
Abstract
Macular and cone/cone-rod dystrophies (MD/CCRD) demonstrate a broad genetic and phenotypic heterogeneity, with retinal alterations solely or predominantly involving the central retina. Targeted next-generation sequencing (NGS) is an efficient diagnostic tool for identifying mutations in patient with retinitis pigmentosa, which shows similar genetic heterogeneity. To detect the genetic causes of disease in patients with MD/CCRD, we implemented a two-tier procedure consisting of Sanger sequencing and targeted NGS including genes associated with clinically overlapping conditions. Disease-causing mutations were identified in 74% of 251 consecutive MD/CCRD patients (33% of the variants were novel). Mutations in ABCA4, PRPH2 and BEST1 accounted for 57% of disease cases. Further mutations were identified in CDHR1, GUCY2D, PROM1, CRX, GUCA1A, CERKL, MT-TL1, KIF11, RP1L1, MERTK, RDH5, CDH3, C1QTNF5, CRB1, JAG1, DRAM2, POC1B, NPHP1 and RPGR. We provide detailed illustrations of rare phenotypes, including autofluorescence and optical coherence tomography imaging. Targeted NGS also identified six potential novel genotype-phenotype correlations for FAM161A, INPP5E, MERTK, FBLN5, SEMA4A and IMPDH1. Clinical reassessment of genetically unsolved patients revealed subgroups with similar retinal phenotype, indicating a common molecular disease cause in each subgroup.
Similar content being viewed by others


Introduction
Macular and cone/cone-rod dystrophies (MD/CCRD) comprise diverse inherited retinal diseases with progressive degeneration, dysfunction and vision loss of the central retina1. Characteristic symptoms include decreasing visual acuity, reading difficulties, photophobia and dyschromatopsia. Later, variable loss of rod function with reduced night vision and loss of peripheral visual field may occur1. Full-field electroretinography (ffERG) allows classifying patients into those with MD (normal ffERG), CD (only reduced photopic responses) and CRD (reduced photopic and scotopic responses).
Genotype-phenotype correlations of MD/CCRD are often complex. For instance, different mutations in one gene can cause highly diverse phenotypes, and mutations in different genes can cause very similar phenotypes. Furthermore, there is a considerable clinical overlap of CCRD with MD and RP, and the classification may change from MD to CCRD with disease progression1,2,3. Although more than 30 disease-causing genes have been reported for MD/CCRD (RetNet, https://sph.uth.edu/retnet), the genetic disease cause in a substantial number of patients is currently unknown.
As novel therapeutic options including gene therapy are being developed, the identification of the individual mutation has gained importance. Next-generation sequencing (NGS) has become a very efficient diagnostic tool, as exemplified in patients with RP4,5,6,7,8. However, only few studies – most of them with limited patient numbers – have reported the performance of targeted NGS for defining the molecular basis of unselected patient cohorts with MD/CCRD2,9,10,11.
Here, we report results from a cohort of 251 unrelated, consecutive and clinically well characterized MD/CCRD patients who underwent extensive molecular genetic analysis. The results provide insights into the diversity of the mutational spectrum of MD/CCRD and indicate potential novel or uncommon genotype-phenotype correlations. Moreover, we reviewed the retinal phenotype of patients in whom the molecular disease cause remained unexplained and propose phenotypic subgroups which may correspond to specific yet unknown genetic disease causes.
Methods
All methods were carried out in accordance with the approved guidelines. All data generated or analyzed during this study are included in this article (and its Supplementary Information files).
Patients
This retrospective single-center cross-sectional study included 251 consecutive MD/CCRD patients (235 Caucasian, 139 female) seen between summer 2012 and summer 2015 at the Department of Ophthalmology, University of Bonn, Germany. Inclusion criteria were 1) the clinical diagnosis of a MD/CCRD by one of the senior clinicians (P.C.I., P.H.) based on the patient’s history of visual symptoms, clinical examination, retinal imaging, and – where available – electrophysiology, and 2) that the patient underwent genetic testing for their retinal disease. Patients were excluded if they had an obvious syndromic retinal disease, age-related macular degeneration, central serous retinopathy, autoimmune retinopathy, or retinal vascular disease. Also, patients were excluded if they had the clinical diagnosis of achromatopsia (cone dysfunction syndromes were differentiated from retinal dystrophies, and genotyping was performed in a different lab), X-linked retinoschisis (genetic testing performed in different labs), and North Carolina macular dystrophy (genetic basis not published before 2016). The study was in adherence with the Declaration of Helsinki. Institutional review board approval (Ethics Committee, Medical Faculty, University of Bonn) and patients’ informed consent were obtained. Asymptomatic family members received genetic counselling prior to genetic testing.
A general medical history was obtained from each patient and a pedigree was assembled based on a detailed family history with regards to ocular diseases and visual symptoms. The mode of inheritance was assumed to be autosomal recessive (a.r.) in case of parental consanguinity or if only siblings were affected, autosomal dominant (a.d.) if the family history was suggestive for the same inherited retinal disease in at least 3 successive generations, and X-linked if the disease occurred in different generations without male-to-male transmission and with only males being severely affected while females were normal or with only minor symptoms. If other family members were affected but the pedigree was not suggestive for any of the above patterns, inheritance was classified as inconclusive. Disease in patients without other affected family members and no parental consanguinity was considered as sporadic.
Image acquisition and functional testing
All patients underwent a standardized clinical examination and retinal imaging, including spectral domain optical coherence tomography (OCT), fundus autofluorescence (AF) imaging (both, Spectralis HRA + OCT, Heidelberg Engineering, Heidelberg, Germany), fundus photography (Zeiss, Visucam, Oberkochen, Germany) and wide-field fundus imaging (Optos PLC, Dunfermline, United Kingdom). To assess retinal function, best corrected visual acuity (BCVA), ffERG, electroocoulogram (EOG) and visual field testing (different devices and protocols depending on availability and patient needs) were performed. In total, ffERG was acquired from 223 patients (89%) and EOG from 48 patients (19%, mainly those with vitelliform lesions).
Molecular genetic analysis
Genomic DNA was extracted from blood lymphocytes by a standard protocol. A two-tier procedure was implemented to identify the molecular cause of disease: If the retinal phenotype was highly suggestive for mutations in ABCA4, PRPH2 or BEST1, Sanger sequencing (and, in case of ABCA4, multiplex ligation-dependent probe amplification) for the respective gene was performed. If the result of this initial molecular testing was negative, or if the phenotype was not clearly suggestive for retinopathy associated with these three genes, then 48 genes whose mutations were known to cause MD/CCRD at the time of panel design (2015; ABCA4, ACBD5, ADAM9, AIPL1, BEST1, C1QTNF5, C21orf2, C8orf37, CABP4, CACNA1F, CACNA2D4, CDH3, CDHR1, CERKL, CNGA3, CNGB3, CNNM4, CRX, CTNNA1, ELOVL4, GUCA1A, GUCA1B, GUCY2D, IFT140, KCNV2, MFSD8, NR2E3, PCYT1A, PDE6C, PITPNM3, POC1B, PRDM13, PROM1, PRPH2, RAB28, RAX2, RBP3, RDH5, RGS9, RGS9BP, RIMS1, RP1L1, RPGR, RPGRIP1, SEMA4A, TIMP3, TTLL5, UNC119) were enriched using Roche/NimbleGen sequence capture technology, sequenced on an Illumina HiSeq. 1500 system and bioinformatically evaluated (including analysis for copy number variations) as described previously8. To detect X-linked mutations, we added NGS of amplicons comprising RPGR ORF15 (to be described elsewhere) to panel-NGS of the remaining exons of RPGR. The NGS panel additionally included all the genes known to be associated with clinically overlapping conditions such as rod-cone dystrophy, Leber’s congenital amaurosis (LCA) and several syndromes with retinal dystrophies. This allowed for an extended genetic assessment if no mutation was found in the “core genes”, without need for additional experimental efforts. Verification of mutations identified in NGS and segregation analyses were carried out by PCR and subsequent Sanger sequencing.
Variants were filtered against dbNSFP v2.0, dbSNP v137, gnomAD (exomes) and the Human Gene Mutation Database (HGMD® Professional 2017.3). The cut-off for the maximum minor allele frequency (MAF) was set to 1%12. Nonsense, frameshift, large deletions and canonical splice site variants were regarded pathogenic. Rare non-synonymous single nucleotide variations were considered likely pathogenic when at least half of the algorithms of used in silico prediction software tools predicted that the variant is probably damaging and when it was predicted as conserved with conservation prediction algorithms. The impact of splice site variants was assessed using specific splice site prediction programs.
Genotype-phenotype correlation
After molecular testing, all patients with identified variants were re-evaluated to survey if their retinal phenotype was compatible with previous descriptions of the retinopathy related to the mutated gene. In case of inconsistency, the respective phenotypes were considered as potential novel genotype-phenotype correlations. Where possible, segregation analysis was performed, although the number of available family members was often small and included mostly the parents and, occasionally, siblings.
Results
Mutations in genes known to be involved in MD/CCRD pathogenesis
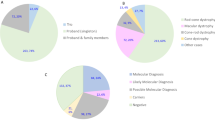
Disease-causing or likely disease-causing mutations were identified in 185 out of 251 patients (74%) with MD/CCRD (Supplementary Table 1). Hereof, 64 (33%) mutations were novel at the time of manuscript submission (Supplementary Table 2). Overall, the 193 different mutations were distributed across 22 genes that encode proteins from diverse pathways and cellular compartments (Fig. 1A,B, Supplementary Table 3).

Mutational spectrum (A) Spectrum of variants identified in 251 patients affected by macular and cone/cone-rod dystrophies. (B) Functional categorization of variants identified in our study. (C) Inheritance based on the genetic findings. Percentages refer to patients with mutations in the considered causative gene, pathway or mode of inheritance.
In 57% of all MD/CCRD cases (77% of the 185 solved cases), retinal disease was explained by mutations in ABCA4 (n = 94, 37%), PRPH2 (n = 29, 12%) or BEST1 (n = 19, 8%; including 5 a.r. and 14 a.d. mutations). Further mutations were identified in CDHR1 (n = 6), GUCY2D (n = 6), PROM1 (n = 6), CRX (n = 4), GUCA1A (n = 3), CERKL (n = 2), MT-TL1 (n = 2), KIF11 (n = 2), RP1L1 (n = 2), MERTK (n = 1), RDH5 (n = 1), CDH3 (n = 1), C1QTNF5 (n = 1), CRB1 (n = 1), JAG1 (n = 1), DRAM2 (n = 1), POC1B (n = 1), NPHP1 (n = 1) and RPGR (n = 1) (Fig. 1A, Supplementary Table 1). Although there was considerable phenotypic heterogeneity even between patients with mutations in the same disease-causing gene, the phenotype was compatible with the previously described gene-associated retinal phenotype(s) in all instances. In the 185 solved cases, inheritance based on the genetic findings was autosomal recessive (n = 119, 64%), autosomal dominant (n = 63, 34%), X-linked (n = 1, 1%) and mitochondrial (n = 2, 1%) (Fig. 1C).
If the pedigree fulfilled the criteria for a.r. or a.d. inheritance (see Methods), a molecular diagnosis was achieved in 89% (n = 31 out of 35) or 84% (n = 31 out of 37), respectively, and then confirmed the expected mode of inheritance (Table 1). The pedigree of 3 patients (#54, #63, #83, Supplementary Table 1) with only 2 affected generations was inconclusive, and pseudo-dominant inheritance due to mutations in ABCA4 explained this finding at the molecular level. The mutation detection rate was lower (68%; n = 120 out of 176) in patients with sporadic MD/CCRD. In these 120 patients, the molecular diagnosis specified inheritance as a.r. in 85 patients (71%), a.d. in 32 (27%) patients, X-linked in 1 (1%) patients and mitochondrial in 2 patients (2%) (Table 1). Out of the 66 unsolved cases, 56 (85%) were sporadic and 10 (15%) had a family history clearly suggestive for a.d. (n = 6) or a.r. (n = 4) inheritance.
Clinical description of uncommonly observed but characteristic genotype-phenotype correlations
CRB1
A 57 year-old female patient (#117, Supplementary Table 1, Fig. 2A) with macular dystrophy reported first symptoms (reduced visual acuity) at the age of 49 years. Now, BCVA was 20/50 and 20/400 in the right and left eye, respectively. Retinal imaging revealed a (para-) central loss of photoreceptors and reduction of fundus AF (Fig. 2A). Full-field ERG recordings were within normal limits. We identified a homozygous in frame deletion in CRB1 previously described in patients with early-onset retinal dystrophy13,14. There were no further affected individuals in the family history, and no family members were available for segregation analysis.

Uncommonly observed but characteristic genotype-phenotype correlations. Retinal phenotype associated with mutations in CRB1 [#117] (A), KIF11 [#179] (B), JAG1 [#184] (C), DRAM2 [#119] (D), POC1B [#120] (E), NPHP1 [#115] (F), RPGR [#189] (G). Fundus color image (first column), fundus AF with 488 nm excitation light (second column), and horizontal spectral-domain OCT (third and fourth column) are shown. Patient numbers refer to Supplementary Table 1. Only one eye is shown due to high symmetry between eyes.
KIF11
Figure 2B shows the retinal phenotype in an apparently non-syndromic 11 year-old patient (#179, Supplementary Table 1) with a heterozygous in frame deletion in the KIF11 gene. Mutations in this gene are otherwise known to cause an autosomal dominant syndrome including microcephaly, mental retardation, lymphedema and focal chorioretinal atrophy at the ocular fundus15,16,17, and familial exudative vitreoretinopathy18,19. Fundus AF imaging revealed areas with a decreased signal surrounded by a line of increased AF and associated with thinning of the photoreceptor layer (Fig. 2B). Similar but more progressed findings were identified in an additional patient (#180, Supplementary Table 1) with a heterozygous nonsense mutation in KIF11. A detailed characterization of KIF11-associated retinopathy including these patients has recently been reported20. The retinopathy was initially classified as non-syndromic due to the very mild systemic manifestations that may be too subtle to be noted in routine ophthalmologic workup.
JAG1
Figure 2C shows the phenotype of a 24 year-old patient (#184, Supplementary Table 1) with Bull’s eye retinopathy associated with a heterozygous 4-bp deletion in JAG1, a gene commonly associated with autosomal dominant Alagille syndrome21. The retinal phenotype of Alagille syndrome is variable and includes granular pigmentary changes and maculopathy22,23,24. The patient experienced a mild reduction in visual acuity (BCVA now 20/25 in the right eye and 20/32 in the left eye) and dark adaption problems. In addition, she also had diverse heart defects including a double-outlet-right-ventricle (cardiac surgery at the age of six years), pulmonary stenosis, and a severe scoliosis for which she underwent spinal surgery. These heart defects had not been seen in a syndromic context with her retinal changes before genetic testing, and sequencing of PTPN11 and KRAS for suspected Noonan syndrome had revealed no mutation. In her family, no visual problems or other diseases associated with Alagille syndrome were known. Only the mother was available for segregation analysis and did not carry the JAG1 mutation. Notably, mutations in JAG1 frequently occur de novo25,26,27.
DRAM2
Figure 2D illustrates the retinal phenotype of a 53 years-old patient (#119, Supplementary Table 1) with compound heterozygous missense mutations in DRAM2. DRAM2 mutations have been found to cause a retinal dystrophy with early macular involvement28,29. The patient experienced dark adaption difficulties starting in her third decade and visual field defects for the past two years. Increased glare and reduced visual acuity were noted for one year. Epiretinal membrane and cataract surgery were performed in her left eye some months ago without functional success. BCVA was now 20/32 and 20/800 in the right and left eye, respectively. Funduscopy revealed a granular macular appearance, and fundus AF showed a paracentral hyperautofluorescent ring with central thinning of the photoreceptor layer. Scotopic and photopic responses on ffERG recordings were reduced to about 2/3 of the lower normal limits. There were no further affected individuals in the family history, and no family members were available for segregation analysis.
POC1B
Figure 2E shows the retinal phenotype of a non-syndromic 49 years-old patient (#120, Supplementary Table 1) with a homozygous missense mutation in the POC1B gene (parents are second degree cousins). Mutations in POC1B have been associated with severe and slowly progressive CRD, and with Joubert syndrome with Leber congenital amaurosis (LCA)30,31,32. The patient experienced reduced visual acuity since childhood, and BCVA now was 20/200 in the right eye and 20/400 in the left eye. Increased glare and color vision abnormalities were noted at 18 years of age, but she had no visual difficulties in dim lightening. Fundus AF imaging revealed mild granular irregularities associated with thinning of the photoreceptor layer on OCT imaging. In the mid periphery, there was a faintly noticeable whitish patchy/flecked appearance. Photopic and scotopic responses on full-field ERG testing were undetectable. There were no further affected individuals in the family history, and no family members were available for segregation analysis.
NPHP1
Figure 2F shows the retinal phenotype of a 49 years-old patient (#115, Supplementary Table 1) with a homozygous deletion of all coding exons of NPHP1, a gene commonly associated with non-syndromic nephronophthisis, Senior-Loken or Joubert syndrome33,34,35. Since childhood, the patient experienced reduced visual acuity, glare and, over the last ten years, progressive loss of visual acuity. BCVA now was 20/63 and 20/40 in the right and left eye, respectively. Fundus AF imaging was relatively unremarkable, but OCT imaging revealed diffuse thinning of the outer retinal layers. Scotopic responses on ffERG testing were reduced to about ½ of the lower normal limits and photopic responses were almost extinguished. Only after the genetic test result was discussed with the patient, she disclosed that kidney transplantation had been performed in adolescence due to juvenile-onset nephronophthisis, allowing the diagnosis of a Senior-Loken syndrome. No family members were available for clinical examination or segregation analysis.
RPGR
Figure 2G shows the retinal phenotype of a patient with sporadic CRD and mutation in RPGR (Supplementary Table 1; hemizygous readthrough mutation in patient #189), a gene usually associated with X-linked RP36,37,38 and less commonly with CRD39,40,41,42. The 41 year-old patient reported reduced visual acuity starting in his 5th decade of life. BCVA was now 20/40 in the right eye and counting fingers due to amblyopia in the left eye. On ffERG testing, scotopic and photopic responses were reduced to about 1/3 of the lower normal limits. No family members were available for segregation analysis or clinical examination.
Potential novel genotype-phenotype correlations
Potentially disease-causing mutations were detected in six genes which have as yet not been described in association with the observed MD/CCRD phenotype: FAM161A, INPP5E, MERTK, FBLN5, SEMA4A and IMPDH1 (n = 1 in all cases). Inclusion of these potential novel genotype-phenotype correlations would increase the diagnostic yield to 76% (n = 191).
FAM161A
A 54-year-old patient (#122, Supplementary Table 1) reported reduced visual acuity and reading problems around 50 years of age. Two years later, she also noticed dark adaption problems and alterations in color vision. Her visual acuity was 20/32 in both eyes. Scotopic responses on ffERG testing were reduced to about ½ of the lower normal limits and photopic responses reduced to about 1/10 of the lower limit. On OCT imaging, a perifoveal loss of the myoid zone and thinning of the photoreceptor layer was detected, and fundus AF imaging revealed a slightly increased perifoveal autofluorescence (Fig. 3A). NGS identified a novel homozygous 1-basepair deletion in the FAM161A gene. The parents of the patient and the only child were deceased. The twelve years older brother had no ocular problems, but was not available for clinical examination or genetic testing.

Potential novel genotype-phenotype correlations. Potential novel genotype-phenotype correlations in FAM161A [#122] (A), INPP5E [#121] (B), MERTK [#113] (C), FBLN5 [#187] (D), SEMA4A [#188] (E) and IMPDH1 [#186] (F). Fundus color image (first column), fundus AF with 488 nm excitation light (second column), and horizontal spectral-domain OCT (third and fourth column) are shown. Patient numbers refer to Supplementary Table 1. Only one eye is shown due to high symmetry between eyes.
INPP5E
A 57 years-old patient (#121, Supplementary Table 1) with a pattern dystrophy reported reduced visual acuity and increased sensitivity to light for the past 3 years. Funduscopy showed macular subretinal yellowish deposits which were associated with a pattern of increased and decreased fundus AF (Fig. 3B). Visual acuity was 20/20 in both eyes, and ERG examination revealed responses within the normal range. NGS identified compound heterozygosity for a nonsense mutation and a missense variant (c.844 G > A; p.Gly282Arg) affecting an evolutionarily highly conserved amino acid in INPP5E. The minor allele frequency of the missense variant is low (0.01%), and no homozygotes have been documented in the general population. The patient was otherwise healthy, specifically without signs of Joubert syndrome, which has previously been associated with mutations in INPP5E43,44,45,46. Only the patient’s mother was available for segregation analysis and carried only the nonsense mutation.
MERTK
A 39 years-old patient (#113, Supplementary Table 1) was affected by increased sensitivity to light and dark adaption problems for about the past 3 years. At initial presentation, BCVA was 20/20 and 20/25 in the right and left eye, respectively, but decreased in the left eye to 20/2000 over a period of two years. Funduscopy showed central hypo- and hyperpigmentations (L > R) and, in the far periphery temporal inferior, scattered bone spicule pigmentations. Fundus AF imaging revealed an uncommon pattern with central granular irregularities and a hyperfluorescent area mainly temporal to the macula with thinning of the photoreceptor layer (Fig. 3C) which progressed over a 5-years observation period. Scotopic and photopic responses on ffERG recordings were reduced to about 2/3 of the lower normal limits. NGS revealed two novel missense variants in MERTK (c.1801G > C/p.Val601Leu and c.2360 G > A/p.Gly787Asp) in compound heterozygous state (segregation analysis of the parents), both affecting highly conserved protein residues. Only p.Val601Leu has a (very low) documented minor allele frequency (0.00041%).
FBLN5
A 78 years-old patient (#187, Supplementary Table 1) reported reduced visual acuity over the past year, and BCVA now was 20/50 in both eyes. Funduscopy showed mild drusen at the vascular arcades and foveal yellowish subretinal deposits with associated increased fundus AF (vitelliform lesion; Fig. 3D). The Arden ratio was 2.0 in both eyes. The patient carried a rare (minor allele frequency of 0.0037%) heterozygous and potentially pathogenic missense variant (c.1093 A > G; p.Ile365Val) in FBLN5, affecting an evolutionarily conserved residue of the FBLN5 protein47. The patient’s deceased mother and a maternal cousin also had late onset visual problems, but no fundus images and no DNA samples were available for segregation analysis.
SEMA4A
A novel heterozygous frameshift insertion in the SEMA4A gene was identified in a 48-year-old patient (#188, Supplementary Table 1) whose first symptoms were reduced visual acuity noted about four years ago. BCVA was 20/63 and 20/40 in the right and left eye, respectively. Funduscopy and OCT imaging showed an atrophic macular lesion associated with decreased fundus AF (Fig. 3E). On ffERG recording, scotopic responses were reduced to about 2/3 and photopic to about ½ of the lower normal limits. The Arden ratio was 1.8 in both eyes. The patient’s parents were deceased and therefore, segregation analysis was not possible.
IMPDH1
A 58-year-old patient (#186, Supplementary Table 1) was affected by increasing metamorphopsia for several months and increased glare for decades. BCVA was 20/20 in both eyes and ffERG recordings were within normal limits. Fundus examination, OCT and fundus AF imaging revealed features of a mild pattern dystrophy (Fig. 3F). We identified a novel, heterozygous, synonymous mutation in the IMPDH1 gene which affects the second-to-last nucleotide of exon 2, predicted to impair the adjacent donor splice site (minor allele frequency 0.05%). Mutations in IMPDH1 have previously been associated with a.d. retinitis pigmentosa48,49,50. The mother of the patient also had reduced vision beginning in the fourth decade of life. However, the patient’s close relatives were all deceased and therefore, segregation analysis was not possible.
Phenotype of unsolved patients
Out of the remaining 60 patients, 7 patients (12%) revealed one potentially disease-causing variant in a gene causing recessive retinopathy in combination with a phenotype usually associated with mutations in the respective gene (ABCA4, n = 5; CDH3, n = 1; CERKL, n = 1; Supplementary Table 4). Seven out of the remaining 53 patients carried at least one previously described ABCA4 mutation, but showed no phenotype characteristics for ABCA4-associated retinopathy.
All 60 genetically unsolved patients were reviewed in order to identify groups with common phenotypic characteristics on fundus photography, OCT- and fundus AF imaging. This allowed to cluster 41 patients into 4 subgroups, each with a phenotypic spectrum resembling 1) mitochondrial retinopathy (n = 10), 2) adult vitelliform or pattern dystrophy (n = 15), 3) late-onset retinal dystrophy (LORD) or Sorsby fundus dystrophy (n = 7) or 4), PRPH2- or ABCA4-associated retinopathy (n = 9). The remaining 19 patients showed less frequent phenotypes and could not be assigned to any of these subgroups.
All patients with a phenotype typical for mitochondrial retinopathies showed changes compatible with those described for the MT-TL1 mutation (m.3243 A > G)51, such as pigmentary abnormalities, flecks of increased AF with borders of decreased AF and/or chorioretinal atrophy. However, none of these 10 patients carried the m.3243 A > G variant. A muscle biopsy in 3 of the 10 patients revealed characteristics of a mitochondrial disease, and two patients exhibited further clinical characteristics such as deafness, cardiac problems and a family history suggestive for a mitochondrial disease.
Discussion
Our study demonstrates a high detection rate of disease-causing mutations in MD/CCRD patients using targeted NGS and confirms the extensive genetic heterogeneity of this disease group in 251 MD/CCRD patients. The study’s unique feature is the in-depth clinical characterization of all patients regardless of the genetic testing results, including the “unsolved”. Moreover, the unselected cohort of consecutive patients provides a realistic insight into the distribution of MD/CCRD in a routine clinical setting.
The overall diagnostic yield of 74% achieved in our study is in the upper range compared to previous reports of targeted NGS in MD/CCRD patients2,9,10. However, the study by Stone et al. had a significantly higher detection rate in the subgroup of patients with macular dystrophies11. Reasons for discrepancy between yields may include different cohort sizes and populations, variable inclusion criteria and the investigation of specific subgroups. Moreover, efficiency of molecular genetic testing and data analysis may differ between studies.
Mutations in genes known to be involved in MD/CCRD pathogenesis
Mutations in ABCA4, PRPH2 and BEST1 were predominant, explaining the retinal disease in 57% of the entire cohort and 74% of the solved cases. Although referral patterns might have biased our cohort towards a slightly exaggerated frequency of ABCA4-associated retinopathy, previous studies have likewise indicated that mutations in ABCA4 are the most common cause of central retinal dystrophies2,11,52,53,54.
In a large French CCRD cohort, PRPH2 mutations were found in 3.4% of all cases (in 11.8% of a.d. patients)2, compatible with other studies9,55. Thus, the high proportion of MD/CCRD patients with PRPH2 mutations (12% of all cases) in this study is surprising. Many patients with PRPH2 mutation present with a phenotype similar to late-onset Stargardt disease. Such patients may often be preferentially tested only for ABCA4 mutations, or be diagnosed with age-related macular degeneration and therefore receive no genetic diagnosis.
Some of the genes whose mutations are associated with isolated or syndromic MD/CCRD have been reported only rarely, and sometimes without in-depth characterization of the retinal phenotype. Targeted NGS allows to include these genes in the molecular analysis with only minor additional resources when compared with Sanger sequencing. Phenotypic re-evaluation after identification of the (likely) molecular basis of the disease revealed in the vast majority of cases that the respective retinal phenotype was in line with previously published and often characteristic genotype-phenotype correlations on multi-modal imaging (Fig. 2). This may not only increase the confidence in a correct molecular diagnosis, but also confirms previous rare phenotypic disease characterizations for genes infrequently involved in the pathophysiology of MD/CCRD.
Such rare genotype-phenotype correlations were confirmed in this report in patients with mutations in CRB1, DRAM2, KIF11, JAG1, POC1B, NPHP1 and RPGR. Mutations in CRB1 mainly account for LCA and RP13,56,57,58,59 and have only recently been described in association with isolated MD60,61. The herein reported homozygous CRB1 mutation was previously reported in a patient with EOSRD13,62. Occurrence in a patient with late-onset MD (Fig. 2A) may suggest that other (most likely genetic) factors might influence the disease manifestation. The patients with mutations in genes associated with syndromic MD/CCRD all showed a retinal phenotype compatible with the previously described syndrome-associated retinopathy. These patients either revealed no other obvious systemic abnormalities (POC1B), showed only very mild syndromic changes that may easily escape attention (KIF11)20, or received their precise syndromic diagnosis subsequent to the identification of the mutation causing their retinopathy (JAG1, NPHP1). Thus, analysis of genes causing syndromic retinal dystrophies in patients who are primarily diagnosed with an isolated retinopathy may clarify the exact clinical diagnosis.
The identification of a patient with a mutation in RPGR – mostly causing X-linked RP36,37,38 – confirms its rarer association with MD/CCRD39,40,41. Sporadic RPGR-associated CRD is rare63, and our case supports the importance to consider RPGR mutations in this specific subgroup. Two additional patients in our cohort were found to have either an in-frame deletion (c.2203_2226del) or duplication (c.2919_2939dup) in ORF15 of RPGR, which were not interpreted as disease-causing due to the relatively frequent occurrence of such variants in the normal population, although the patient with the duplication revealed a phenotype compatible with RPGR-related retinopathy. Retinal phenotyping of obligate female carriers and segregation analysis would be desirable for confirming pathogenicity of identified mutations in future studies, but this was not possible in our patients.
Potential novel genotype-phenotype correlations
One of the advantages of targeted NGS is the possibility to screen a large number of genes involved in the pathophysiology of retinal dystrophies independent of the exact phenotype. In 6 (2%) patients, potentially disease-causing variants were detected in genes for which an association with the observed MD/CCRDs phenotype has not previously been established. In these cases, it remains challenging to confirm or exclude the pathogenicity of identified variants in a routine clinical setting. In recessive traits, with nowadays small and highly mobile Western families, only one affected disease carrier is often identified which limits the confidence to establish novel genotype-phenotype correlations, even when parental segregation analysis is performed. In dominant traits, mostly in those with a late-onset of the disease, small families with no further living disease carriers also limit the interpretability of the genetic findings. In absence of functional assessment and/or animal models for these individual genetic variants, future observations may eventually confirm or disprove such potential novel genotype-phenotype correlations. We hope to facilitate such future comparisons by providing essential phenotype information.
We identified patients exhibiting features of a pattern dystrophy (IMPDH1), or non-syndromic CRD (INPP5E and FAM161A). These genes are otherwise associated with a.r. (FAM161A64,65,66,67,68,69) or a.d. (IMPDH148,49,50) retinitis pigmentosa, or Joubert syndrome (INPP5E43,44,45). Of note, the patient with FAM161A mutations had a late onset of first symptoms (reduced visual acuity at the age of 50 years) which was followed 2 years later by dark adaption problems. Although ffERG currently indicated CRD, the phenotype might be classified as RP later in the disease course. There is little published information on the INPP5E-related retinal phenotype, but the report by Hardee et al. on INPP5E-related Joubert syndrome indicates a retinal dystrophy that primarily affects the central retina/cone system46. There is a lack of detailed phenotypic description of a.d. SEMA4A-associated retinopathy70,71, and the as yet reported MERTK-associated phenotype72,73,74 differs on retinal imaging and with regards to the age-of-onset from the patient reported herein. Despite the less consistent pathogenicity prediction of the FBLN5 variant, the phenotype may be in line with previous reports47.
Unsolved cases
There are still a substantial number of patients in which targeted NGS is not effective in confirming the clinical diagnosis on a molecular level. Common phenotypes in a subset of these patients may indicate that specific mutations, genes or pathways are involved. Recognizing such phenotypic patterns in mutation negative patients might be helpful to collaboratively build more substantial cohorts despite the rarity of the individual cases for identifying novel disease causes not detected with the current diagnostic approach.
We identified four phenotypic subgroups similar to 1) mitochondrial retinopathy, 2) adult vitelliform or pattern dystrophy, 3) fundus dystrophy similar to LORD or Sorsby fundus dystrophy or 4) PRPH2- or ABCA4-related retinopathy. Other phenotypes with less frequent and possibly specific fundus features might represent retinal dystrophies with as yet unknown, rare genetic cause.
Summary
We demonstrate a high detection rate of mutations in a representative MD/CCRD cohort of 251 consecutive patients and confirm the genetic heterogeneity of MD/CCRD. Targeted NGS is efficient in detecting even unexpected or rare genetic disease causes and might facilitate the identification of novel genotype-phenotype correlations.
References
Hamel, C. P. Cone rod dystrophies. Orphanet J Rare Dis 2, 7, https://doi.org/10.1186/1750-1172-2-7 (2007).
Boulanger-Scemama, E. et al. Next-generation sequencing applied to a large French cone and cone-rod dystrophy cohort: mutation spectrum and new genotype-phenotype correlation. Orphanet J Rare Dis 10, 85, https://doi.org/10.1186/s13023-015-0300-3 (2015).
Thiadens, A. A. et al. Clinical course, genetic etiology, and visual outcome in cone and cone-rod dystrophy. Ophthalmology 119, 819–826, https://doi.org/10.1016/j.ophtha.2011.10.011 (2012).
Neveling, K. et al. Next-generation genetic testing for retinitis pigmentosa. Hum Mutat 33, 963–972, https://doi.org/10.1002/humu.22045 (2012).
O’Sullivan, J. et al. A paradigm shift in the delivery of services for diagnosis of inherited retinal disease. J Med Genet 49, 322–326, https://doi.org/10.1136/jmedgenet-2012-100847 (2012).
Shanks, M. E. et al. Next-generation sequencing (NGS) as a diagnostic tool for retinal degeneration reveals a much higher detection rate in early-onset disease. Eur J Hum Genet 21, 274–280, https://doi.org/10.1038/ejhg.2012.172 (2013).
Glöckle, N. et al. Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur J Hum Genet 22, 99–104, https://doi.org/10.1038/ejhg.2013.72 (2014).
Eisenberger, T. et al. Increasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: the example of retinal dystrophies. PLoS One 8, e78496, https://doi.org/10.1371/journal.pone.0078496 (2013).
Oishi, M. et al. Next-generation sequencing-based comprehensive molecular analysis of 43 Japanese patients with cone and cone-rod dystrophies. Mol Vis 22, 150–160 (2016).
Huang, L. et al. Exome sequencing of 47 chinese families with cone-rod dystrophy: mutations in 25 known causative genes. PLoS One 8, e65546, https://doi.org/10.1371/journal.pone.0065546 (2013).
Stone, E. M. et al. Clinically focused molecular investigation of 1000 consecutive families with inherited retinal disease. Ophthalmology 124, 1314–1331, https://doi.org/10.1016/j.ophtha.2017.04.008 (2017).
Bamshad, M. J. et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 12, 745–755, https://doi.org/10.1038/nrg3031 (2011).
Corton, M. et al. High frequency of CRB1 mutations as cause of Early-Onset Retinal Dystrophies in the Spanish population. Orphanet J Rare Dis 8, 20, https://doi.org/10.1186/1750-1172-8-20 (2013).
Vallespin, E. et al. Gene symbol: CRB1. Disease: Leber congenital amaurosis. Accession #Hd0510. Hum Genet 118, 774 (2006).
Jones, G. E. et al. Microcephaly with or without chorioretinopathy, lymphoedema, or mental retardation (MCLMR): review of phenotype associated with KIF11 mutations. Eur J Hum Genet 22, 881–887, https://doi.org/10.1038/ejhg.2013.263 (2014).
Mirzaa, G. M. et al. Congenital microcephaly and chorioretinopathy due to de novo heterozygous KIF11 mutations: five novel mutations and review of the literature. Am J Med Genet A 164A, 2879–2886, https://doi.org/10.1002/ajmg.a.36707 (2014).
Ostergaard, P. et al. Mutations in KIF11 cause autosomal-dominant microcephaly variably associated with congenital lymphedema and chorioretinopathy. Am J Hum Genet 90, 356–362, https://doi.org/10.1016/j.ajhg.2011.12.018 (2012).
Robitaille, J. M. et al. Phenotypic overlap between familial exudative vitreoretinopathy and microcephaly, lymphedema, and chorioretinal dysplasia caused by KIF11 mutations. JAMA Ophthalmol 132, 1393–1399, https://doi.org/10.1001/jamaophthalmol.2014.2814 (2014).
Hu, H. et al. KIF11 mutations are a common cause of autosomal dominant familial exudative vitreoretinopathy. Br J Ophthalmol, https://doi.org/10.1136/bjophthalmol-2015-306878 (2015).
Birtel, J. et al. Novel insights into the phenotypical spectrum of KIF11-associated retinopathy, including a new form of retinal ciliopathy. Invest Ophthalmol Vis Sci 58, 3950–3959, https://doi.org/10.1167/iovs.17-21679 (2017).
Turnpenny, P. D. & Ellard, S. Alagille syndrome: pathogenesis, diagnosis and management. Eur J Hum Genet 20, 251–257, https://doi.org/10.1038/ejhg.2011.181 (2012).
Hingorani, M. et al. Ocular abnormalities in Alagille syndrome. Ophthalmology 106, 330–337, https://doi.org/10.1016/S0161-6420(99)90072-6 (1999).
Wells, K. K. et al. Ophthalmic features of Alagille syndrome (arteriohepatic dysplasia). J Pediatr Ophthalmol Strabismus 30, 130–135 (1993).
Kim, B. J. & Fulton, A. B. The genetics and ocular findings of Alagille syndrome. Semin Ophthalmol 22, 205–210, https://doi.org/10.1080/08820530701745108 (2007).
Crosnier, C. et al. Mutations in JAGGED1 gene are predominantly sporadic in Alagille syndrome. Gastroenterology 116, 1141–1148 (1999).
Spinner, N. B. et al. Jagged1 mutations in Alagille syndrome. Hum Mutat 17, 18–33, https://doi.org/10.1002/1098-1004 (2001).
Krantz, I. D. et al. Spectrum and frequency of jagged1 (JAG1) mutations in Alagille syndrome patients and their families. Am J Hum Genet 62, 1361–1369, https://doi.org/10.1086/301875 (1998).
El-Asrag, M. E. et al. Biallelic mutations in the autophagy regulator DRAM2 cause retinal dystrophy with early macular involvement. Am J Hum Genet 96, 948–954, https://doi.org/10.1016/j.ajhg.2015.04.006 (2015).
Sergouniotis, P. I. et al. Disease expression in autosomal recessive retinal dystrophy associated with mutations in the DRAM2 gene. Invest Ophthalmol Vis Sci 56, 8083–8090, https://doi.org/10.1167/iovs.15-17604 (2015).
Durlu, Y. K., Koroglu, C. & Tolun, A. Novel recessive cone-rod dystrophy caused by POC1B mutation. JAMA Ophthalmol 132, 1185–1191, https://doi.org/10.1001/jamaophthalmol.2014.1658 (2014).
Roosing, S. et al. Disruption of the basal body protein POC1B results in autosomal-recessive cone-rod dystrophy. Am J Hum Genet 95, 131–142, https://doi.org/10.1016/j.ajhg.2014.06.012 (2014).
Beck, B. B. et al. Mutation of POC1B in a severe syndromic retinal ciliopathy. Hum Mutat 35, 1153–1162, https://doi.org/10.1002/humu.22618 (2014).
Caridi, G. et al. Renal-retinal syndromes: association of retinal anomalies and recessive nephronophthisis in patients with homozygous deletion of the NPH1 locus. Am J Kidney Dis 32, 1059–1062, doi:S0272638698003643 (1998).
Parisi, M. A. et al. The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am J Hum Genet 75, 82–91, https://doi.org/10.1086/421846 (2004).
Simms, R. J., Eley, L. & Sayer, J. A. Nephronophthisis. Eur J Hum Genet 17, 406–416, https://doi.org/10.1038/ejhg.2008.238 (2009).
Fahim, A. T. et al. Polymorphic variation of RPGRIP1L and IQCB1 as modifiers of X-linked retinitis pigmentosa caused by mutations in RPGR. Adv Exp Med Biol 723, 313–320, https://doi.org/10.1007/978-1-4614-0631-0_41 (2012).
Sharon, D. et al. RP2 and RPGR mutations and clinical correlations in patients with X-linked retinitis pigmentosa. Am J Hum Genet 73, 1131–1146, https://doi.org/10.1086/379379 (2003).
Vervoort, R. et al. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat Genet 25, 462–466, https://doi.org/10.1038/78182 (2000).
Yang, Z. et al. Mutations in the RPGR gene cause X-linked cone dystrophy. Hum Mol Genet 11, 605–611 (2002).
Demirci, F. Y. et al. X-linked cone-rod dystrophy (locus COD1): identification of mutations in RPGR exon ORF15. Am J Hum Genet 70, 1049–1053, https://doi.org/10.1086/339620 (2002).
Ebenezer, N. D. et al. Identification of novel RPGR ORF15 mutations in X-linked progressive cone-rod dystrophy (XLCORD) families. Invest Ophthalmol Vis Sci 46, 1891–1898, https://doi.org/10.1167/iovs.04-1482 (2005).
Bellingrath, J. S. et al. High Symmetry of Visual Acuity and Visual Fields in RPGR-Linked Retinitis Pigmentosa. Invest Ophthalmol Vis Sci 58, 4457–4466, https://doi.org/10.1167/iovs.17-22077 (2017).
Bielas, S. L. et al. Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat Genet 41, 1032–1036, https://doi.org/10.1038/ng.423 (2009).
Wang, X. et al. Comprehensive molecular diagnosis of 179 Leber congenital amaurosis and juvenile retinitis pigmentosa patients by targeted next generation sequencing. J Med Genet 50, 674–688, https://doi.org/10.1136/jmedgenet-2013-101558 (2013).
Xu, Y. et al. Mutation analysis in 129 genes associated with other forms of retinal dystrophy in 157 families with retinitis pigmentosa based on exome sequencing. Mol Vis 21, 477–486 (2015).
Hardee, I. et al. Defective ciliogenesis in INPP5E-related Joubert syndrome. Am J Med Genet A 173, 3231–3237, https://doi.org/10.1002/ajmg.a.38376 (2017).
Stone, E. M. et al. Missense variations in the fibulin 5 gene and age-related macular degeneration. N Engl J Med 351, 346–353, https://doi.org/10.1056/NEJMoa040833 (2004).
Wada, Y. et al. Screen of the IMPDH1 gene among patients with dominant retinitis pigmentosa and clinical features associated with the most common mutation, Asp226Asn. Invest Ophthalmol Vis Sci 46, 1735–1741, https://doi.org/10.1167/iovs.04-1197 (2005).
Bowne, S. J. et al. Spectrum and frequency of mutations in IMPDH1 associated with autosomal dominant retinitis pigmentosa and leber congenital amaurosis. Invest Ophthalmol Vis Sci 47, 34–42, https://doi.org/10.1167/iovs.05-0868 (2006).
Kennan, A. et al. Identification of an IMPDH1 mutation in autosomal dominant retinitis pigmentosa (RP10) revealed following comparative microarray analysis of transcripts derived from retinas of wild-type and Rho(−/−) mice. Hum Mol Genet 11, 547–557 (2002).
de Laat, P., Smeitink, J. A., Janssen, M. C., Keunen, J. E. & Boon, C. J. Mitochondrial retinal dystrophy associated with the m.3243A>G mutation. Ophthalmology 120, 2684–2696, https://doi.org/10.1016/j.ophtha.2013.05.013 (2013).
Riveiro-Alvarez, R. et al. Outcome of ABCA4 disease-associated alleles in autosomal recessive retinal dystrophies: retrospective analysis in 420 Spanish families. Ophthalmology 120, 2332–2337, https://doi.org/10.1016/j.ophtha.2013.04.002 (2013).
Maugeri, A. et al. Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am J Hum Genet 67, 960–966, https://doi.org/10.1086/303079 (2000).
Kitiratschky, V. B. et al. ABCA4 gene analysis in patients with autosomal recessive cone and cone rod dystrophies. Eur J Hum Genet 16, 812–819, https://doi.org/10.1038/ejhg.2008.23 (2008).
Kohl, S. et al. Genes and mutations in autosomal dominant cone and cone-rod dystrophy. Adv Exp Med Biol 723, 337–343, https://doi.org/10.1007/978-1-4614-0631-0_44 (2012).
Lotery, A. J. et al. Mutations in the CRB1 gene cause Leber congenital amaurosis. Arch Ophthalmol 119, 415–420 (2001).
den Hollander, A. I. et al. CRB1 mutation spectrum in inherited retinal dystrophies. Hum Mutat 24, 355–369, https://doi.org/10.1002/humu.20093 (2004).
Bernal, S. et al. Study of the involvement of the RGR, CRPB1, and CRB1 genes in the pathogenesis of autosomal recessive retinitis pigmentosa. J Med Genet 40, e89 (2003).
Henderson, R. H. et al. Phenotypic variability in patients with retinal dystrophies due to mutations in CRB1. Br J Ophthalmol 95, 811–817, https://doi.org/10.1136/bjo.2010.186882 (2011).
Tsang, S. H. et al. Whole exome sequencing identifies CRB1 defect in an unusual maculopathy phenotype. Ophthalmology 121, 1773–1782, https://doi.org/10.1016/j.ophtha.2014.03.010 (2014).
Shah, N. et al. Isolated maculopathy associated with biallelic CRB1 mutations. Ophthalmic Genet 38(2), 190–193 (2016).
Beryozkin, A. et al. Mutations in CRB1 are a relatively common cause of autosomal recessive early-onset retinal degeneration in the Israeli and Palestinian populations. Invest Ophthalmol Vis Sci 54, 2068–2075, https://doi.org/10.1167/iovs.12-11419 (2013).
Branham, K. et al. Mutations in RPGR and RP2 account for 15% of males with simplex retinal degenerative disease. Invest Ophthalmol Vis Sci 53, 8232–8237, https://doi.org/10.1167/iovs.12-11025 (2012).
Venturini, G. et al. Molecular genetics of FAM161A in North American patients with early-onset retinitis pigmentosa. PLoS One 9, e92479, https://doi.org/10.1371/journal.pone.0092479 (2014).
Langmann, T. et al. Nonsense mutations in FAM161A cause RP28-associated recessive retinitis pigmentosa. Am J Hum Genet 87, 376–381, https://doi.org/10.1016/j.ajhg.2010.07.018 (2010).
Gu, S., Kumaramanickavel, G., Srikumari, C. R., Denton, M. J. & Gal, A. Autosomal recessive retinitis pigmentosa locus RP28 maps between D2S1337 and D2S286 on chromosome 2p11-p15 in an Indian family. J Med Genet 36, 705–707 (1999).
Bandah-Rozenfeld, D. et al. Homozygosity mapping reveals null mutations in FAM161A as a cause of autosomal-recessive retinitis pigmentosa. Am J Hum Genet 87, 382–391, https://doi.org/10.1016/j.ajhg.2010.07.022 (2010).
Zobor, D., Balousha, G., Baumann, B. & Wissinger, B. Homozygosity mapping reveals new nonsense mutation in the FAM161A gene causing autosomal recessive retinitis pigmentosa in a Palestinian family. Mol Vis 20, 178–182 (2014).
Rose, A. M. et al. Diverse clinical phenotypes associated with a nonsense mutation in FAM161A. Eye (Lond) 29, 1226–1232, https://doi.org/10.1038/eye.2015.93 (2015).
Abid, A., Ismail, M., Mehdi, S. Q. & Khaliq, S. Identification of novel mutations in the SEMA4A gene associated with retinal degenerative diseases. J Med Genet 43, 378–381, https://doi.org/10.1136/jmg.2005.035055 (2006).
Tajiguli, A. et al. Next-generation sequencing-based molecular diagnosis of 12 inherited retinal disease probands of Uyghur ethnicity. Sci Rep 6, 21384, https://doi.org/10.1038/srep21384 (2016).
Mackay, D. S. et al. Novel mutations in MERTK associated with childhood onset rod-cone dystrophy. Mol Vis 16, 369–377 (2010).
Charbel Issa, P. et al. Characterisation of severe rod-cone dystrophy in a consanguineous family with a splice site mutation in the MERTK gene. Br J Ophthalmol 93, 920–925, https://doi.org/10.1136/bjo.2008.147397 (2009).
Ostergaard, E., Duno, M., Batbayli, M., Vilhelmsen, K. & Rosenberg, T. A novel MERTK deletion is a common founder mutation in the Faroe Islands and is responsible for a high proportion of retinitis pigmentosa cases. Mol Vis 17, 1485–1492 (2011).
Acknowledgements
This work was supported by the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre (BRC; PCI); the Pro Retina Deutschland (PCI), and the BONFOR research program of the University of Bonn (PLM, grant No O-137.0023). No sponsor or funding agency had any involvement in the design, collection, analysis and interpretation of the data, manuscript writing or the decision to submit the manuscript for publication. The authors alone are responsible for the content and writing of the paper. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
Author information
Authors and Affiliations
Contributions
J.B., E.M., H.J.B. and P.C.I. wrote the manuscript. J.B., M.G., P.L.M., P.H., F.G.H. and P.C.I. carried out detailed clinical investigation of the patients. T.E., C.B., D.Z., C.N., S.L. and H.J.B. analyzed and interpreted NGS data. J.B., P.C.I. and H.J.B. designed the study.
Corresponding authors
Ethics declarations
Competing Interests
Dres. Eisenberger, Betz, Zahnleiter, Neuhaus, Lenzner and Bolz (until the end of 2016) are employees of Bioscientia, a publicly traded diagnostic company. The other authors have no competing interests
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Birtel, J., Eisenberger, T., Gliem, M. et al. Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone-rod dystrophy. Sci Rep 8, 4824 (2018). https://doi.org/10.1038/s41598-018-22096-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-22096-0
This article is cited by
-
Clinical exome analysis and targeted gene repair of the c.1354dupT variant in iPSC lines from patients with PROM1-related retinopathies exhibiting diverse phenotypes
Stem Cell Research & Therapy (2024)
-
Retinal primary cilia and their dysfunction in retinal neurodegenerative diseases: beyond ciliopathies
Molecular Medicine (2024)
-
The central retinal thickness and its related genotype in ABCA4-related retinopathy
Eye (2024)
-
Therapeutic Extracellular Vesicles from Tonsil-Derived Mesenchymal Stem Cells for the Treatment of Retinal Degenerative Disease
Tissue Engineering and Regenerative Medicine (2023)
-
Inherited retinal disorders: a genotype–phenotype correlation in an Indian cohort and the importance of genetic testing and genetic counselling
Graefe's Archive for Clinical and Experimental Ophthalmology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
