
Abstract
Excessive alcohol consumption leads to severe alcoholic hepatitis (sAH) or chronic alcoholic pancreatitis (CAP) only in a subset of patients. We aimed to characterize the intestinal microbiota profiles of alcoholic patients according to the presence and nature of the complications observed: sAH or CAP. Eighty two alcoholic patients were included according to their complications: CAP (N = 24), sAH (N = 13) or no complications (alcoholic controls, AC, N = 45). We analyzed the intestinal microbiota by high-throughput sequencing. Bacterial diversity was lower in patients with CAP, who had a global intestinal microbiota composition different from that of AC. The intestinal microbiota composition of these two groups differed for 17 genera, eight of which were more frequent in patients with CAP (e.g. Klebsiella, Enterococcus and Sphingomonas). There was no significant difference in bacterial diversity between the sAH and CAP groups. However, 16 taxa were more frequent in sAH patients, and 10 were more frequent in CAP patients. After adjustment for confounding factors sAH patients were found to have higher levels of Haemophilus. For alcoholic patients, specific intestinal microbiota signatures are associated with different complications. Patients with CAP and sAH also display specific dysbiosis relative to AC.
Similar content being viewed by others

Introduction
Chronic alcohol consumption is the major cause of pancreatitis and liver disease1. There is currently no available treatment, other than weaning off alcohol, for alcoholic pancreatitis or alcoholic hepatitis (AH).
The factors determining susceptibility to alcohol toxicity in a given individual remain unclear. Genetic variability and environmental exposure to various factors have been shown to play a role in the tissue predilection of alcohol toxicity2,3, but these factors explain only a fraction of the cases observed, suggesting the likely involvement of other factors.
The intestinal microbiota was recently identified as a major factor in AH4, and we have shown that individual susceptibility to the development of AH depends on intestinal microbiota profile in an animal model of AH5,6. Moreover, alcohol intake induces dysbiosis, triggering intestinal inflammation7. An increase in intestinal permeability, leading to a high load of pro-inflammatory bacterial products in the blood and the portal vein, is frequently observed in alcoholics and in animal models of AH, and these features contribute to liver inflammation and disease progression8,9,10,11.
Limited data are available concerning the intestinal microbiota in chronic alcoholic pancreatitis (CAP). A recent study reported a decrease in Faecalibacterium prausnitzii and Ruminococcus bromii levels in patients with chronic pancreatitis12. It has also been suggested that the oral microbiota may play a role in pancreatic cancer and chronic pancreatitis (CP), consistent with an impact of the microbiota on the pancreas13,14. Nevertheless, no data concerning intestinal microbiota modifications in patients with alcoholic pancreatitis have been published. Moreover, despite similar levels of exposure to alcohol, the specific consequences for the intestinal microbiota of AH and CAP are unknown.
In this study, our aim was to investigate the intestinal microbiota profile of alcoholic patients according to the presence and nature of the complications observed: AH or CAP.
Results
Clinical characteristics of the study population
Eighty-two patients were recruited between September 2008 and July 2016. The patients’ characteristics are summarized in Table 1. Patients with sAH had higher body mass index (BMI), a lower quantity of alcohol intake but for longer durations as compared to CAP and AC. Proton pump inhibitors intake was lower in patients with CAP as compared to AC and sAH patients.
Intestinal microbiota profile in the study population
Intra-individual bacterial diversity (alpha diversity) according to the complication
Patients with CAP had a lower alpha diversity than AC suggesting a lower richness and evenness in the intestinal microbiota (Fig. 1A). This decrease in intestinal microbiota richness was also associated with a decrease in the amount of bacteria in feces, as shown by the quantification of bacterial DNA in stools (Fig. 1B). There was no difference in the alpha-diversity between CAP and sAH patients or between sAH and AC patients.

Intestinal microbiota profile in alcoholic patients without complications (AC) and with chronic alcoholic pancreatitis (CAP) or severe alcoholic hepatitis (sAH). (A) Box plots showing differences in microbiota alpha diversity based on the Shannon Index. (B) Concentration of total bacterial DNA in feces, and 16S rRNA DNA concentration (µg total DNA/mg; qPCR). P-values were calculated using a nonparametric ANOVA-test and Dunn post-hoc test for multiple comparisons. The rarefaction depth was 7,000. *p < 0.05, ***p < 0.001. (C) Principal coordinate analysis of the weighted UNIFRAC (p = 0.001) and (D) Unweighted UNIFRAC (p = 0.001) distances of the three groups.
Inter-individual diversity in the intestinal microbiota of the study population (beta diversity) according to the complication
We compared the global composition of the intestinal microbiota in the study population according to the complication by calculating UniFrac distances and comparing them between the three groups. Principal coordinate analysis (PCoA) of weighted (Fig. 1C) and unweighted (Fig. 1D) distances identified three clusters, corresponding to the three groups of patients (unweighted, p = 0.001 and weighted, p = 0.001, respectively). These results suggest that the intestinal microbiota has a different composition (unweighted) and structure (weighted) in our study population. Therefore we performed comparisons between each group (CAP vs. AC, CAP vs. sAH and sAH vs. AC) in order to investigate the specific differences between our study population.
Bacterial dysbiosis in patients with chronic alcoholic pancreatitis
Inter-individual diversity in patients with chronic alcoholic pancreatitis and alcoholic controls (beta diversity)
When comparing the global composition of the intestinal microbiota in CAP and AC patients, principal coordinate analysis (PCoA) of weighted (Fig. 2A) and unweighted (Fig. 2B) distances identified two clusters, corresponding to the two groups of patients (unweighted, p corr = 0.003 and weighted, p corr = 0.003, respectively). These results suggest that the intestinal microbiota has a different composition (unweighted) and structure (weighted) in CAP patients and AC.

Intestinal microbiota analysis in patients with chronic alcoholic pancreatitis (CAP) and alcoholic controls (AC). (A) Weighted UniFrac distances (quantitative method reflecting differences in the structure of the intestinal microbiota between the two groups) and (B) Unweighted UniFrac distances (qualitative method reflecting differences in the composition of the intestinal microbiota). Each point represents a subject and the distance between the points is proportional to the similarity in the intestinal microbiota. The distances between groups are significantly different (P-value < 0.050 in the ANOSIM test). (C) Cladogram showing the taxa with the largest differences in relative abundance between CAP (green) and AC (red) patients. Circle sizes in the cladogram plot are proportional to bacterial abundance. From inside to outside, the circles represent phylum, class, order, family and genus. Only taxa with a LDA score >2 and a p < 0.05 in the Wilcoxon signed rank test are shown.
Bacterial taxonomic differences between chronic alcoholic pancreatitis patients and alcoholic controls
We then investigated the specific differences in the intestinal microbiota at different taxonomic levels in these two groups. At phylum level, Proteobacteria were found to be more abundant in CAP patients than in AC (p corr = 0.02), whereas Bacteroidetes and Fusobacteria were less abundant (p corr = 0.006 and 0.03) (Supplementary Fig. 1). Fifty genera were found to differ in abundance between the two groups (p corr < 0.2) (Table 2 and Supplementary Fig. 1), and 43 of these genera were confirmed to be discriminant in LEfSe analysis (Table 2). Cladogram and LDA analyses identified the taxa specifically associated with CAP (Fig. 2C). After adjustment for sex, age, BMI, alcohol intake, smoking status, diabetes and proton-pump inhibitors use (using MaAsLin), 17 genera were found to differ in abundance between the two groups, with Klebsiella, Enterococcus and Sphingomonas specifically overrepresented in CAP patients (Table 2).
Differences in the intestinal microbiota between patients with chronic alcoholic pancreatitis and severe alcoholic hepatitis
Inter-individual diversity in chronic alcoholic pancreatitis and severe alcoholic hepatitis (beta diversity)
Weighted distances did not differ between the two groups (p corr = 0.23, Fig. 3A). However, there was a trend towards two different clusters based on unweighted distances (p corr = 0.07, Fig. 3B). These results indicate an absence of difference in the relative abundances of specific OTUs between CAP and sAH patients, but suggest that CAP and sAH patients may differ in terms of the presence/absence of some OTUs.

Intestinal microbiota analysis in patients with chronic alcoholic pancreatitis (CAP) and severe alcoholic hepatitis (sAH). (A) Weighted UniFrac distances (quantitative method) showing no difference in the structure of the intestinal microbiota between the two groups (p > 0.050); (B) Unweighted UniFrac distances (qualitative method), showing no difference in the composition of the intestinal microbiota between patients with chronic alcoholic pancreatitis (red) and patients with severe alcoholic hepatitis (blue), (p > 0.050). Each point represents a subject and the distance between the points is proportional to the similarity in their intestinal microbiota. (C) Cladogram showing the taxa with the largest differences in relative abundance between CAP (red) and sAH (green) patients. Circle sizes in the cladogram plot are proportional to bacterial abundance. From inside to outside, the circles represent phylum, class, order, family and genus. Only taxa with a LDA score > 2 and a p < 0.05 in the Wilcoxon signed rank test are shown.
Bacterial taxonomic differences between chronic alcoholic pancreatitis and severe alcoholic hepatitis
No differences in the intestinal microbiota bacterial composition at phylum level were found between CAP and sAH patients (Supplementary Fig. 1). We found differences in abundance between CAP and sAH patients for 29 genera (p < 0.05, p corr < 0.20) (Table 3). For these genera, the abundance of Haemophilus, Sutterella, Campylobacter, Lactobacillus, Faecalibacterium, Prevotella, Paraprevotella and Fusobacterium was higher in sAH patients and that of Serratia, Acinetobacter, Pseudomonas and Enterococcus was higher in CAP patients. LEfSe analysis showed differences between the two groups for 26 taxa (10 being more abundant in CAP and 16 in sAH, Fig. 3C and Table 3). However, after adjustment for sex, age, BMI, alcohol intake, smoking status, diabetes and proton pump inhibitors use (by MaAsLin), only one taxon remained statistically significant between the two groups (Table 3). The relative abundance of Haemophilus was 100 times higher in sAH patients than in CAP patients (p corr = 0.002). We were able to trace 85% of the Haemophilus reads to a single species: Haemophilus parainfluenzae.
Differences in intestinal microbiota profile between alcoholic patients without complications and patients with severe alcoholic hepatitis
Principal coordinate analysis (PCoA) of unweighted (Supplemental Fig. 2A) distances revealed two clusters, corresponding to the two groups of patients (p corr = 0.03). There was a trend towards a difference in the weighted distances (p corr = 0.07, Supplemental Fig. 2B). These results suggest a different composition (unweighted distances) and a trend towards a different structure (weighted distances) of the intestinal microbiota between the two groups. When comparing the specific taxa, differences between sAH patients and AC (p < 0.05, p corr < 0.20) were observed for 31 genera. LEfSe analysis revealed 23 taxa that were differed between the two groups (10 were more abundant in sAH patients and 13 were more abundant in AC, Supplementary Fig. 2C).
Specific differences in taxa between groups
When analyzing the taxa that were different between the three groups (in LEfSe, CAP vs. AC, sAH vs. AC and sAH vs. CAP), seven taxa differed only for comparisons of sAH and CAP, 17 differed in comparisons of CAP and AC and six differed in comparisons of sAH and AC. (Fig. 4). For taxa displaying differences only between CAP and sAH, the abundances of Favobacterium, SMB53 and Anaerotruncus were higher in CAP patients and those of Dialister, Clostridium, Campylobacter and a member of the S24–7 family were higher in sAH patients, suggesting an effect independent of alcohol consumption on the development of theses complications.

Venn diagram for the significant taxa (genus level) differing in all three analyses (CAP vs. AC, CAP vs. sAH and AC vs. sAH), showing the taxa displaying modifications specific to a particular complication (LDA score >2 and a p < 0.05 determined in a Wilcoxon signed rank test). CAP, chronic alcoholic pancreatitis; AC, alcoholic controls; sAH, severe alcoholic hepatitis.
Discussion
In the present study, we analyzed the intestinal microbiota associated with CAP and its specificity. As alcohol intake alters intestinal microbiota composition7, we used a group of alcoholic patients without alcohol-related organ complications as the control group. Furthermore, as patients with excessive alcohol consumption may develop different complications, we investigated the intestinal microbiota in patients with CAP and with alcoholic cirrhosis presenting a sAH. We show that the intestinal microbiota associated with CAP has a different structure and composition as compared to AC and we identified several members of the intestinal microbiota as significantly associated with CAP. We also observed several differences in the bacterial composition of the intestinal microbiota between patients with CAP and sAH.
CAP patients had a lower microbial diversity than AC, whereas microbial diversity did not differ between CAP vs. sAH and AC vs. sAH patients. High bacterial diversity is usually associated with a healthy state and is thought to be crucial for the maintenance of immune system homeostasis15. A lower alpha diversity in patients with chronic pancreatitis has been reported before but only 25% of the patients included had alcoholic pancreatitis12. Moreover, blockade of the acinar cell exocytose of antimicrobial peptides in mice is associated with intestinal dysbiosis, inflammation and systemic bacterial translocation; this suggests that the secretion of antimicrobial peptides by pancreatic acinar cells regulates intestinal microbiota composition16. In AH, antimicrobial peptide levels are also low and are dependent on intestinal microbiota composition. Moreover, intestinal microbiota manipulation by fecal microbiota transplantation or prebiotics administration, prevents the decrease in antimicrobial peptide levels in the intestine and has hepatoprotective effects6. Overall, our results suggest that the decrease in intestinal microbiota richness and diversity may be due to the low antimicrobial peptide levels and exocrine pancreatic insufficiency observed in chronic pancreatitis.
The relative abundances of several potential pathogenic taxa (Klebsiella, Enterococcus, Pseudomonas) associated with systemic inflammations and infection17 were higher in CAP patients than in AC.
Previous studies have suggested that some bacteria may be involved in the pathogenesis of pancreatic diseases. Granulicatella adiacens and Streptococcus mitis from the oral microbiota have been shown to be associated with chronic pancreatitis13 while Helicobacter pylori infection has been shown to be associated with autoimmune pancreatitis18. Microorganisms may infect the pancreas through ascending gastric infections or retrograde transfer from the small intestine19. The intestinal dysbiosis observed in alcoholic patients was associated with an increase in intestinal permeability that facilitates the translocation of intestinal microbiota components and contributes to liver injury20. Moreover, a fluorescence in situ hybridization (FISH) study in the pancreatic duct biopsy specimens of patients with pancreatitis revealed the presence of a bacterial biofilm, including members of the Enterobacteriaceae family21.
We found that CAP patients had a higher relative abundance of Enterococcus. In a study that investigated intestinal microbiota using qPCR, Enterococcus was increased in patients with acute pancreatitis and positively correlated to plasma endotoxin levels22. E. faecalis is associated with impaired intestinal permeability via its gelatinase that alters the epithelial barrier and contributes to intestinal inflammation23. This could facilitate bacterial translocation and bile colonization, which may then come into contact with the pancreas. Conversely, Enterococci produce antimicrobial compounds24. This may account for decreased bacterial richness in CAP patients observed here, and the increases in Enterococcus, Klebsiella, Pseudomonas and Serratia, which are pathogenic in humans.
Haemophilus parainfluenzae (Pasteurellaceae family) abundance was increased in sAH patients. This commensal bacteria may also act as an opportunistic pathogen, causing invasive infections. Moreover, higher abundance of Pasteurellaceae was associated with acute-on-chronic liver failure and were found also to be an independent predictor of mortality in these patients25.
The dysbiosis observed in CAP was associated with a lower abundance of Faecalibacterium in CAP as compared to both AC and sAH patients. A decrease in the abundance of Faecalibacterium prausnitzii has also been reported in patients with chronic pancreatitis12, suggesting that this difference may be related to the pancreatitis itself rather than its cause. F. prausnitzii has anti-inflammatory activity26 and stimulates mucin and tight-junction protein synthesis, thereby improving intestinal barrier function27.
Interestingly, CAP patients also had lower levels of Lactobacillus than sAH patients. The abundances of Lactobacillus species are low in humans with alcoholic cirrhosis and in animal models of this disease28 and they can attenuate the features of alcoholic liver disease29. Lactobacilli secrete lactic acid, which reduces intestinal pH, the growth of commensal microbiota30 and the amounts of luminal bacterial products passing into the systemic circulation. Based on our results, we hypothesize that the lower levels of Lactobacillus in CAP patients than in sAH patients may underlie the higher abundances of pathogenic bacteria, such as Pseudomonas, Klebsiella and Serratia. However, Lactobacilli use in severe acute pancreatitis patients did not reduce the risk of infectious complications and was associated with an increase risk of mortality31.
In our study, there were less proton pump inhibitors users among patients with CAP as compared to the other two groups. This could have an impact on the intestinal microbiota as their use was associated with a decrease in the Bacteroidetes and an increase in Firmicutes at the phylum level and an increase in Holdemania filiformis and decrease in Pseudoflavonifractor capillosus at the species level32. Therefore, to take into account these differences, we adjusted for this potential confounding factor in the MaASLin model. Thus the changes observed in our populations were independent of the use of proton-pump inhibitors.
Our study has several limitations. Firstly, the cross-sectional design of the study precludes investigations of the underlying mechanisms and of the time-sequence of the association. Secondly, we lack an external validation cohort that could confirm our results. Nevertheless, we clearly demonstrate that, despite a similar amount of alcohol consumption, a specific intestinal microbiota is associated to the nature of alcohol-induced complication. Finally, the low power of this study, due to the small number of patients included, may also account for the smaller number of genera identified as differing between groups in MaAsLin than in the other analyses. However, there is no method to estimate a sample size for microbiota studies.
In conclusion, a specific intestinal microbiota signature is associated with the nature of the complication in alcoholic patients. However, it remains unclear whether CAP dysbiois is the cause or simply a consequence of pancreatitis. Further studies are required, with a longitudinal design, in which the microbiota of alcoholic patients can be assessed regularly and correlations with outcome can be assessed, together with studies in humanized animal models. Moreover, the specific microbial signature could be useful in identifying patients at risk to develop alcohol related complications and thus improve the management of these patients. These results are a key first step towards understanding why only 5% of alcoholic patients develop alcoholism-related pancreatic disorders.
Methods
Patients
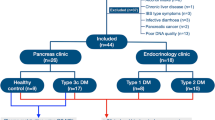
For this prospective study, patients admitted in two tertiary hospitals for CAP or the management of excessive drinking were included in this study. Alcoholic patients were eligible for inclusion if they were between 17 and 75 years old and had been consuming at least 60 g of alcohol/day for more than five years, and were negative for hepatitis B surface antigens, hepatitis C or any other cause of liver disease. The exclusion criteria were gastrointestinal bleeding, bacterial infection, hepatocellular carcinoma, any other carcinoma, other severe associated disease, presence of anti-HIV antibodies, antibiotic intake in the last three months and refusal to undergo liver biopsy if required (abnormal liver function).
Three groups of patients were recruited:
-
CAP patients (n = 24, recruited at Beaujon University Hospital, Clichy, France) admitted for an acute bout of pancreatitis or chronic pain. The additional inclusion criteria for this group were: 1) patients with CAP admitted for an acute bout of pancreatitis or chronic pain; 2) no other cause of chronic pancreatitis identified, despite an exhaustive search; 3) absence of pancreatic or extra-pancreatic tumors and 4) absence of liver disease. CAP was diagnosed on the basis of the presence of at least one of the following: pancreatic calcifications visible on CT scan or endoscopic ultrasonography; moderate-to-severe pancreatic ductal lesions on endoscopic retrograde or magnetic resonance pancreatography (Cambridge classification)33; histological features typical of pancreatitis on an adequate surgical specimen of the pancreas or recurrent acute alcoholic pancreatitis. Recurrent acute pancreatitis was defined by more than two bouts of acute abdominal pain with increases in serum pancreatic enzyme levels to more than three times the upper limit of the normal range.
-
Alcoholic controls (AC, n = 45, recruited at Antoine-Béclère University Hospital, Clamart, France) admitted for the management of excessive drinking. The additional inclusion criteria for this group were: 1) admission for the management of excessive drinking, 2) no signs of acute pancreatitis or CAP (based on symptoms and morphological features), 3) no signs of liver disease (as described previously5).
-
Alcoholic patients with severe alcoholic hepatitis and alcoholic cirrhosis (sAH, n = 13, recruited at the Antoine-Béclère University Hospital, Clamart, France) as defined by a histological score for alcoholic hepatitis ≥ 6 and neutrophilic infiltration on a liver biopsy34. The additional inclusion criteria for this group were: 1) admission for the management of excessive drinking, 2) no signs of acute pancreatitis or CAP (based on symptoms and morphological features). Feces collection was done before any steroid therapy.
General demographic and clinical characteristics were recorded for all patients at inclusion. Also a standardized questionnaire was used to collect information about alcohol consumption35.
We choose to compare the patients with acute bouts of CAP or pain exacerbation to sAH patients with alcoholic cirrhosis in order to have 2 groups of patients with an acute manifestation of an underlying chronic condition.
This study received IRB approval from the Ile de France VII ethics committee (Bicêtre Hospital, 94270 le Kremlin-Bicêtre, France). All methods were performed in accordance with the relevant local guidelines and regulations and all patients provided written informed consent for participation in the study.
Collection of feces
Feces from the first bowel movement after admission were collected. An anaerobiosis generator (Anaerocult, Merck, Darmstadt, Germany) was used to favor the preservation of anaerobic bacteria. The samples were processed within 24 h and frozen for bacterial preservation at −80 °C and DNA extraction, as previously described6. Briefely, Bacterial DNA was obtained by homogenizing cecal content in a Guanidinium thiocyanate containing lysis buffer using a Fast Prep homogenizer. High quality bacterial DNA was extracted by successive steps of purification and precipitation using “Laboratory-made” buffers36. PCR were performed to prepare amplicons using V3-V4 oligonucleotides (PCR1F_460: 5′CTTTCCCTACACGACGCTCTTCCGATCTACGGRAGGCAGCAG3′, PCR1R_460: 5′GGAGTTCAGACGTGTGCTCTTCCGATCTTACCAGGGTATCTAATCCT3′). Amplicon quality was verified by gel electrophoresis and they were sent to the GenoToul plateform (Toulouse, France) for sequencing. All the samples were collected and processed in the same way.
Analysis of the intestinal microbiota by 16s ribosomal RNA sequencing
The composition of the intestinal microbiota was analyzed by high-throughput sequencing with Illumina MiSeq technology, targeting the 16S ribosomal DNA V3-V4 region. Data was analyzed using QIIME v1.9.0. Paired-end reads were assembled with PANDAseq v 2.7, to generate the sequence of a 450-base pair amplicon37. Reads were demultiplexed and processed with the quantitative insights into microbial ecology (QIIME v1.9.0) pipeline, using its default parameters38. Chimeric sequences were identified de novo, on the basis of references, and were removed with usearch6139. The non-chimeric sequences were then clustered into operational taxonomic units (OTUs) displaying at least 97.0% sequence similarity, by a closed reference-based picking approach in UCLUST software applied to the Greengenes 13.8 database of bacterial 16S rDNA sequences40. The mean number of quality-controlled reads was 26008 ± 8106.69 (mean ± SD) per sample (minimum count: 8370.0, maximum count: 79884.0). After rarefaction at 7,000 reads per sample, bacterial alpha diversity was estimated on the basis of the Shannon’s index. OTUs with a prevalence <5% were removed from the analysis.
We assessed beta diversity with weighted and unweighted UniFrac distances. The weighted Unifrac metric is weighted by the difference in the abundance of OTUs from each community, whereas unweighted UniFrac considers only the absence/presence of OTUs, providing different information. Both are phylogenetic beta diversity metrics.
We investigated the OTUs not identified by QIIME further, using the BLASTN program (vBLAST + 2.6.0) from NCBI Blast, against the NCBI 16 s Microbial database. Only hits with an overall sequence identity of at least 97% were considered.
Statistical analyses
The results are expressed as means ± SEM. Alpha diversity comparisons were performed with nonparametric Student’s t-tests and Monte Carlo permutations in QIIME.
Taxa were compared using Mann–Whitney U-tests and Adonis test was used to compare distance matrices in QIIME. Benjamini–Hochberg false discovery rate (FDR) correction (p corr) was used to correct for multiple hypothesis testing, when applicable. A FDR corrected p value < 0.2 was considered as statistical significant in these comparisons due to the exploratory design of the study.
LDA effect size (LEfSe) analysis was performed to identify the taxa displaying the largest differences in abundance in the microbiota between groups41. Only taxa with an LDA score >2 and a significance of α < 0.05, as determined in Wilcoxon signed-rank tests, are shown. The sizes of the circles in the cladogram plot are proportional to bacterial abundance.
MaAsLin (multivariate analysis by linear models) was used to identify associations between clinical metadata and microbial community abundance or function42. Sex, age, BMI, alcohol intake, smoking status, diabetes and proton pump intake were included as covariates in the MaAsLin. Relative abundances were subjected to arcsine root transformation before regression analyses using MaAsLin. Both LEfSe and MaAsLin were accessed online (http://huttenhower.sph.harvard.edu/galaxy/). For the MaAsLin compariason, a corrected p value < 0.2 (p corr) was considerd statisicaly significant.
The remaining comparisons were performed with R software v2.14.1. Unpaired t-tests or Mann–Whitney U-tests were used to compare continuous data between groups, according to data distribution. Chi2 or Fisher’s exact tests were used to compare discrete parameters between groups. A p-value < 0.05 was considered to be statistically significant, unless specified.
References
Yang, A. L., Vadhavkar, S., Singh, G. & Omary, M. B. Epidemiology of alcohol-related liver and pancreatic disease in the United States. Arch. Intern. Med. 168, 649–656 (2008).
O’Shea, R. S., Dasarathy, S. & McCullough, A. J. Practice Guideline Committee of the American Association for the Study of Liver Diseases & Practice Parameters Committee of the American College of Gastroenterology. Alcoholic liver disease. Hepatol. Baltim. Md 51, 307–328 (2010).
Whitcomb, D. C. et al. Common genetic variants in the CLDN2 and PRSS1-PRSS2 loci alter risk for alcohol-related and sporadic pancreatitis. Nat. Genet. 44, 1349–1354 (2012).
Chen, P. et al. Microbiota Protects Mice Against Acute Alcohol-Induced Liver Injury. Alcohol. Clin. Exp. Res. 39, 2313–2323 (2015).
Llopis, M. et al. Intestinal microbiota contributes to individual susceptibility to alcoholic liver disease. Gut 65, 830–839 (2016).
Ferrere, G. et al. Fecal microbiota manipulation prevents dysbiosis and alcohol-induced liver injury in mice. J. Hepatol. 66, 806–815 (2017).
Mutlu, E. A. et al. Colonic microbiome is altered in alcoholism. Am. J. Physiol. Gastrointest. Liver Physiol. 302, G966–978 (2012).
Mutlu, E. et al. Intestinal dysbiosis: a possible mechanism of alcohol-induced endotoxemia and alcoholic steatohepatitis in rats. Alcohol. Clin. Exp. Res. 33, 1836–1846 (2009).
Mathurin, P. et al. Exacerbation of alcoholic liver injury by enteral endotoxin in rats. Hepatol. Baltim. Md 32, 1008–1017 (2000).
Puri, P. et al. The circulating microbiome signature and inferred functional metagenomics in alcoholic hepatitis. Hepatol. Baltim. Md, https://doi.org/10.1002/hep.29623 (2017).
Perlemuter, G. Circulating bugs in blood in alcoholic liver disease! Hepatol. Baltim. Md, https://doi.org/10.1002/hep.29624 (2017).
Jandhyala, S. M. et al. Altered intestinal microbiota in patients with chronic pancreatitis: implications in diabetes and metabolic abnormalities. Sci. Rep. 7, 43640 (2017).
Farrell, J. J. et al. Variations of oral microbiota are associated with pancreatic diseases including pancreatic cancer. Gut 61, 582–588 (2012).
Fan, X. et al. Human oral microbiome and prospective risk for pancreatic cancer: a population-based nested case-control study. Gut, https://doi.org/10.1136/gutjnl-2016-312580 (2016).
Ley, R. E., Peterson, D. A. & Gordon, J. I. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 124, 837–848 (2006).
Ahuja, M. et al. Orai1-Mediated Antimicrobial Secretion from Pancreatic Acini Shapes the Gut Microbiome and Regulates Gut Innate Immunity. Cell Metab. 25, 635–646 (2017).
Marshall, J. C., Christou, N. V., Horn, R. & Meakins, J. L. The microbiology of multiple organ failure. The proximal gastrointestinal tract as an occult reservoir of pathogens. Arch. Surg. Chic. Ill 1960 123, 309–315 (1988).
Rieder, G., Karnholz, A., Stoeckelhuber, M., Merchant, J. L. & Haas, R. H pylori infection causes chronic pancreatitis in Mongolian gerbils. World J. Gastroenterol. 13, 3939–3947 (2007).
Nilsson, H.-O., Stenram, U., Ihse, I. & Wadstrom, T. Helicobacter species ribosomal DNA in the pancreas, stomach and duodenum of pancreatic cancer patients. World J. Gastroenterol. 12, 3038–3043 (2006).
Parlesak, A., Schäfer, C., Schütz, T., Bode, J. C. & Bode, C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J. Hepatol. 32, 742–747 (2000).
Swidsinski, A. et al. Bacterial biofilm within diseased pancreatic and biliary tracts. Gut 54, 388–395 (2005).
Tan, C. et al. Dysbiosis of Intestinal Microbiota Associated With Inflammation Involved in the Progression of Acute Pancreatitis. Pancreas 44, 868–875 (2015).
Steck, N. et al. Enterococcus faecalis metalloprotease compromises epithelial barrier and contributes to intestinal inflammation. Gastroenterology 141, 959–971 (2011).
Karimaei, S. et al. Antibacterial potential and genetic profile of Enterococcus faecium strains isolated from human normal flora. Microb. Pathog. 96, 67–71 (2016).
Chen, Y. et al. Gut dysbiosis in acute-on-chronic liver failure and its predictive value for mortality. J. Gastroenterol. Hepatol. 30, 1429–1437 (2015).
Rossi, O. et al. Faecalibacterium prausnitzii Strain HTF-F and Its Extracellular Polymeric Matrix Attenuate Clinical Parameters in DSS-Induced Colitis. PloS One 10, e0123013 (2015).
Wrzosek, L. et al. Bacteroides thetaiotaomicron and Faecalibacterium prausnitzii influence the production of mucus glycans and the development of goblet cells in the colonic epithelium of a gnotobiotic model rodent. BMC Biol. 11, 61 (2013).
Yan, A. W. & Schnabl, B. Bacterial translocation and changes in the intestinal microbiome associated with alcoholic liver disease. World J. Hepatol. 4, 110–118 (2012).
Chen, P. et al. Supplementation of saturated long-chain fatty acids maintains intestinal eubiosis and reduces ethanol-induced liver injury in mice. Gastroenterology 148, 203–214.e16 (2015).
Aiba, Y., Suzuki, N., Kabir, A. M., Takagi, A. & Koga, Y. Lactic acid-mediated suppression of Helicobacter pylori by the oral administration of Lactobacillus salivarius as a probiotic in a gnotobiotic murine model. Am. J. Gastroenterol. 93, 2097–2101 (1998).
Besselink, M. G. et al. Probiotic prophylaxis in predicted severe acute pancreatitis: a randomised, double-blind, placebo-controlled trial. Lancet Lond. Engl. 371, 651–659 (2008).
Clooney, A. G. et al. A comparison of the gut microbiome between long-term users and non-users of proton pump inhibitors. Aliment. Pharmacol. Ther. 43, 974–984 (2016).
Conwell, D. L. et al. American Pancreatic Association Practice Guidelines in Chronic Pancreatitis: evidence-based report on diagnostic guidelines. Pancreas 43, 1143–1162 (2014).
Alcoholic liver disease: morphological manifestations. Review by an international group. Lancet Lond. Engl. 1, 707–711 (1981).
Williams, G. D., Proudfit, A. H., Quinn, E. A. & Campbell, K. E. Variations in quantity-frequency measures of alcohol consumption from a general population survey. Addict. Abingdon Engl. 89, 413–420 (1994).
Tomas, J. et al. Primocolonization is associated with colonic epithelial maturation during conventionalization. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 27, 645–655 (2013).
Masella, A. P., Bartram, A. K., Truszkowski, J. M., Brown, D. G. & Neufeld, J. D. PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics 13, 31 (2012).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336 (2010).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinforma. Oxf. Engl. 26, 2460–2461 (2010).
DeSantis, T. Z. et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072 (2006).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60 (2011).
Morgan, X. C. et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 13, R79 (2012).
Acknowledgements
The authors thank the NED team (Olivier Zemb, Béatrice Gabinaud) and the GeT-PlaGe platform for their help with the sequencing data. This work was supported by INSERM, Univ Paris-Sud, SNFGE (Société Nationale Francophone de Gastro-entérologie), AFEF (French Association for the Study of Liver), IREB/FRA (Institut de Recherches sur les Boissons/Fondation pour la recherche en alcoologie), IRIS (Institut de Recherches Internationales Servier), ERAB (The European Foundation for Alcohol Research), FRM (Fondation pour la Recherche Médicale) and Biocodex. INSERM U996 is a member of the Laboratory of Excellence LERMIT supported by a grant from the Agence Nationale de la Recherche (ANR-10-LABX-33). DC was supported by research fellowship from INSERM.
Author information
Authors and Affiliations
Contributions
D.C. designed the project, performed the post-sequencing data processing and the statistical analysis, and wrote the manuscript. V.R. designed the project, included patients, supervised experiments and wrote the paper. A.M.C. designed, performed and supervised experiments and wrote the manuscript. LW designed the project and performed presequencing preparation procedures. C.S.V. designed the project and included patients. V.P. performed presequencing preparation procedures. G.P. designed the project, included patients, supervised experiments and wrote the paper. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing Interests
D.C. has received travel funds from Biocodex. G.P. has, in the last three years, received travel funds from Abbvie, Gilead, Biocodex and Adare, consulting fees from Biocodex, Gilead, Servier, Adare and Pileje, and royalties from Elsevier-Masson, John Libbey Eurotext, Solar and Flammarion/Versilio.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ciocan, D., Rebours, V., Voican, C.S. et al. Characterization of intestinal microbiota in alcoholic patients with and without alcoholic hepatitis or chronic alcoholic pancreatitis. Sci Rep 8, 4822 (2018). https://doi.org/10.1038/s41598-018-23146-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-23146-3
This article is cited by
-
Alcoholic liver disease: a new insight into the pathogenesis of liver disease
Archives of Pharmacal Research (2022)
-
Impaired autophagy increases susceptibility to endotoxin-induced chronic pancreatitis
Cell Death & Disease (2020)
-
Identification of Dysfunctional Gut Microbiota Through Rectal Swab in Patients with Different Severity of Acute Pancreatitis
Digestive Diseases and Sciences (2020)
-
Demystifying the manipulation of host immunity, metabolism, and extraintestinal tumors by the gut microbiome
Signal Transduction and Targeted Therapy (2019)
-
Alcohol-induced changes in the gut microbiome and metabolome of rhesus macaques
Psychopharmacology (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
