and macrophages were stained with phycoerythrin (PE)–conjugated anti–Ly6G (clone: 1A8, Beckton Dickinson) and eFluor450–conjugated anti–F4/80 (clone: BM8, eBioscience) antibodies, respectively. Samples were analyzed on a FACSCanto II flow cytometer (Becton Dickinson). Data analysis was conducted by FlowJo (TreeStar) software.
HIF1α Stabilization in Primary PTC
Primary proximal tubular cells isolated from WT and Trpv4 KO mice were treated with 300 μM CoCl2 to stabilize HIF1α or left untreated in a low-glucose DMEM medium (D6046, Sigma) for 24 hours. Low glucose medium was used because it is known that cell death in proximal tubular cell cultures is HIF1α-dependent only when glucose availability is limited55. After 24 hours medium was removed and cells were harvested for further analysis. To count Annexin V and DAPI-positive WT and Trpv4 KO PTC, cells were seeded at a density of 104 cells/well in a chamber slide with a removable 8 well silicone chamber (Ibidi). Untreated PTC and PTC treated with 300 μM CoCl2 for 24 hours were stained with Annexin V (100 µL binding buffer +5 µL Annexin V/well) after washing with PBS. After 15 minutes incubation in the dark at room temperature the staining solution was removed and PTC were fixed with pre-warmed (37 °C) 4% formaldehyde (200 µL/well) for 30 minutes in the dark at room temperature. Thereafter the PTC were washed, the chamber was removed and the slide was subjected to microscopic analysis.
Caspase-Glo 3/7 and CellTiter-Glo Luminescent Cell Viability Assays
96-well clear plates were seeded with 1 × 104 WT or Trpv4 KO PTC/well. Cells were allowed to attach overnight. On average, cells were at 50% confluency before addition of 300 μM CoCl2. Twenty-four hours after CoCl2 treatment the cells were assayed by the Caspase-Glo 3/7 or by the CellTiter-Glo Luminescent Cell Viability Kit according to manufacturer’s instructions (Promega). After 10 min on a plate shaker at room temperature, 90% of the lysate volume was transferred to a 96-well solid-white plate (Nunc). Cell lysates were analysed in a SpectraFluor Plus microplate reader (Tecan), and blank values were subtracted from data points. Relative fluorescence intensity of treated cells was normalized by their respective untreated control cells to normalize for possible seeding density differences between WT and Trpv4 KO PTC. In case of the CellTiter-Glo Luminescent Cell Viability Assay an ATP standard curve was measured in every experiment.
Statistics
Statistical analysis was performed using GraphPad Prism 5.04 (GraphPad Software). Normality of the data was evaluated by Kolmogorov–Smirnov test. Grubbs’ test was used to detect outliers. Data were analysed by repeated measurement or regular two-way ANOVA with Sidak’s multiple comparisons test, as appropriate. Data with two groups were tested by two-sided unpaired t–test (data with normal distribution). Data are presented as boxplots, showing median, 25 and 75 percentiles and minimum and maximum. Detailed tubular injury score data are presented as mean ± SD. P values < 0.05 were considered as statistically significant.
Data availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information files). Raw data is available to interested parties upon request.
References
Montell, C. & Rubin, G. M. Molecular characterization of the Drosophila trp locus: a putative integral membrane protein required for phototransduction. Neuron 2, 1313–1323 (1989).
Kaneko, Y. & Szallasi, A. Transient receptor potential (TRP) channels: a clinical perspective. Br J Pharmacol 171, 2474–2507 (2014).
Nilius, B. & Owsianik, G. The transient receptor potential family of ion channels. Genome Biol 12, 218 (2011).
Pochynyuk, O., Zaika, O., O’Neil, R. G. & Mamenko, M. Novel insights into TRPV4 function in the kidney. Pflugers Arch 465, 177–186 (2013).
Strotmann, R., Harteneck, C., Nunnenmacher, K., Schultz, G. & Plant, T. D. OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nature cell biology 2, 695–702 (2000).
Liedtke, W., Tobin, D. M., Bargmann, C. I. & Friedman, J. M. Mammalian TRPV4 (VR-OAC) directs behavioral responses to osmotic and mechanical stimuli in Caenorhabditis elegans. Proc Natl Acad Sci USA 100(Suppl 2), 14531–14536 (2003).
Liedtke, W. et al. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell 103, 525–535 (2000).
Mizuno, A., Matsumoto, N., Imai, M. & Suzuki, M. Impaired osmotic sensation in mice lacking TRPV4. Am J Physiol Cell Physiol 285, C96–101 (2003).
Gao, X., Wu, L. & O’Neil, R. G. Temperature-modulated diversity of TRPV4 channel gating: activation by physical stresses and phorbol ester derivatives through protein kinase C-dependent and -independent pathways. J Biol Chem 278, 27129–27137 (2003).
Vriens, J. et al. Cell swelling, heat, and chemical agonists use distinct pathways for the activation of the cation channel TRPV4. Proc Natl Acad Sci USA 101, 396–401 (2004).
Watanabe, H. et al. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature 424, 434–438 (2003).
Nilius, B. & Voets, T. The puzzle of TRPV4 channelopathies. EMBO Rep 14, 152–163 (2013).
Yin, J. et al. Negative-feedback loop attenuates hydrostatic lung edema via a cGMP-dependent regulation of transient receptor potential vanilloid 4. Circ Res 102, 966–974 (2008).
Moore, C. et al. UVB radiation generates sunburn pain and affects skin by activating epidermal TRPV4 ion channels and triggering endothelin-1 signaling. Proc Natl Acad Sci USA 110, E3225–3234 (2013).
Kanju, P. & Liedtke, W. Pleiotropic function of TRPV4 ion channels in the central nervous system. Experimental physiology 101, 1472–1476 (2016).
Kassmann, M. et al. Transient receptor potential vanilloid 1 (TRPV1), TRPV4, and the kidney. Acta Physiol (Oxf) 207, 546–564 (2013).
Liedtke, W. & Friedman, J. M. Abnormal osmotic regulation in trpv4-/- mice. Proc Natl Acad Sci USA 100, 13698–13703 (2003).
Tian, W. et al. Renal expression of osmotically responsive cation channel TRPV4 is restricted to water-impermeant nephron segments. Am J Physiol Renal Physiol 287, F17–24 (2004).
Woudenberg-Vrenken, T. E., Bindels, R. J. & Hoenderop, J. G. The role of transient receptor potential channels in kidney disease. Nat Rev Nephrol 5, 441–449 (2009).
Berrout, J. et al. Function of transient receptor potential cation channel subfamily V member 4 (TRPV4) as a mechanical transducer in flow-sensitive segments of renal collecting duct system. J Biol Chem 287, 8782–8791 (2012).
Cabral, P. D., Capurro, C. & Garvin, J. L. TRPV4 mediates flow-induced increases in intracellular Ca in medullary thick ascending limbs. Acta Physiol (Oxf) 214, 319–328 (2015).
Galizia, L. et al. Functional interaction between AQP2 and TRPV4 in renal cells. Journal of cellular biochemistry 113, 580–589 (2012).
Earley, S., Heppner, T. J., Nelson, M. T. & Brayden, J. E. TRPV4 forms a novel Ca2+ signaling complex with ryanodine receptors and BKCa channels. Circ Res 97, 1270–1279 (2005).
Kohler, R. et al. Evidence for a functional role of endothelial transient receptor potential V4 in shear stress-induced vasodilatation. Arterioscler Thromb Vasc Biol 26, 1495–1502 (2006).
Chen, L. et al. Functional TRPV1 and TRPV4 channels along different segments of the renal vasculature. Acta Physiol (Oxf) 213, 481–491 (2014).
Sonkusare, S. K. et al. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science 336, 597–601 (2012).
Rath, G. et al. Vascular hypoxic preconditioning relies on TRPV4-dependent calcium influx and proper intercellular gap junctions communication. Arterioscler Thromb Vasc Biol 32, 2241–2249 (2012).
Weinberg, J. M. The cell biology of ischemic renal injury. Kidney Int 39, 476–500 (1991).
Molitoris, B. A. & Nelson, W. J. Alterations in the establishment and maintenance of epithelial cell polarity as a basis for disease processes. J Clin Invest 85, 3–9 (1990).
Schrier, R. W., Arnold, P. E., Van Putten, V. J. & Burke, T. J. Cellular calcium in ischemic acute renal failure: role of calcium entry blockers. Kidney Int 32, 313–321 (1987).
Lieberthal, W., Wolf, E. F., Rennke, H. G., Valeri, C. R. & Levinsky, N. G. Renal ischemia and reperfusion impair endothelium-dependent vascular relaxation. Am J Physiol 256, F894–900 (1989).
Brodsky, S. V. et al. Endothelial dysfunction in ischemic acute renal failure: rescue by transplanted endothelial cells. Am J Physiol Renal Physiol 282, F1140–1149 (2002).
Conger, J. D., Robinette, J. B. & Guggenheim, S. J. Effect of acetylcholine on the early phase of reversible norepinephrine-induced acute renal failure. Kidney Int 19, 399–409 (1981).
Marko, L., Mannaa, M., Haschler, T. N., Kramer, S. & Gollasch, M. Renoprotection: focus on TRPV1, TRPV4, TRPC6 and TRPM2. Acta Physiol (Oxf) 219, 589–612 (2017).
Daemen, M. A. et al. Inhibition of apoptosis induced by ischemia-reperfusion prevents inflammation. J Clin Invest 104, 541–549 (1999).
Mehta, R. L. et al. International Society of Nephrology’s 0by25 initiative for acute kidney injury (zero preventable deaths by 2025): a human rights case for nephrology. Lancet 385, 2616–2643 (2015).
Rayamajhi, S., Contractor, T. & Wang, D. H. The potential of TRPV1 agonists for treating ischemia/reperfusion-induced renal injuries. Curr Opin Investig Drugs 10, 963–970 (2009).
Chen, L. et al. Role of TRPV1 channels in ischemia/reperfusion-induced acute kidney injury. PLoS One 9, e109842 (2014).
Randhawa, P. K. & Jaggi, A. S. TRPV4 channels: physiological and pathological role in cardiovascular system. Basic research in cardiology 110, 54 (2015).
Janas, S. et al. TRPV4 is associated with central rather than nephrogenic osmoregulation. Pflugers Arch 468, 1595–1607 (2016).
Paragas, N. et al. The Ngal reporter mouse detects the response of the kidney to injury in real time. Nat Med 17, 216–222 (2011).
Padanilam, B. J. Cell death induced by acute renal injury: a perspective on the contributions of apoptosis and necrosis. Am J Physiol Renal Physiol 284, F608–627 (2003).
Zhan, L. et al. Transient receptor potential vanilloid 4 inhibits rat HSC-T6 apoptosis through induction of autophagy. Mol Cell Biochem 402, 9–22 (2015).
Hatano, N. et al. Hypoxia-inducible factor-1alpha (HIF1alpha) switches on transient receptor potential ankyrin repeat 1 (TRPA1) gene expression via a hypoxia response element-like motif to modulate cytokine release. J Biol Chem 287, 31962–31972 (2012).
Jang, H. R. & Rabb, H. Immune cells in experimental acute kidney injury. Nat Rev Nephrol 11 (2015).
Butenko, O. et al. The increased activity of TRPV4 channel in the astrocytes of the adult rat hippocampus after cerebral hypoxia/ischemia. PLoS One 7, e39959 (2012).
Wu, Q. F. et al. Activation of transient receptor potential vanilloid 4 involves in hypoxia/reoxygenation injury in cardiomyocytes. Cell death & disease 8, e2828 (2017).
Dalsgaard, T., Sonkusare, S. K., Teuscher, C., Poynter, M. E. & Nelson, M. T. Pharmacological inhibitors of TRPV4 channels reduce cytokine production, restore endothelial function and increase survival in septic mice. Scientific reports 6, 33841 (2016).
Basile, D. P., Anderson, M. D. & Sutton, T. A. Pathophysiology of acute kidney injury. Comprehensive Physiology 2, 1303–1353 (2012).
Wei, X. et al. Activation of TRPV4 by dietary apigenin antagonizes renal fibrosis in deoxycorticosterone acetate (DOCA)-salt-induced hypertension. Clinical science 131, 567–581 (2017).
Sharfuddin, A. A. & Molitoris, B. A. Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol 7, 189–200 (2011).
Seth, R. K. et al. TRPV4 activation of endothelial nitric oxide synthase resists nonalcoholic fatty liver disease by blocking CYP2E1-mediated redox toxicity. Free radical biology & medicine 102, 260–273 (2017).
Marko, L. et al. Tubular Epithelial NF-kappaB Activity Regulates Ischemic AKI. J Am Soc Nephrol 27, 2658–2669 (2016).
Lee, H. T. et al. Isoflurane protects against renal ischemia and reperfusion injury and modulates leukocyte infiltration in mice. Am J Physiol Renal Physiol 293, F713–722 (2007).
Breggia, A. C. & Himmelfarb, J. Primary mouse renal tubular epithelial cells have variable injury tolerance to ischemic and chemical mediators of oxidative stress. Oxidative medicine and cellular longevity 1, 33–38 (2008).
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) (M.G. and D.N.M.), DAAD (German Academic Exchange Service) and Dr. Werner Jackstädt–Stiftung (to M.G.). M.G. and M.K. were further supported by the DZHK (German Centre for Cardiovascular Research) and by the BMBF (German Ministry of Education and Research. We thank Dr. Arnd Heuser MD, PhD, Dr. Sabine Bartel PhD, May–Britt Köhler, Martin Taube, Stefanie Schelenz and Max Müller for their help and expert technical assistance.
Author information
Authors and Affiliations
Contributions
M.M. and L.M. designed the experiments, performed qPCRs, analysed data and drafted the manuscript. A.B. performed the flow-cytometry experiments. E.V. performed the histological evaluation of kidneys. G.N. performed ischaemia-reperfusion surgery and collected blood and tissues. M.K. performed genotyping of the mice and revised the manuscript. L.M. and U.W. performed immunofluorescence staining. K.S.-O. supervised the histological analysis and revised the manuscript. W.B.L. revised the manuscript critically for important intellectual content. Y.H. revised the manuscript critically for important intellectual content. D.N.M. supervised the experimental work, revised the manuscript critically for important intellectual content. W.M.K. and M.G. supervised the experimental work, contributed to the drafting of the manuscript and revised the manuscript critically for important intellectual content. Final approval of the version to be published was obtained. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mannaa, M., Markó, L., Balogh, A. et al. Transient Receptor Potential Vanilloid 4 Channel Deficiency Aggravates Tubular Damage after Acute Renal Ischaemia Reperfusion. Sci Rep 8, 4878 (2018). https://doi.org/10.1038/s41598-018-23165-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-23165-0
This article is cited by
-
Calcium signalling and transport in the kidney
Nature Reviews Nephrology (2024)
-
The role of TRPV4 in programmed cell deaths
Molecular Biology Reports (2024)
-
Role of TRPC6 in kidney damage after acute ischemic kidney injury
Scientific Reports (2022)
-
In vivo silencing of amphiregulin by a novel effective Self-Assembled-Micelle inhibitory RNA ameliorates renal fibrosis via inhibition of EGFR signals
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
