
Abstract
Soil microbes provide important ecosystem services. Zoige Plateau wetland, the largest alpine peat wetland in the world, has suffered from serious degradation in the past 30 years. We studied the composition of the Zoige Plateau alpine wetland soil microbiota and relations among specific taxa using 16S rRNA amplicon sequencing combined with association network analysis. Compared to the pristine swamp soil, taxons DA101, Aeromicrobium, Bradyrhizobium, and Candidatus Nitrososphaera were enriched and several methanogenic Euryarchaeota were depleted in the moderately degraded meadow soil and highly degraded sandy soil. Soil total potassium contents in soils with different degradation levels were significantly different, being the highest in meadow soil and lowest in swamp soil. The association network analysis showed that total potassium positively correlated with specific bacterial and archaeal taxa. Jiangella, Anaerolinea, Desulfobulbus, Geobacter, Flavobacterium, Methanobacterium and Methanosaeta were identified as the keystone genera in the networks. Soil degradation affected soil properties, and caused changes in the bacterial and archaeal community composition and the association patterns of community members. The changes could serve as early warning signals of soil degradation in alpine wetlands.
Similar content being viewed by others

Introduction
Despite the increased understanding of the important roles of wetlands in terrestrial ecosystems, severe land loss and degradation occur in the wetlands of China due to population growth, land reclamation projects and over-grazing1. The alpine wetlands are more vulnerable to climate change than those in other regions2. The Zoige alpine wetland on the Qinghai-Tibetan Plateau is the highest and largest alpine wetland in the world. Zoige wetland plays important roles in maintaining water balance, carbon sequestration, and biodiversity preservation3. It covers an area of 1.2 × 104 km2, occupying about 0.1% of the total global peatland area4. In the past 40 years, numerous studies have focused on the degradation of Zoige wetland that has resulted from increasing temperature, anthropogenic perturbations, and overgrazing by cattle. The studies have shown significant impacts of these activities on the landscape, hydrology, biodiversity, and ecosystem degradation, including severe peat deterioration. The mean annual temperature has increased at a rate of 0.16 °C per decade in this area, resulting in the drying of the wetlands5. The wetland area in Zoige has decreased by approximately 30% from the 1970s to the 2000s due to the decline of water table6, resulting in a transition from pristine swamp soil to degraded meadow soil, and even aeolian sandy soil at severely deteriorated sites7,8. Deterioration has also changed the composition of plant communities from humidogenes and aquatics to mesophytes and xerophytes9, and resulted in the decomposition of soil organic matter, loss of humus and peat horizon, and decrease in nutrient content10,11.
Soil microbes are critical in biogeochemical processes and organic material decomposition, and quickly respond to environmental disturbances12,13. At high taxonomic ranks, for example at phylum or class level, bacteria and archaea exhibit ecological coherence, and community composition may be valuable in predicting ecosystem functioning14. Based on nutritional requirements and growth rates, microbes can be classified as either copiotrophs or oligotrophs at the phylum level, and applying this classification may contribute to further understanding of microbial community dynamics in response to environmental interferences15. Alpine wetland degradation could directly alter the composition of specific bacterial and archaeal phyla. For example, Bacteroidetes and Flavobacterium were widely distributed in the pristine swamp soil in Zoige wetland, while Alphaproteobacteria were more dominant in the degraded sandy soil16. However, knowledge on the response of bacterial and archaeal taxa at low taxonomical level (such as genus or species) to the alpine wetland degradation remains limited. It is likely that the loss of humus due to wetland degradation would select for specific taxa that depend primarily on organic substrates, resulting in changes in microbial community composition and soil nutrient status17,18. Taxa that are affected by alpine wetland degradation should be associated with edaphic properties like soil organic carbon, which could then be used as valuable indicators of alpine wetland soil nutrient status and development16. Microbial communities in soil are complex, including many potentially interacting taxa, and a full exploration of these interactions is important to allow an integrated understanding of soil microbial community structure and the ecological rules that govern them19. Co-abundance network analyses can be carried out using data generated from high-throughput sequencing to provide information on the community composition and interactions within communities19. Furthermore, co-presence links reflect shared niches while exclusion links suggest niche segregation.
Knowledge on the effects of wetland degradation on bacterial and archaeal communities is still scarce. To address this, we applied 16S rRNA gene amplicon sequencing to detect microbial community variation in microhabitats associated with the succession of degradation, and determined factors associated with variation. The effects of wetland degradation on specific bacterial and archaeal taxa and their associations with soil nutrients were analyzed by analyzing differences in abundant taxa and by co-abundance network analysis. Our purpose was to determine (1) the variation in bacterial and archaeal communities, (2) the specific taxa affected by wetland degradation in the three soils, and (3) the possible relationships between specific taxa and soil properties. We hypothesized that (i) the bacterial and archaeal communities in the pristine soil would significantly differ with those found in the degraded soil due to the inherent differences in nutritional conditions, (ii) that taxa related to carbon and nitrogen cycling would either be enriched/or depleted, and (iii) the specific taxa that were substantially changed would form distinct association patterns.
Results
Soil properties and community composition
Soil properties were different in the three soils associated with the degraded Zoige wetland (Table 1). Soil total potassium content (TK: 6.79 to 23.13 g kg−1) was highest in the meadow soil and lowest in swamp soil (Table 1). Principal component analysis (PCA) showed clear separation of the three soils indicating that wetland degradation resulted in distinct microhabitats (ANOSIM: R > 0.9, P < 0.001) (Fig. 1A). Proteobacteria (24.6–31.6%), Acidobacteria (12.3–19.7%), and Chloroflexi (10.8–19.7%) were the dominant phyla in all soil types. In the swamp soil, the relative abundance of Proteobacteria was highest, while that of Acidobacteria was lower than in the other soils. Chloroflexi was more abundant in the swamp and sandy soils than in the meadow (Fig. 1B). Within Proteobacteria, the Delta subdivision was the most abundant group (34.7 to 40.0%), followed by Betaproteobacteria (26.9 to 29.4%), Alphaproteobacteria (16.5–22.6%), and Gammaproteobacteria (14.0–15.4%) (Fig. S1). The relative abundances of Deltaproteobacteria and Betaproteobacteria were higher in the swamp soil than in the other soils. The relative abundance of Alphaproteobacteria was highest in the swamp soil, and that of Gammaproteobacteria in the sandy soil. The relative abundances of ten families were higher than 0.5% of the total abundance (Fig. S2). Compared to the pristine swamp soil, the relative abundance of Nitrososphaeraceae, Sphingomonadaceae, Xanthomonadaceae, and Chitinophagaceae were significantly higher in the degraded soils, while those of Sinobacteraceae and Comamonadaceae were lower (P < 0.05). Moreover, the relative abundances of Myxococcaceae (P < 0.05) and Rhodocyclaceae were lowest in the meadow soil, while those of Syntrophobacteraceae and Rhodospirillaceae were lowest in the swamp soil (Fig. S2).

Principal component analysis (PCA) of soil edaphic properties (A) and microbial community composition in Zoige plateau wetland soils (B). SW: swamp soil; MD: meadow soil; and SD: sandy soil.
Differentially abundant taxa
Enriched taxa (e-taxa) and depleted taxa (d-taxa), representing taxa with relative abundances twice higher or lower (P = 0.05) in the swamp soil than in the meadow and sandy soils, were identified by differential abundance analysis. Using taxa counts from swamp soil as a control and an FDR-adjusted P < 0.1, eleven and 89 differentially abundant taxa were detected at the phylum and genus levels, respectively (Fig. 2).

Volcano plots illustrating taxa that were significantly enriched (red) and depleted (green) at phylum (A,B and C) and genus level (D,E and F). SW: swamp soil; MD: meadow soil; and SD: sandy soil.
At the phylum level, Euryarchaeota, WWE1, OP8 and GOUTA4 were enriched in the swamp soil when compared with the meadow soil (Figs 2A and S3A). Compared with the sandy soil, WWE1, Euryarchaeota, OP8, GOUTA4 and Bacteroidetes were enriched, and TM7 and AD3 were depleted in the swamp soil (Figs 2B and S3B). At the genus level, additional 35 e-taxa and 16 d-taxa were detected in the swamp soil when compared with the meadow soil (Fig. 2D). The top five e-taxa were DA101, Crocinitomix, Aeromicrobium, Bradyrhizobium, and Candidatus Nitrososphaera (phylum Crenarchaeota), and the top five d-taxa were Syntrophus, Methanobacterium, Methanosarcina, Chloronema, and Methanosaeta (Dataset S1). When compared with the sandy soil, 31 e-taxa and 6 d-taxa were detected in the swamp soil (Fig. 2E), among which DA101, Aeromicrobium, Sporichthya, JG37-AG-70, and Zoogloea were the top five e-taxa and Chryseobacterium, Syntrophus, Chloronema, Oryzihumus, and Methanosarcina the top five d-taxa (Dataset S2). When comparing sandy soil with the meadow soil, only one d-taxa was detected at the genus level (Fig. 2C,F).
The Mantel test showed significant correlations between differentially abundant phyla and multiple edaphic factors. Soil TK correlated negatively with most of the differentially abundant phyla (P = 0.001) (Fig. S3C,D).
Community structure, variation, and determinants
Principal coordinate analysis (PCoA) with weighted and unweighted UniFrac distances clearly separated the swamp communities from the communities in degraded soils (Fig. 3A,B). Weighted UniFrac takes the abundance of taxa into account whereas unweighted UniFrac is based on the presence and absence of species, making it more sensitive to rare taxa (Fig. 3B). The clear separation between the swamp soil and the degraded soils in unweighted Unifrac-based PCoA indicates that wetland degradation affects especially the rare taxa. In the distance-based redundancy analysis (dbRDA), soil TK was the strongest driver of microbial community structure among the constrained nine edaphic factors (Fig. 3C,D). In addition, Mantel test showed that soil TK correlated positively and significantly with soil microbial community composition (Fig. S4A,B). These results further suggest that the total potassium content of the soils was the key factor in shaping the microbial community composition in the wetland soils.

Microbial community variation across the Zoige plateau wetland soils. (A,B) Principal coordinate analysis (PCoA) plots of OTU-based weighted (A) and unweighted (B) UniFrac distances between samples; (C,D) distance-based redundancy analysis (dbRDA) of weighted (C) and unweighted (D) UniFrac distances quantifying the impacts of edaphic factors on microbial community composition. SW: swamp soil; MD: meadow soil; and SD: sandy soil. WC: water content; SOC: soil organic carbon; TN: total nitrogen; TP: total phosphorus; TK: total potassium; AN: available nitrogen; OP: Olsen phosphorus (available phosphorus); AK: available potassium. PDA: Potential denitrifying activity. *SW: swamp soil; MD: meadow soil; SD: sandy soil.
Network associations
Based on differential taxa abundance analysis, association networks were generated to characterize bacterial and archaeal communities in the soils (Fig. 4). The topological features of the network are presented in Table S2. In total, 1893 edges connected the 229 differentially abundant taxa in the network. Many of the differentially abundant taxa correlated with each other positively (Fig. 4), indicating that these taxa likely respond similarly to changes associated with wetland degradation. Actinobacteria, Bacteroidetes, Chloroflexi, Euryarchaeota and Proteobacteria showed higher degree centrality in the network, suggesting that these taxa are strongly associated with the other members of the community (Fig. 5). Taxa belonging to Actinobacteria, Bacteroidetes, Chloroflexi, Proteobacteria and Planctomycetes had higher betweenness centrality compared to the other taxa, indicating that these taxa likely influence the interactions among taxa in the community (Fig. 6).

The abundance association network of differentially abundant taxa in the Zoige plateau wetland soils. Purple and red lines represent negative and positive correlation, respectively.

Network level topological features in the Zoige plateau wetland soils.

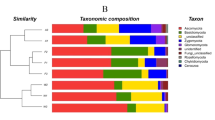
The taxonomic composition at the phylum level of the network modules in the Zoige platerau wetland soils.
Based on greedy modularity optimization, three potential functional modules (M1, M2, and M3) were detected according to association strength among the differentially abundant taxa (Fig. 4). Module M1 included Jiangella (phylum Actinobacteria), Anaerolinea (phylum Chloroflexi), Desulfobulbus (Proteobacteria), Flavobacterium (Bacteroidetes), and Methanobacterium and Methanosaeta (phylum Euryarchaeota). The abundances of these taxa were significantly higher in the swamp soil than in the degraded soils (Fig. 6, Table 2). With the exception of DA101 in M2 and Acidovorax in M3, most of the differentially abundant taxa in modules M2 and M3 were not identified at genus level. The relative abundances of almost all taxa in each module were distinct in different soils (Fig. 6). The variation in abundance and taxonomic composition may provide clues on the functional modules that potentially rapidly and reversibly respond to environmental changes induced by wetland degradation through species sorting (plastic adjustment). Although the microhabitats in the meadow and sandy soils were distinct, the composition of the functional modules was similar, especially in modules M1 and M2 (Fig. 6).
PCoA result showed significant (p = 0.001) correlations between network modules and soil properties, with soil TK ranking highest (Fig. S5A–C). This was supported by Mantel test, which showed high correlations between soil TK and these three potential functional modules, further indicating that soil TK drives structuring in these functional modules (Fig. S5D).
Discussion
We studied shifts in bacterial and archaeal community composition in three soil types associated with wetland degradation: pristine swamp soil, moderately degraded meadow soil and highly degraded sandy soil. As in previous studies based on denaturing gradient gel electrophoresis16,20, the bacterial and archaeal community compositions in the three soils differed significantly, but all were dominated by the phyla Proteobacteria, Acidobacteria and Chloroflexi. Proteobacteria are prevalent in various ecosystems and involved in many biogeochemical processes21. As in Röskeet al.22, Proteobacterial communities were mainly composed of Deltaproteobacteria and Betaproteobacteria, both classified as copiotrophs stimulated by increased nutrients in the environment15, which could partially explain why these two groups were more abundant in the swamp soil with higher organic matter content. Acidobacteria include many oligotrophs23,24, and their relative abundance was higher in the degraded soils that contained less organic matter, thus possibly favoring these oligotrophs. Interestingly, Chloroflexi that are known to survive on plant residues and grow even under nutrient limitation25,26 were more abundant in the sandy soil which had less nutrients than the pristine swamp soil.
Understanding the environmental variables that shape soil microbial community structuring is one of the key goals of microbial ecology. Soil water content was previously found to be the major factor connected with Zoige wetland degradation, with significant impacts on the decrease of fungal community abundance and prokaryotic diversity20,27. With the lowering of water table, some of the wetland plant species that are essential in swamp soil formation are being replaced by meadow vegetation28. Different vegetation types may release different types of root exudates: the exudates are significant sources of carbon, nitrogen and energy of the soil microbiota29. Different plant species also have diverse nutritional needs, promoting the development of particular soil microbiome depending on the growth substrate30. In our study, water content, organic carbon content, and the total and available nitrogen and phosphorus contents all decreased along with the wetland degradation, making them valuable indicators for evaluating wetland degradation. Soil total potassium content was found to be an important factor shaping the bacterial and archaeal communities. Similarly, Pereira et al.31 found that the bacterial diversity and community composition in the soil were highly affected by the total potassium content. Potassium is easily leached because of its mobility in soils, and thus directly affected by soil degradation.
The microbial community structure in the pristine swamp soil differed from those in the meadow and sandy soils. Consistent with our hypothesis, more taxa were enriched in the swamp soil, indicating that wetland degradation decreased the abundance of these bacterial and archaeal taxa. Methanogenic Euryarchaeota is mainly distributed in anoxic environments, for example in wetlands, paddy soils and marine sediments, and is involved in greenhouse gas production32. In this study, as expected, the abundances of Euryarchaeota were significantly higher in the pristine soils than in the degraded soils. Previous studies indicated that Zoige wetland was one of the hotspots of methane emissions in the Qinghai-Tibetan plateau, harboring a unique methanogen community33. In contrast, there were hardly any methanogens in the meadow soil in Zoige wetland34, probably due to the decreased soil water table and content that had lead to less anaerobic conditions. Although we did not measure the oxygen content in the soils, soil water content in the degraded soils had decreased significantly and water table was lower. A change in water table and content could directly affect the oxic–anoxic interfaces, increase the thickness of the boreal peatland aerobic layer, and ultimately change the microbial community composition and abundances35. Moreover, low water tables affect soil organic carbon (SOC) by changing the vegetation cover and SOC decomposition rate which is positively correlated with the methanogenic archaea community36. At the genus level, the enrichment of Methanobacterium, Methanosarcina and Methanosaeta in the swamp soil indicate their potential roles as ‘keystone species’ in methanogenesis in this ecosystem.
Syntrophy, defined as a long-term stable relation of organisms which can be either beneficial or unbeneficial, plays a key role in many anaerobic ecosystems, even in systems where metabolism is sustained by the reduction of electron acceptors such as sulphate or nitrogen oxides37. Species related to Syntrophobacteraceae generally constitute consortia with methanogenic Archaea to syntrophically convert propionate to methane, which is a typical metabolic process during anaerobic degradation. It is likely that the relative abundance of Syntrophobacteraceae affiliated species in this study would similarly vary with the Methanogenic Euryarchaeota in response to wetland degradation. However, phylogenetic analysis of the community showed that contrary to the decreased abundance of the methanogenic Euryarchaeota, the abundances of members of the Syntrophobacteraceae actually increased. Syntrophobacteraceae are extremely diverse in their nutritional transformation capabilities, with some members being able to use a wide variety of electron donors and carbon sources syntrophically or in pure cultures, and in the presence or absence of sulfate as electron acceptor38. This metabolic peculiarity confers these microorganisms with adaptability to changing environmental conditions, and may explain the increase of Syntrophobacter-related species in the degraded meadow and sandy microcosms.
DA101 can occupy various ecological niches and perform oligotrophic life strategy in soil39. Aeromicrobium are lignocellulose decomposers and play important roles in organic matter turnover in soil40. Bradyrhizobium are beneficial for plant growth due to their nitrogen-fixing ability. Candidatus Nitrososphaera thrives in organic-rich habitats41. The enrichment of these taxa in the degraded meadow and sandy soils suggest their subtle differences in habitat adaption, which also contributes differently to the carbon and nitrogen cycling in this specific ecosystem.
Network analysis provides understanding of potential interactions such as cooperation, competition and niche partitioning in bacterial and archaeal communities, and may identify keystone populations42. A node with high betweenness centrality value is in a core location in the network, and may affect other interactions in the community43. Keystone species with maximum betweenness centrality values are likely to serve as gatekeepers in ecosystem functions44,45. In our study, most archaeal keystone species in the networks were affiliated with Euryarchaeota that participate in wetland carbon cycling as discussed above, while most bacterial keystone species were from Actinobacteria, Proteobacteria and Bacteroidetes. Of all the keystone species detected in our study, Desulfobulbus are the only ones involved in the sulfur biogeochemical cycle, particularly in the reduction of sulfate to sulfide46. Geobacter are able to oxidize recalcitrant organic carbon47, and Flavobacterium are stimulated in organic matter rich environments48. The organic matter content in the swamp soil was higher than in the degraded soils, which may explain the significantly higher abundance of Flavobacterium in the swamp soil. Taken altogether, the keystone species may be pivotal determinants of the succession of bacterial and archaeal taxa during the wetland degradation.
In summary, wetland degradation changed soil properties, which further affected specific bacterial and archaeal taxa involved in nutrient cycling resulting in distinct association patterns. Proteobacteria, Acidobacteria, and Chloroflexi were the most abundant phyla. Soil total potassium strongly affected the shaping of bacterial and archaeal communities. Interpretation of the interaction networks in soil bacterial and archaeal community may open new avenues for understanding the ecological roles of bacterial and archaeal taxa in the degradation of the Zoige alpine wetland soil. The observed changes may serve as early warning signals of soil degradation in alpine wetlands.
Materials and Methods
Study area and soil sampling
The Zoige Plateau is in the northeast of Qinghai-Tibetan Plateau (101°30′E–103°30′E, 32°20′N–34°00′N) at an average altitude of 3500 m (Table S1). The mean annual precipitation in this area is 654 mm49. Eighty-five percent of precipitation is delivered from April to September. The mean annual air temperature is 1.1 °C33, with the average temperatures ranging from −10.1 °C in January to 10.7 °C in July.
Swamp soil (SW), meadow soil (MD) and sandy soil (SD) in Zoige were sampled in August 2014. The swamp was covered with water with an annual average water table of 5.87 cm above ground. The vegetation was dominated by hydrophytes and sedge hydro-mesophytes (Carexmuliensis, Carexlasiocarpa and Carexmeyeriana). The meadow surface was in a humid state and the vegetation was dominated by mesophytes and hydro-mesophytes (Kobresiatibetica), and the water table was approximately 4.26 cm below ground. The sandy soil surface was continuously dry and had little or only few Psammophytes as vegetation cover50, and the water table was at least 21 cm below ground. The soil texture was peat soil in SW, sandy loam in soil MD, and sandy soil in SD. The parent materials of swamp and meadow soils were homogeneous silt clay and Triassic slate residues, and sandstones and siltstone, respectively51. In the sandy soil, the top layer (0–30 cm) was mostly sand and rest was composed of aeolian parent material52. Based on the FAO soil taxonomy system53, the SW soil is classified as Histosol, the MD soil as Haplic Calcisol, while the SD was Arenosol.
For each soil type, six sampling plots of at least 50 × 50 m2 with a minimum distance of 100 m from each other were selected. At each sampling plot, three soil cores from the top 20 cm below the litter layer were collected using a 6.5 cm diameter steel push corer, and mixed into one approximately 1.5 kg homogenized composite sample. Visible plant roots and residues were removed before mixing. The corer was sterilized using 70% ethanol between samplings. Samples were stored in plastic bags on ice and divided into two portions. Sub-samples for physicochemical analyses and DNA extraction were stored at 4 °C and at −80 °C, respectively.
Soil physicochemical properties
Soil pH and soil organic carbon (SOC) were determined using a soil-to-water ratio of 1:5 and dichromate oxidization method, respectively54. Soil gravimetric water content (WC) was determined by oven-drying at 105 °C for 48 h54. Soil total nitrogen (TN) and available nitrogen (AN), total phosphorus (TP) and available phosphorus (AP), total potassium (TK) and available potassium (AK) were determined by Kjeldahl digestion, alkaline hydrolysis diffusion method, and molybdenum blue method and flame photometry, respectively54.
DNA extraction, amplification and sequencing
DNA was extracted from 0.75–0.91 g fresh weight soils (corresponding to 0.50 g dry weight) using the FastDNA Spin Kit for Soil (MP Biomedicals, Solon, OH, USA) following the manufacturer’s instructions. The quality of DNA was examined using the Nano-200 spectrophotometer (Aosheng, Hangzhou, China). DNA samples were stored at −20 °C.
PCR amplification was carried out using the primers 515F (5′-GTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-GGACTACVSGGGTATCTAAT-3′) targeting the V4 hyper variable region in the bacterial and archaeal 16S rRNA gene55. The reverse primer was combined with the adapter and barcode sequences for multiplexing. Amplification was done in 50 μl-reaction volumes containing 3 U of TaKaRa Ex Taq HS (TaKARA Shuzo Co., Shiga, Japan), 5 mM dNTP Mixture (TaKARA), 2.0 mM MgCl2, 5 μl of 10× Ex Taq Buffer (TaKARA), 0.5 mM of each primer and 2.5 ng of soil DNA. The PCR procedure included an initial denaturation at 94 °C for 4 min, 30 cycles of 15S at 94 °C, 15S at 55 °C and 30S at 72 °C, and a final extension at 72 °C for 10 min.
PCR products were purified using PCR Clean-up Purification Kit (MP Biomedicals) and quantified using Qubit 2.0 fluorimeter (Invitrogen, Carlsbad, CA, USA). Purified amplicons were pooled in equimolar concentrations and sequenced using MiSeq Reagent Kit V2 for Illumina MiSeq. The raw sequence data were submitted to NCBI Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra/) with accession number SRS2127453.
Bioinformatics analysis
Sequence reads were quality-filtered in QIIME (v1.9.1)56. Sequences with phred-quality score over 20 and longer than 300 bps were kept for downstream analyses while those could not be assembled were discarded. Chimeric sequences were removed using UCHIME against a reference alignment55. Sequences were clustered into operational taxonomic units (OTUs) at 97% using the UPARSE pipeline57. The RDP classifier was used to assign OTU representative sequences at 70% threshold56. For inter-group comparison, OTUs were randomly resampled at a depth of 12822 sequences. Singletons were eliminated during resampling, results in 35,014 OTUs. The phylogenetic tree was generated based on the representative sequences and used for UniFrac distance calculations.
Statistical analysis
We applied principal component analysis (PCA) to assess differences in properties between soils. Analysis of similarity (ANOSIM) was used to test significant differences among abundances of taxa. The relative abundances of taxa at phylum and family levels were analyzed using R58. Two-way ANOVA was performed to test differences in the abundances of taxa at family level at P < 0.05. Principal coordinate analysis (PCoA) based on weighted and unweighted UniFrac distances were carried out in the R package vegan59,60. Distance based redundancy analysis (dbRDA) was performed to investigate factors driving community variation. Significance of factors was calculated by the permu test function in vegan over the dbRDA model using a maximum of 999 permutations. The goodness of fit for each edaphic factor was estimated by applying the envfit function in vegan (999 permutations). Mantel test was used to determine correlations between soil properties and microbial community composition.
The R package DESeq2 was used to analyze differential log2-fold transformed abundances at phylum and genus levels61. Differential abundance analysis was performed by fitting a generalized linear model with a negative binomial distribution to the normalized value for each type of taxa, and testing for differential abundance using a Wald test61. P-values were adjusted for multiple testing following the procedure of Benjamini and Hochberg62, and a false discovery rate (FDR) of 10% was selected to denote statistical significance63. Taxa with significant changes in abundance were defined as those that had differential abundance >1.0 and FDR-adjusted P < 0.1. Correlations between taxa with significant changes in abundance and edaphic features were determined using Spearman’s rank correlation.
Network analysis
To reduce the complexity of the network, only OTUs with significant abundance variation among groups were considered for co-abundance network construction, which was then visualized in Cytoscape v3.3.064. Firstly, a Spearman’s rank correlation coefficient matrix was calculated using “scipy” Python package for adjacency matrix construction60. To get robust links among nodes in the network, we further corrected all P-values using the Benjamini and Hochberg false discovery rate (FDR) as performed in “multtest” R package58,65. The cutoffs of correlation were determined as ±0.75 based on random theory-based methods66. The networks were generated using the “igraph” Python package based on cutoffs of correlation (FDR-P < 0.001)67. The network topological properties were calculated to evaluate the importance of each OTU (nodes). OTUs that were strongly correlated were screened using greedy modularity optimization algorithm68. To identify the primary driving force of modularity, we fit taxon abundance and edaphic factors to Bray-Curtis dissimilarities of modules to find the group of taxa and edaphic factors with highest correlation coefficient. The goodness of fit for each taxon and edaphic factor were estimated by the envfit function in vegan R package (999 permutations). Relations between the nodes of networks and soil properties were evaluated by Mantel test and partial Mantel test.
Data accessibility
Sequencing data were submitted to NCBI Sequence Read Archive database (https://www.ncbi.nlm.nih.gov/sra/) with accession number SRS2127453. The OTU table is accessible in Dryad database with doi:10.5061/dryad.ks767.
References
Bai, J. H. et al. Landscape pattern evolution processes of alpine wetlands and their driving factors in the Zoige Plateau of China. J Mt Sci-Eng l10, 54–67 (2013).
Xue, Z. S. et al. Predicted areas of potential distributions of alpine wetlands under different scenarios in the Qinghai-Tibetan Plateau, China. Global Planet Change 123, 77–85 (2014).
Zhang, X., Liu, H. & Xing, Z. Challenges and solutions for sustainable land use in Ruoergai—the highest altitude peatland in Qinhai-Tibetan Plateau, China. Energy Procedia 5, 1019–1025 (2011).
Chen, H. G. et al. The carbon stock of alpine peatlands on the Qinghai-Tibetan Plateau during the Holocene and their future fate. Quaternary Sci Rev 95, 151–158 (2014).
Wu, P. F., Zhang, H. & Wang, Z. Y. The response of soil macroinvertebrates to alpine meadow degradation in the Qinghai-Tibetan Plateau, China. Appl Soil Ecol 90, 60–67 (2015).
Dong, Z. B., Hu, G. Y., Yan, C. Z., Wang, W. L. & Lu, J. F. Aeolian desertification and its causes in the Zoige Plateau of China’s Qinghai-Tibetan Plateau. Environ Earth Sci 59, 1731–1740 (2010).
Guo, X., Du, W., Wang, X. & Yang, Z. F. Degradation and structure change of humic acids corresponding to water decline in Zoige peatland, Qinghai-Tibet Plateau. Sci Total Environ 445–446C, 231–236 (2013).
Luan, J. et al. Soil carbon stocks and quality across intact and degraded alpine wetlands in Zoige, east Qinghai-Tibet Plateau. Wetl Ecol Manag 22, 427–438 (2014).
Han, D. Y., Yang, Y. X. & Yang, Y. Changes of plant species diversity and interspecific correlation in a degraded swamp community along drainage gradients on the Zoige Plateau of China. Chin J Plant Ecol 36, 411–419 (2012).
Xiang, S., Guo, R. Q., Wu, N. & Sun, S. C. Current status and future prospects of Zoige Marsh in Eastern Qinghai-Tibet Plateau. Ecol Eng 35, 553–562 (2009).
Shang, Z. H., Feng, Q. S., Wu, G. L., Ren, G. H. & Long, R. J. Grasslandification has significant impacts on soil carbon, nitrogen and phosphorus of alpine wetlands on the Tibetan Plateau. Ecol Eng 58, 170–179 (2013).
Deng, J. et al. Shifts of tundra bacterial and archaeal communities along a permafrost thaw gradient in Alaska. Mol Ecol 24, 222–234 (2015).
Jiao, S. et al. Temporal dynamics of microbial communities in microcosms in response to pollutants. Mol Ecol 26, 923–936 (2017).
Philippot, L. et al. The ecological coherence of high bacterial taxonomic ranks. Nat Revs Microbiol 8, 523–529 (2010).
Fierer, N., Bradford, M. A. & Jackson, R. B. Toward an ecological classification of soil bacteria. Ecology 88, 1354–1364 (2007).
Tang, J. et al. Effects of wetland degradation on bacterial community in the Zoige Wetland of Qinghai- Tibetan Plateau (China). World J Microbiol Biot 28, 649–657 (2012).
Marschner, P., Kandeler, E. & Marschner, B. Structure and function of the soil microbial community in a long-term fertilizer experiment. Soil Biol Biochem 35, 453–461 (2003).
Cederlund, H. et al. Soil carbon quality and nitrogen fertilization structure bacterial communities with predictable responses of major bacterial phyla. Appl Soil Ecol 84, 62–68 (2014).
Faust, K. & Raes, J. Microbial interactions: from networks to models. Nat Rev Microbiol 10, 538–550 (2012).
Wu, L. S., Nie, Y. Y., Yang, Z. R. & Zhang, J. Responses of soil inhabiting nitrogen-cycling microbial communities to wetland degradation on the Zoige Plateau, China. J Mt Sci-Eng l13, 2192–2204 (2016).
Zhang, L. & Xu, Z. H. Assessing bacterial diversity in soil. J Soil Sediment 8, 379–388 (2008).
Röske, K., Sachse, R., Scheerer, C. & Röske, I. Microbial diversity and composition of the sediment in the drinking water reservoir Saidenbach (Saxonia, Germany). Syst Appl Microbiol 35, 35–44 (2012).
Nemergut, D. R., Cleveland, C. C., Wieder, W. R., Washenberger, C. L. & Townsend, A. R. Plot-scale manipulations of organic matter inputs to soils correlate with shifts in microbial community composition in a lowland tropical rain forest. Soil Biol Biochem 42, 2153–2160 (2010).
Pascault, N. et al. Stimulation of different functional groups of bacteria by various plant residues as a driver of soil priming effect. Ecosystems 16, 810–822 (2013).
Hug, L. A. et al. Community genomic analyses constrain the distribution of metabolic traits across the chloroflexi phylum and indicate roles in sediment carbon cycling. Microbiome 1, 22 (2013).
Barton, H. A. et al. Microbial diversity in a Venezuelan orthoquartzite cave is dominated by the Chloroflexi (class Ktedonobacterales) and Thaumarchaeota group i.1c. Front Microbiol 5, 615 (2014).
Zhong, Q. P. et al. Water table drawdown shapes the depth-dependent variations in prokaryotic diversity and structure in Zoige peatlands. FEMS Microbiol Ecol 93, 049 (2017).
Ma, Q. F. et al. Aboveground and belowground biomass relationships in the Zoige Peatland, Eastern Qinghai–Tibetan Plateau. Wetlands 37, 461–469 (2017).
Conrad, R., Klose, M., Noll, M., Kemnitz, D. & Bodelier, P. L. E. Soil type links microbial colonization of rice roots to methane emission. Global Change Biol 14, 657–669 (2008).
Pii, Y. et al. The interaction between iron nutrition, plant species and soil type shapes the rhizosphere microbiome. Plant Physiol Biochem 99, 39–48 (2016).
Pereira, L. B., Vicentini, R. & Ottoboni, L. M. M. Changes in the bacterial community of soil from a neutral mine drainage channel. PLoS ONE 9, e96605 (2014).
Liu, Y. & Whitman, W. Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. Ann NY Acad Sci 1125, 171–189 (2008).
Cui, X. & Graf, H. Recent land cover changes on the Tibetan Plateau: a review. Climatic Change 94, 47–61 (2009).
Deng, Y., Cui, X., Hernández, M. & Dumont, M. G. Microbial diversity in hummock and hollow soils of three wetlands on the Qinghai-Tibetan Plateau revealed by 16S rRNA pyrosequencing. PLoS ONE 9, e103115 (2014).
Jaatinen, K. et al. Effects of short- and long-term water-level drawdown on the populations and activity of aerobic decomposers in a boreal peatland. Global Change Biol 13, 491–510 (2007).
Tian, J. Q. et al. Effects of drought on the archaeal community in soil of the Zoige wetlands of the Qinghai-Tibetan plateau. Eur J Soil Biol 52, 84–90 (2012).
Wang, Y. et al. Time-resolved analysis of a denitrifying bacterial community revealed a core microbiome responsible for the anaerobic degradation of quinoline. Sci Rep-UK 7, 14778 (2017).
Harmsen, H. J. M. et al. Syntrophobacter fumaroxidans sp. nov., a syntrophic propionate- degrading sulfate- reducing bacterium. Int J Syst Evol Microbial 48, 1383–1387 (1998).
Shen, C. C., Ge, Y., Yang, T. & Chu, H. Y. Verrucomicrobial elevational distribution was strongly influenced by soil pH and carbon/nitrogen ratio. J Soil Sediment, 1–8 (2017).
Abdulla, H. M. & El-Shatoury, S. A. Actinomycetes in rice straw decomposition. Waste Manage 27, 850–853 (2007).
Bouali, M. et al. The structure and spatio-temporal distribution of the Archaea in a horizontal subsurface flow constructed wetland. Sci Total Environ 435–436, 465–71 (2012).
Layeghifard, M., Hwang, D. M. & Guttman, D. S. Disentangling Interactions in the microbiome: A network perspective. Trends Microbiol 25, 217–228 (2016).
Greenblum, S., Turnbaugh, P. J. & Borenstein, E. Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. PNAs 109, 594–599 (2012).
Vick-Majors, T. J., Priscu, J. C. & Amaral-Zettler, L. A. Modular community structure suggests metabolic plasticity during the transition to polar night in ice-covered Antarctic lakes. ISME J 8, 778–789 (2014).
Banerjee, S. et al. Network analysis reveals functional redundancy and keystone taxa amongst bacterial and fungal communities during organic matter decomposition in an arable soil. Soil Biol Biochem 97, 188–198 (2016).
Motteran, F., Braga, J. K., Silva, E. L. & Varesche, M. B. A. Influence of sucrose on the diversity of bacteria involved in nonionic surfactant degradation in fluidized bed reactor. Water, Air, & Soil Poll 228, 21 (2017).
Brenner D. J., Garrity G., Krieg N. R. & Staley J. R. In Brenner, Krieg. D. J., Staley. N. R., Garrity. J. T., Boone. D. R. & De Vos, P. (Eds), Bergey’s manual of systematic bacteriology. Boston: Springer US (2005).
Bernardet, J. F. & Bowman, J. P. The Genus Flavobacterium. The Prokaryotes pp, 481–531 (2016).
Lehmkuhl, F. & Liu, S. An outline of physical geography including Pleistocene glacial landforms of eastern Tibet (Provinces Sichuan and Qinghai). Geo-J 34, 7–29 (1994).
Zhang, G. S. et al. Methanogen community in Zoige wetland of Tibetan plateau and phenotypic characterization of a dominant uncultured methanogen cluster ZC-I. Environ Microbiol 10, 1850–1860 (2008).
Zhang, X. Y., Lv, X. G. & Shen, S. P. Dynamic changes of Ruoergai Plateau wetland ecosystem service value. Chin. J Appl Ecol 20, 1147–1152 (2009).
Liang, Y. X. et al. The research of the relationship between shrink age of Zoige’s wetlands, deterioration and desertification of Zoige’s grasslands and north sandy arid region. Chin J Nat 29, 233–238 (2007).
WRB. World reference base for soil resources 2014, update 2015. International soil classification system for naming soils and creating legends for soil maps. World Soil Resources Reports No. 106, FAO, Rome (2015).
Lu, R. K. Soil and Agro-chemical Analytical Methods. ChinaAgricultural Science and Technology Press, Beijing, China, pp 146–195 (1999).
Caporaso, J. G. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6, 1621–1624 (2012).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat Med 7, 335–336 (2010).
Edgar, R. C. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10, 996–998 (2013).
Pollard K. S., Gilbert H. N., Ge Y., Taylor S. & Dudoit S. Multtest: resampling-based multiple hypothesis testing. R package version 2.17.0. R Development Core Team. 2010. R: A language and environment for statistical computing. Available: http://cran.r-project.org (2013).
Lozupone, C. & Knight, R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71, 8228–8235 (2005).
Oksanen J. et al. Vegan: Community Ecology Package. R packageversion 2.4-4, http://CRAN.R-project.org /package=vegan (2017).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq. 2. Genome Biol 15, 550 (2014).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57, 289–300 (1995).
Whitman, T. et al. Dynamics of microbial community composition and soil organic carbon mineralization in soil following addition of pyrogenic and fresh organic matter. ISME J 10, 2918–2930 (2016).
Li, F., Chen, L., Zhang, J. B., Yin, J. & Huang, S. M. Bacterial community structure after long-term organic and inorganic fertilization reveals important associations between soil nutrients and specific taxa involved in nutrient transformations. Front Microbiol 8, 187 (2017).
Benjamini, Y., Krieger, A. M. & Yekutieli, D. Adaptive linear step-up procedures that control the false discovery rate. Biometrika 93, 491–507 (2006).
Luo, F., Zhong, J., Yang, Y., Scheuermann, R. H. & Zhou, J. Z. Application of random matrix theory to biological networks. Phys Lett A 357, 420–423 (2006).
Csardi, G., & Nepusz, T. The igraph software package for complex network research. Int J Compl Syst, 1695 (2006).
Newman, M. Finding community structure in networks using the eigenvectors of matrices. Phys Rev E 74, 036104 (2006).
Acknowledgements
The study was funded by the National Natural Science Foundation of China (grant No. 41201256).
Author information
Authors and Affiliations
Contributions
Y.F.G. and Q.C. conceived the ideas and designed study. Y.B., Q.J.X., X.M.Y., and K.Z. performed the experiments and collected data. Y.F.G. and Y.B. the data. Y.F.G. and Z.X.P. interpreted the data and wrote the paper. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gu, Y., Bai, Y., Xiang, Q. et al. Degradation shaped bacterial and archaeal communities with predictable taxa and their association patterns in Zoige wetland at Tibet plateau. Sci Rep 8, 3884 (2018). https://doi.org/10.1038/s41598-018-21874-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-21874-0
This article is cited by
-
Short-term warming decreased soil DOM content and microbial species in alpine wetlands but increased soil DOM content and hydrolase activity in alpine meadows on the Tibetan Plateau
Biogeochemistry (2024)
-
Performance evaluation of modified Living Wall garden for treating septic tank effluent
Environmental Science and Pollution Research (2024)
-
Distribution of microbial communities in seasonally frozen soil layers on the Tibetan Plateau and the driving environmental factors
Environmental Science and Pollution Research (2023)
-
Environmental DNA Sequencing to Monitor Restoration Practices on Soil Bacterial and Archaeal Communities in Soils Under Desertification in the Brazilian Semiarid
Microbial Ecology (2023)
-
Soil macrofaunal communities develop a habitat-specific trophic structure dependent on the degree of degradation of alpine wetlands
Soil Ecology Letters (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
