
Abstract
To analyse the function of the duck plague virus (DPV) glycoprotein J homologue (gJ), two different mutated viruses, a gJ deleted mutant ΔgJ and a gJR rescue mutant gJR with US5 restored were generated. All recombinant viruses were constructed by using two-step of RED recombination system implemented on the duck plague virus Chinese virulent strain (DPV CHv) genome cloned into a bacterial artificial chromosome. DPV-mutants were characterized on non-complementing DEF cells compared with parental virus. Viral replication kinetics of intracellular and extracellular viruses revealed that the ΔgJ virus produce a 10-fold reduction of viral titers than the gJR and parental virus, which especially the production of extracellular infectivity was affected. In addition, the ΔgJ virus produced viral plaques on DEF cells that was on average approximately 11% smaller than those produced by the gJR and parental viruses. Electron microscopy confirmed that although DPV CHv without gJ could efficiently carry out viral replication, virion assembly and envelopment within infected cells, the ΔgJ virus produced and accumulated high levels of anuclear particles in the nuclear and cytoplasm. These results show that the gJ slightly impaired in viral replication, virion assembly and cell-to-cell spread, and is not essential in virion envelopment.
Similar content being viewed by others


Introduction
Duck plague(DP), also called duck virus enteritis (DVE), is one of the major acute, fatal and contagious diseases of duck, geese, and swans, characterized by vascular damage, tissue hemorrhages, digestive mucosal eruptions, and lesions of lymphoid organs. Due to high mortality, morbidity as well as decreased egg production and hatchability, DP caused significant economic losses around the world1,2,3,4. DPV, which belong to the genus Mardivirus, subfamily Alpha-herpesvirinae, and family Herpesviridae, is the pathogen of DP disease3,4. The entire DNA sequence of the DPV has been determined few years ago5. However, not too many genes and gene products have been characterized in term of relevance or functional cooperation in viral lifecycle6,7,8.
Herpesvirus glycoproteins paly important roles in the different stages of viral lifecycle, such as enter the target cells, direct cell-to-cell spread, and the egress of virions from infected cells9,10, and 12 glycoproteins have been identified and designated gB, gC, gD, gE, gG, gH, gI, gJ, gK, gL, gM and gN in herpes simplex virus type 1 (HSV-1)11. DPV glycoproteins are named in accordance with the nomenclature used for the HSV-1 glycoproteins. However, previously studies about DPV focused on the epidemiology and prevention, instead of the studying of molecular biology and the function of glycoproteins5. To date only a few gens such as gC and gE from DPV have been addressed by studying mutant viruses12,13. gC involved in alphaherpesvirus adsorption were present in the DPV genome and relatively conserved, but the function of DPV gC also makes a difference with other alphaherpesvirus, which is independent of interaction with heparin sulfate of cell surface12,14. Therefore, although alphaherpesvirus share many common strategies for their replication cycle15, they also keep some individually patterns to invade and infect target cells.
The US5 gene of alpha-herpesvirus encodes glycoprotein J (gJ), which shares lowly nucleotide and amino acid similarity among alpha-herpesvirus sub-family. Exploring the functions of gJ is far behind other glycoproteins. The only previously reported function of alpha-herpesvirus gJ was its ability to inhibit apoptosis and viral egress, and the mechanisms of these functions have not been fully revealed16,17,18,19,20. DPV glycoprotein J (gJ) is encoded by US5 in the viral genome which is positional homologue in the HSV-1 genome. Comparisons of nucleotide and deduced amino acid sequences uncovered that gJ is lowly conserved throughout the alpha-herpesvirus subfamily, but highly conserved among the different strain of DPV.
In the present study, to clarify roles of gJ in DPV lifecycle, we describe the construction of a DPV mutant virus, based on the infectious BAC clone by using two-step of RED recombination for generation of US5 deletion and revertant mutant in E.coli. Viral particles lacking gJ in the envelope were produced in non-complementing DEF cells, which demonstrated that US5 gene of DPV is nonessential for virus replication. We determined kinetics of virus growth, relative plague morphology, and conducted ultrastructural visualization of gJ-deficient mutant and parental virions in the same DPV genetic background to gain an understanding of the glycoprotein J contribution in infectious virion replication cycle. The results firstly show that gJ is nonessential for DPV virion replication and slightly impaired in viral replication, virion assembly and cell-to-cell spread.
Results
Construction and molecular analysis of recombinant virus
The DPV genome has been cloned into a BAC by our laboratory, which enables the rapid and efficient genetic manipulation of the DPV genome in E. Coli DH10B. To investigate of the relative role of DPV gJ, the gJ deletion mutant and its revertant was generated using the two-step Red recombination mutagenesis system implemented on the pBeloBAC11 bacterial artificial chromosome carrying the DPV genome, as described in Materials and Methods. The DPV CHv-BAC-ΔgJ virus is the deletion of the entire gJ open reading frame (ORF) (Fig. 1). Meanwhile, to eliminate the small possibility that this deletion causes indirect epigenetic effects on foreign DNA insertion, we constructed the DPV CHv-BAC-gJR virus which is the revertant of the entire gJ ORF.

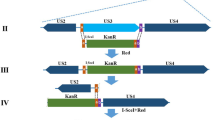
Genomic map of mutated genes. (a) Represents the prototypic arrangement of the DPV genome with the unique long (UL) and unique short (US) regions flanked by the terminal repeat (TR) and internal repeat (IR) regions. (b) Shows expanded genomic regions of the US4, US5, and US6 open reading frames. (c) Shows the deletion of the whole US5 open reading frames.
The engineered mutations were confirmed via diagnostic PCR, restriction fragment length polymorphism (RFLP) analysis and DNA sequencing (data not shown). Specifically, the BAC DNAs of DPV CHv-BAC-ΔgJ and DPV CHv-BAC-gJR were extracted, and then were identified by primer UL48 (2555 bp), gJ (1820 bp), sopB (966 bp) and repA (681 bp), among which UL48 is a DPV conversed gene to identify the existence of DPV genome and sopB as well as repA are two important genes to identify BAC-Mini-F sequence (Fig. 2A). As a result, we found repA, sopB and UL48 can be detected in all recombinant viruses, which indicated the presence of BAC component and DPV genome in recombinant viruses. What’s more, the whole fragment of gJ gene can be detected in the parental virus of DPV CHv-BAC and the revantant virus of DPV CHv-BAC-gJR, but cannot be found in the DPV CHv-BAC-ΔgJ, which showed that the construction of gJ-deleted DPV virus was successful.

Identification of the recombinant viruses. (A) PCR analysis of the gJ deletion mutant. The BAC DNAs of DPV CHv-BAC, DPV CHv-BAC-ΔgJ and DPV CHv-BAC-ΔgJR were extracted and amplified by PCR using the indicated primers. The positions of DNA molecular size markers are shown on the lift side. (B) Restriction fragement length polymorphism analysis of recombinant virus. Indicated the orientation and real Gel analysis of DPV CHv-BAC, DPV CHv-BAC-ΔgJ and DPV CHv-BAC-gJR digested by BamHI, respectively. The asterisk was made to show the different band, and the image on the right was obtained by software simulation.
Furthermore, the BAC DNA of recombinant virus extracted from the PCR identified clones was digested with BamHI for RFLP analysis. As a result, the restriction patterns of BamHI disgestion products of DPV CHv-BAC, DPV CHv-BAC-ΔgJ and DPV CHv-BAC-gJR were as same as we prediction, respectively (Fig. 2B).
Rescue and confirm of the recombinant virus
To generate virus stocks from the mutant BAC genomic constructs, individual BAC DNAs were transfected into DEF cells. Passage of these viruses up to three times in DEF cells eliminated the effects of transient transfection of plasmid containing EGFP and did not led to any phenotypic revertants, which also purified and enriched the recombinant viruses (Fig. 3A).

Rescue mutant viruses and Identification of gJ expression. (A) Purification and enrichment of mutant viruses. Purification and enrichment of mutant viruses were obtained by the three times passage after transfection. (B) Immunofluorescence detection of gJ expression. DEF cells were infected at 1000 TCID50, and gJ expression was detected by indirect immunofluorescence at 36 hpi. Rabbit anti-gJ were used as primary antibody, and goat anti-rabbit IgG TRITC were used as secondary antibody. (C) Anti-gJ monoclonal antibody (MAb) was used to detect gJ via western immunoblot analysis. DPV CHv-BAC-ΔgJ infected DEF cells were detected as parental virus.
The mutant viruses of DPV CHv-BAC-ΔgJ and DPV CHv-BAC-gJR were identified indirect immunouorescence assay (IFA) and western blotting. The expression of gJ protein in parental virus of DPV CHv-BAC and derived recombinant DPV CHv-BAC-ΔgJ and DPV CHv-BAC-gJR infected host cells were recongnized by polyclonal rabbit anti-gJ in IFA and WB (Fig. 3B,C).
One-step viral growth kinetics
To examine the effect of the gJ-deleted engineered mutation on virus replication, DEF cells were infected in an MOI of 0.01 with either the parental or each mutant viruses and virus growth kinetics within infected cells and their supernatants were performed as described in Materials and Methods. Compared to the parental virus, gJ-deleted mutant virus and its revantant virus accumulated infectious virions within infected cells at similar rates. As the result (Fig. 4), growth properties of the repaired virus DPV CHv-BAC-gJR resembled those of DPV CHv-BAC, whereas DPV CHv-BAC-ΔgJ presented with a marked growth defect. Within these experiments, the replication of viruses kept quiescence at the first 12 h after infection, then significant increases were observed during the whole observation time in supernatants while it stopped increasing at 72 hpi and slightly dropped after that in cells except DPV CHv-BAC-ΔgJ mutant virus. It was worthy to mention that viral titers in cells were higher than titers in medium at 24 hpi. However, on the contrary, at 48 hip, titers of mature viral particles in supernatant was more than virus in cells. Moreover, DPV CHv-BAC- ΔgJ virus replicated 10 times less efficiently than DPV CHv-BAC- gJR and DPV-CHv-BAC. The conclusion is that although gJ gene is unessential to DPV, the deletion of this gene could influence viral replication at late stage of infection.

Replication kinetics of parental and mutant viruses. Confluent DEF cells monolayers were infected with each virus shown at an MOI of 0.01. Viral titer of infected supernatant, cells and mixture of cells cultures were determined at the indicated time points by measuring TCID50 on DEF cells. All titrations were carried out in three independent experiment. The titers obtained were averaged, and the standard error of the mean was calculated each time point.
Plague morphology of mutant viruses and relative plague area measurements
Envelop proteins of alpha-herpesvirus play an important role on viral cell-to-cell spreading or plaque forming. We performed plaque morphology assays to explore whether the gJ-deleted viruses had some effects on the transmission of viruses between adjacent cells. The plaque morphologies of the ΔgJ and ΔgJR mutant viruses were examined in DEF cells. As expected, the DPV CHv-BAC parental virus and the DPV CHv-BAC-gJR mutant viruses produced plaques that were similar in size to each other and, on average. The plaques produced by DPV CHv-BAC-ΔgJ mutant viruses were approximately 11% samller than the parental virus plaques. To better assess the virus plaque sizes produced by individual mutant viruses, 30 randomly chosen viral plaques were selected and statistically analyzed as described in Materials and Methods. This analysis confirmed that the gJ-deleted mutant virus slightly reduced plaque size in comparison to that of the parental virus. Meanwhile, these data showed a statistically significant difference in mean plaque areas of the parental and recombinant viruses (t-test, p < 0.05) (Fig. 5).

Plaque phenotypes of parental and mutant viruses. (A) Confluent DEF cells monolayers were infected with each virus at a 100 TCID50, and viral plaques were visualized at 48 hpi by immunohistochemistry as described in Materials and Methods. (B) Thirty different viral plaques were randomly selected, imaged, measured, and statistically analyzed as described in Materials and Methods.
Ultrastructural characterization of parental and mutant viruses
The herpesvirus viral life cycle contains the following major steps: entry into the host cell, expression and replication of viral genes, virion assembly, and egress of the new generation of viral particles. The entire process takes approximately 18 to 20 hours in permissive cells3,21. Thus, there was a higher opportunity to observe viral ultrastructural phenotypes and life process stages at 36 hpi. Furthermore, according to the growth curve of the recombinant virus, at 36 hpi, its intracellular titer was similar to the parental virus, but its extracellular titer was significantly lower. Meanwhile, the virus extracellular titer gap narrowed at 48 hpi. Moreover, the cells did not show significant rupture and lesions at 36 hpi, but observed at 48 hpi (not shown). In the state of cell fragmentation, it is not conducive to observe the life process of recombinant virus in cells via transmission electron microscopy.
To test whether the deletion of DPV gJ caused a defect in viral assembly and maturation. The ultrastructural phenotypes of recombination viruses relative to the parental virus were investigated at 36 hpi utilizing transmission electron microscopy, visually examining more than 30 individual cells. As expected, the parental virus did not exhibit any apparent defects in nuclear virion assembly or cytoplasmic virion envelopment, as evidenced by the presence of fully enveloped virions intracellularly. Although the empty capsids could be observed on DPV CHv-BAC infected cells, a large amount of nucleic acids could also be found next to these particles (Fig. 6A,B and C). On the contrary, ultrastructural visualization of DEF cells infected with the gJ-deleted mutant viruses revealed nuclear and cytoplasmic defects in virion assembly and envelopment. The most-pronounced effects, produced by the ΔgJ mutant virus, were that numerous unenveloped and enveloped empty capsids were found in nuclear and cytoplasm of infected cells, which may cause the formation of immature of non-infectious virions (Fig. 6D,E and F).

Ultrastructural morphologies of mutant viruses. Electron micrographs of DEF cells infected at an MOI of 2 with different viruses and processed for electron microscopy at 36 hpi are shown. (A,B,C) Showed the DEF cells infected DPV CHv-BAC. (D,E,F) Showed the DEF cells infected DPV CHv-BAC-ΔUS5. Nucleus (n) and cytoplasm (c) are marked.
The alpha-herpesvirus share some strategies for the lytic replication cycle, which mainly includes the viral DNA replication in nucleus, capsid assembly and egress from the nucleus, the envelopment of viral particles in the cytoplasm and the exocytosis of mature virions. During the viral replication cycle, the viral glycoproteins mainly played important roles on viral envelopment, which interact with the viral tegument protein to drive this budding. Ultrastructural visualization of DEF cells infected with the mutant viruses without the expression of gJ revealed that the process of viral replication could work. As the result showed, the gJ-deleted viral nucleocapsids in the nuclear (Fig. 7A) bound onto the surface of inner nuclear membranes (Fig. 7B), and then these perinuclear enveloped particles fused with outer nuclear membranes (Fig. 7C) to enter the cytoplasm, coated-tegument capsids in the cytoplasm bound onto the surfaces of TNG membranes that contained glycoproteins to produce envelopment virions (Fig. 7D,E and F), and the envelopment particles were released from cells (Fig. 7G,H and I).

Electron micrographs of the steps of gJ-deleted mutant virus lifecycle. (A) Capsids in the nucleus. (B) Primary envelopment, showing the close apposition of the particles and the inner nuclear membrane (INM). (C) Primarily enveloped particles present within the perinuclear space. (D) Particles in the cytoplasm, showing the close apposition of the particles and the Golgi or trans-Golgi network (TGN). (E) Initial steps of secondary envelopment. The unenveloped capsids in the cytoplasm interacted with TGN membrane and was being wrapped in these membranes. (F) Final steps of secondary envelopment. Enveloped particles were present with the TGN-derived membranes. (G) and (H) Virions release. Enveloped virions are transported to cell surface and released. (I) Virion in the extracellular medium.
Discussion
Alpha-herpesvirus encodes at least 12 glycoproteins which play important roles in the virus lifecycle, including virus-induced cell fusion, virion assembly and viral egress9. Over the years, a number of gene-deleted mutant virus based on infectious BAC clone have been constructed and characterized. Previously studies showed that the lack of expression of gB, gH, or gL alone has drastically effect on viral maturation, which is necessary for viral replication, while HSV-1, EHV-1 or other alpha-herpesvirus could replicate and produce infection progeny virus without the expression of other glycoproteins, such as such as gE, gI, gK and so on22,23,24. As a non-conserved gene among the alpha-herpesvirus, the specific characteristic and function of glycoprotein J are just a little known. Previously, studies of HSV-1, ILTV, EHV-1 and EHV-4 gJ suggested that the viruses without expression of gJ could replicate and proliferate25,26,27, but it had different decrease on virus titer except HSV-128.
In this study, based on the construction of infectious BAC clone containing the genome of DPV, experiments were conducted to elucidate the function of the deletion of gene US5, which leads to the absence of gJ from the DPV. The salient findings of these experiments were that the DPV gJ is important for virus growth in vitro, and that gJ is slightly involved in direct cell-to-cell spread and in virion maturation. These above conclusions were drawn based on the finding that the gJ-deleted DPV CHv-BAC mutant, DPV CHv-BAC-ΔgJ, replicated to significantly decreased titers in DEF cells, which exhibited a 10-fold reduction of total titers when compared to parental virus and a 4-fold reduction of endocellular titers, and that especially the production of extracellular infectivity was affected. This means that the DPV gJ did not only have some negatively influence on viral replication, but also slightly mediate viral egress. What’s more, the gJ-deleted mutant virus exhibited an approximately 11% reduction in mean plaque diameters when compared to parental or gJ-revertant virus. Furthermore, more immature virions, non-nuclear particles in nuclear and cytoplasm, were observed by electron microscope, and lower cytoplasmic virion envelopment level. Although numerous immature particles were found, the replication cycle of gJ-deleted mutant virus have still worked.
Ultrastructural examination of cells infected with gJ-deleted mutant viruses revealed that the recombination virus exhibited no appreciable defects in cytoplasmic virion envelopment, as also evidenced by the presence of fully enveloped virions in extracellular spaces and secondary-enveloped process in the cytoplasm of infected cells. Surprisingly, the ΔgJ mutant virus exhibited appreciable defect in viral nuclear assembly. Previously studies showed that glycoproteins of herpesvirus mainly played roles in cytoplasmic virion envelopment10,22. What’s more, gB, gD and the heterodimer gH-gL, also found in the nuclear members, were confirmed that these glycoproteins could participate in de-envelopment29,30. However, there is no evidence that glycoproteins of herpesvirus have effects on viral nuclear assembly21,31. This study is the first to show the result that the absence of DPV gJ exhibited a certain degree of effects in viral nuclear assembly, but the mechanism has not been uncovered and still needs to explore. In summary, a marked replication defect was shown after deleting DPV CHv gJ, slightly influencing the efficiency in cell-to-cell spread and virus egress. In combination with analyses of section of infected cells by electron microscopy, the mechanisms and functions of DPV gJ are complex and still need to be exposed.
Materials and Methods
Cells and Viruses
The duck embryo fibroblast (DEF) monolayer was incubated at 37 °C with 5% CO2 in Minimal Essential Medium (MEM, Gibco, Grand Island, NY) supplemented with 10% newborn calf serum (NBS, Gibco, Grand Island, NY), 100 U/ml penicillin and 100 μl/mg streptomycin. For virus infection, MEM supplemented with 3% NBS was used32. DPV CHv strain was separated and preserved in the laboratory.
Construction of DPV mutant viruses
Mutagenesis was constructed in E. coli DH10B by using the Red recombination mutagenesis system with synthetic oligonucleotides33 implemented on the bacterial artificial chromosome (BAC) plasmid pBeloBAC11 carrying the DPV CHv genome34. The DPV CHv-BAC-ΔgJ virus was constructed by deleting the whole US5 open reading frame (ORF). Moreover, the ΔgJ recombinant virus was used as the backbone for construction of the DPV CHv-BAC-gJR reverse mutant by restoring the US5 ORF. Synthetic oligonucleotides used to mutagenize each targeted gene are shown in Table 1. Specifically, the 5′ end of the forward primer for each mutagenesis contains 56 bp of homologous sequence upstream of the site of mutation, and 20 or 21 bp at the 3′ ends correspond to the kanamycin resistance (KanR) gene (Table 1). A 1590 bp PCR fragment containing the KanR gene flanked on both sides by gJ sequence was amplified from the pKD4 vector by the primers.
Maintenance and mutagenesis of the BAC constructs were performed in E. coli strain DH10B. Firstly, the recombineering plasmid, pKD46, containing a λ prophage encoding recombination enzymes Exo, Beta, and Gam under a heat-inducible promoter was transformed into E.coli strain DH10B. Secondly, Bacteria carrying the target BAC and pKD46 were grown in 50 ml LB cultures with chloramphenicol (25 μl/ml), ampicillin (100 μl/ml), and L-arabinose (100 μl/ml, Sigma) at 30 °C to an OD600 of 0.5~0.6. Electrocompetent bacteria were transformed with 800 ng of the PRC products. Chloramphenicol-resistant (CmR)/KanR transformants were confirmed sufficient resistance, and then screened by PCR using the identification of primer (Table 1). Positive colonies were grown overnight at 42 °C to cure pKD46. To remove the KanR gene, a single CmR/KanR and ampicillin-sensitive colony was transformed with pCP20 and grown overnight at 30 °C on Cm/Amp plates. Some colonies were selected again in Cm plates at 42 °C and then tested for kanamycin sensitivity and loss of ampicillin resistance35. The primarily confirmed colonies were ulteriorly screened by PCR using the identification of primer, and then the PCR products were sequenced to confirm the desired targeting.
Confirmation of the targeted mutations and recovery of infectious virus
DPV CHv-BAC DNAs (plasmids pΔgJ and pgJR) were purified from 100 ml of BAC cultures with a Qiagen Plasmid Midi Kit (Qiagen, Valencia CA). The plasmids pΔgJ and pgJR were confirmed by PCR Using the identification of primers designed to lie outside of the target mutation site.
Viruses were recovered from cells transfected with BACs as follows: DEF cells were grown to 70–90% confluent in 6-well or 12-well plates. Cells were transfected with BAC DNAs mixed with Lipofectamine 3000 in Opti-MEM medium recommended by the manufacturer (Invitrogen). After 6 h of incubation at 37 °C, the medium was removed from the transfected cells, and the cells were washed with phosphate-buffered saline (PBS) and then fresh MEM with 3% NBS was added. After incubating cells for 2–4 days at 37 °C, virus stocks were collected and designated as passage 0 (P0). To remove the transient expression of EGFP, the virus of P0 was blindly passaged. Passage P3 viruses were used for all experiment described in the manuscript.
One-step viral growth kinetics
One-step growth curves were performed as follows. Sub-confluent (85 to 90%) DEF cell monolayers grown in 24-well cell culture dish were infected with each virus at 37 °C at 1000 50% Tissue Culture Infection Does (TCID50). After adsorption at 37 °C for 2 h, the cell monolayers were rinsed with PBS overlaid with 2 ml MEM supplemented with 3% NBS (set as 0 h on the time scale), and then returned to 37 °C humidified incubator (5% CO2). At selected time points (12, 24, 36, 48, 72, and 96 h post-infection [h.p.i]), supernatants and cell pellets were separated at different times post-infection and stored at −20 °C. The samples were frozen and thawed three times, and virus titers were determined by TCID50 on DEF cells. All experiments repeated three times.
Plague morphology of mutant viruses and relative plague area measurements
Near-confluent DEF cell monolayers in 12-well plates were infected with the virus at 100 TCID50. After 2 h at 37 °C, medium was discarded and cells were washed with PBS twice. And then 0.5% methylcellulose was added to cover the cells. After 48 h post-infection, cells were washed three times with PBS to remove methyl cellulose medium and fixed with ice-cold 4% paraformaldehyde for 30 min. Photographs of viral plagues were taken at ×200 magnification on microscope. Thirty randomly selected plagues were imaged in this manner for each of the parental and recombination viruses under consideration.
Electron Microscopy
The ultrastructural morphology of virions within infected cells was examined by transmission electron microscopy essentially as described previously36,37. Cell monolayers were infected with the indicated virus at a multiplicity of infection (MOI) of 2. All samples were prepared for transmission electron microscopy examination according to previously reports38. Briefly, DEF cells were washed with PBS at 36 hpi and fixed with 2.5% glutaraldehyde at 4 °C for 2 h. After that, the fixed adherent cells were collected by scraping from the plates and then centrifuged at 10,000 rpm/min for 1 h. Then the pellets were mixed with 2% low melting-temperature agarose at 37 °C, and centrifuged at 6000 rpm/min for 10 min. Samples were post-fixed in 1.0% osmium tetroxide. After a stepwise dehydration in acetone, samples were embedded in epoxy resin 618 and polymerized at 80 °C for 72 h. Then, 50 nm ultra-thin sections were prepared, collected on grids and stained with uranyl acetate and lead citrate for subsequent examination with the Tecnai G2 F20 transmission electron microscope.
Change history
19 April 2018
A correction to this article has been published and is linked from the HTML and PDF versions of this paper. The error has not been fixed in the paper.
References
Cheng, A. Duck plaque (ed. Cheng A.) 1–4 (Beijing, 2015).
Kaleta, E. F. et al. Outbreak of duck plague (duck herpesvirus enteritis) in numerous species of captive ducks and geese in temporal conjunction with enforced biosecurity (in-house keeping) due to the threat of avian influenza A virus of the subtype Asia H5N1. Dtw Deutsche Tierrztliche Wochenschrift 114, 3–11 (2007).
Dhama, K. et al. Duck virus enteritis (duck plague) - a comprehensive update. Veterinary Quarterly 37, 57 (2017).
Liu, T. et al. RNA-seq comparative analysis of Peking ducks spleen gene expression 24 h post-infected with duck plague virulent or attenuated virus. Veterinary Research 48, 47 (2017).
Wu, Y. et al. Complete genomic sequence of Chinese virulent duck enteritis virus. Journal of virology 86, 5965 (2012).
Liu, C. et al. Regulation of viral gene expression by duck enteritis virus UL54. Scientific reports 7, 1076 (2017).
Hu, X. et al. The duck enteritis virus early protein, UL13, found in both nucleus and cytoplasm, influences viral replication in cell culture. Poult. Sci. 96, 2899–2907 (2017).
He, Q. et al. Replication kinetics of duck enteritis virus UL16 gene in vitro. Virol. J. 9, 281 (2012).
Owen, D. J. & Crump, C. M. & Graham, S. C. Tegument Assembly and Secondary Envelopment of Alphaherpesviruses. Viruses 7, 5084–5114 (2015).
Johnson, D. C. & Baines, J. D. Herpesviruses remodel host membranes for virus egress. Nature Reviews Microbiology 9, 382–394 (2011).
Nishiyama, Y. Herpes simplex virus gene products: the accessories reflect her lifestyle well. Rev. Med. Virol. 14, 33–46 (2004).
Jing et al. Role of duck plague virus glycoprotein C in viral adsorption: Absence of specific interactions with cell surface heparan sulfate. Journal of Integrative Agriculture 16, 1145–1152 (2017).
Sun, K. Distribution characteristics of DNA vaccine encoded with glycoprotein C from duck plague virus in ducks and preliminary study of the biological characteristics of gC-deleted DPV and gE-deleted DPV, Sichuan Agricultral University (2013).
Trybala, E., Liljeqvist, J., Bo, S. & Bergström, T. Herpes Simplex Virus Types 1 and 2 Differ in Their Interaction with Heparan Sulfate. Journal of virology 74, 9106–9114 (2000).
Zeevbenmordehai, T., Hagen, C. & Grünewald, K. A cool hybrid approach to the herpesvirus ‘life’ cycle. Current Opinion in Virology 5, 42–49 (2014).
Mundt, A., Mundt, E., Hogan, R. J. & Garcia, M. Glycoprotein J of infectious laryngotracheitis virus is required for efficient egress of infectious virions from cells. The Journal of general virology 92, 2586–2589, https://doi.org/10.1099/vir.0.031443-0 (2011).
Aubert, M. et al. The antiapoptotic herpes simplex virus glycoprotein J localizes to multiple cellular organelles and induces reactive oxygen species formation. Journal of virology 82, 617–629, https://doi.org/10.1128/jvi.01341-07 (2008).
Aubert, M., Krantz, E. M. & Jerome, K. R. Herpes simplex virus genes Us3, Us5, and Us12 differentially regulate cytotoxic T lymphocyte-induced cytotoxicity. Viral immunology 19, 391–408, https://doi.org/10.1089/vim.2006.19.391 (2006).
Fuchs, W., Wiesner, D., Veits, J., Teifke, J. P. & Mettenleiter, T. C. In vitro and in vivo relevance of infectious laryngotracheitis virus gJ proteins that are expressed from spliced and nonspliced mRNAs. Journal of virology 79, 705–716, https://doi.org/10.1128/jvi.79.2.705-716.2005 (2005).
You, Y. et al. The suppression of apoptosis by alpha-herpesvirus. Cell death & disease 8, e2749, https://doi.org/10.1038/cddis.2017.139 (2017).
Kukhanova, M. K., Korovina, A. N. & Kochetkov, S. N. Human herpes simplex virus: Life cycle and development of inhibitors. Biochemistry Biokhimiia 79, 1635–1652 (2014).
Chouljenko, D. et al. Functional hierarchy of herpes simplex virus 1 viral glycoproteins in cytoplasmic virion envelopment and egress. J. Virol. 86, 4262–4270 (2012).
Lee, H., Chouljenko, V., Chouljenko, D., Boudreaux, M. & Kousoulas, K. The herpes simplex virus type 1 glycoprotein D (gD) cytoplasmic terminus and full-length gE are not essential and do not function in a redundant manner for cytoplasmic virion envelopment and egress. J. Virol. 83, 6115–6124 (2009).
Neubauer, A. & Osterrieder, N. Equine herpesvirus type 1 (EHV-1) glycoprotein K is required for efficient cell-to-cell spread and virus egress. Virology 329, 18–32 (2004).
Mashchenko, A., Riblet, S. M., Zavala, G. & Garcia, M. In ovo vaccination of commercial broilers with a glycoprotein J gene-deleted strain of infectious laryngotracheitis virus. Avian diseases 57, 523–531 (2013).
von Einem, J. et al. The truncated form of glycoprotein gp2 of equine herpesvirus 1 (EHV-1) vaccine strain KyA is not functionally equivalent to full-length gp2 encoded by EHV-1 wild-type strain RacL11. Journal of virology 78, 3003–3013 (2004).
Rudolph, J. & Osterrieder, N. Equine herpesvirus type 1 devoid of gM and gp2 is severely impaired in virus egress but not direct cell-to-cell spread. Virology 293, 356–367, https://doi.org/10.1006/viro.2001.1277 (2002).
Zhou, G., Galvan, V., Campadelli-Fiume, G. & Roizman, B. Glycoprotein D or J delivered in trans blocks apoptosis in SK-N-SH cells induced by a herpes simplex virus 1 mutant lacking intact genes expressing both glycoproteins. Journal of virology 74, 11782–11791 (2000).
Farnsworth, A. et al. Herpes simplex virus glycoproteins gB and gH function in fusion between the virion envelope and the outer nuclear membrane. Proceedings of the National Academy of Sciences of the United States of America 104, 10187 (2007).
Mettenleiter, T. C., Müller, F., Granzow, H. & Klupp, B. G. The way out: what we know and do not know about herpesvirus nuclear egress. Cellular Microbiology 15, 170 (2013).
Cardone, G., Heymann, J. B., Cheng, N., Trus, B. L. & Steven, A. C. Procapsid Assembly, Maturation, Nuclear Exit: Dynamic Steps in the Production of Infectious Herpesvirions. Advances in Experimental Medicine & Biology 726, 423 (2012).
Wu, Y. et al. Establishment of real-time quantitative reverse transcription polymerase chain reaction assay for transcriptional analysis of duck enteritis virus UL55 gene. Virology journal 8, 1–13 (2011).
Datsenko, K. A. & Wanner, B. L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proceedings of the National Academy of Sciences of the United States of America 97, 6640–6645, https://doi.org/10.1073/pnas.120163297 (2000).
Wu, Y. et al. Preliminary study of the UL55 gene based on infectious Chinese virulent duck enteritis virus bacterial artificial chromosome clone. Virology journal 14, 78 (2017).
Cotta-de-Almeida, V., Schonhoff, S., Shibata, T., Leiter, A. & Snapper, S. B. A new method for rapidly generating gene-targeting vectors by engineering BACs through homologous recombination in bacteria. Genome research 13, 2190–2194 (2003).
Guo, Y. et al. Anatid herpesvirus 1 CH virulent strain induces syncytium and apoptosis in duck embryo fibroblast cultures. Vet. Microbiol. 138, 258–265 (2009).
Guiping, Y. et al. Preliminary study on duck enteritis virus-induced lymphocyte apoptosis in vivo. Avian Dis. 51, 546–549 (2007).
Yuan, G. et al. Electron microscopic studies of the morphogenesis of duck enteritis virus. Avian Dis. 49, 50–55 (2005).
Acknowledgements
This research was supported by grants from the National Key Research and Development Program of China (2017YFD0500800), the China Agricultural Research System (CARS-42-17), the Special Fund for Key Laboratory of Animal Disease and Human Health of Sichuan Province (2016JPT0004) and the International S&T Cooperation Program of Sichuan Province (Grant No. 2017HH0026).
Author information
Authors and Affiliations
Contributions
Y.Y. conceived, designed and carried out the experiments, analyzed the data and draft the manuscript; T.L, M.W. and A.C. conceived and designed the experiments, and revised the manuscript; D.Z., S.C., R.J., M.L., Q.Y., Y.W., X.Z., S.Z. and X.C. analyzed and interpreted the data. Y.L, Y.Y, and L.Z provided cells and reagents. All the authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
You, Y., Liu, T., Wang, M. et al. Duck plague virus Glycoprotein J is functional but slightly impaired in viral replication and cell-to-cell spread. Sci Rep 8, 4069 (2018). https://doi.org/10.1038/s41598-018-22447-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-22447-x
This article is cited by
-
Duck plague virus gE serves essential functions during the virion final envelopment through influence capsids budding into the cytoplasmic vesicles
Scientific Reports (2020)
-
The Pivotal Roles of US3 Protein in Cell-to-Cell Spread and Virion Nuclear Egress of Duck Plague Virus
Scientific Reports (2020)
-
Molecular characterization and antiapoptotic function analysis of the duck plague virus Us5 gene
Scientific Reports (2019)
-
US10 Protein Is Crucial but not Indispensable for Duck Enteritis Virus Infection in Vitro
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
