
Abstract
Metabolic resistance to pyrethroid insecticides is widespread in Anopheles mosquitoes and is a major threat to malaria control. DNA markers would aid predictive monitoring of resistance, but few mutations have been discovered outside of insecticide-targeted genes. Isofemale family pools from a wild Ugandan Anopheles gambiae population, from an area where operational pyrethroid failure is suspected, were genotyped using a candidate-gene enriched SNP array. Resistance-associated SNPs were detected in three genes from detoxification superfamilies, in addition to the insecticide target site (the Voltage Gated Sodium Channel gene, Vgsc). The putative associations were confirmed for two of the marker SNPs, in the P450 Cyp4j5 and the esterase Coeae1d by reproducible association with pyrethroid resistance in multiple field collections from Uganda and Kenya, and together with the Vgsc-1014S (kdr) mutation these SNPs explained around 20% of variation in resistance. Moreover, the >20 Mb 2La inversion also showed evidence of association with resistance as did environmental humidity. Sequencing of Cyp4j5 and Coeae1d detected no resistance-linked loss of diversity, suggesting selection from standing variation. Our study provides novel, regionally-validated DNA assays for resistance to the most important insecticide class, and establishes both 2La karyotype variation and humidity as common factors impacting the resistance phenotype.
Similar content being viewed by others
Introduction
Insecticide resistance is a strongly selected, highly heritable trait that represents an excellent model system for the study of contemporary evolution1. Disease-transmitting mosquitoes are often subject to intense insecticidal selection pressure and in many instances there are critical financial and health implications of insecticide resistance evolution. In sub-Saharan Africa, where over 90% of all malaria fatalities occur, cases have fallen dramatically, due largely to the massive scale-up of insecticidal interventions such as treated bednets and indoor residual spraying2. Few insecticides are licenced for vector control and extensive application in public health, agriculture and household pesticide formulations has selected for widespread resistance to pyrethroids, by far the most important class of insecticides which may be used on bednets3. Whilst epidemiological evidence for an impact of resistance on malaria remains sparse4 there exists the worrying spectre that the recent gains in malaria control may be lost5. Preservation of pyrethroid efficacy is thus a primary objective for malaria control programmes, which need to apply effective insecticide resistance management. Insecticide resistance management depends on timely information on changes in resistance and the potential for cross resistance between insecticide options. High throughput molecular diagnostics can provide more sensitive, rapid and geographically-widespread assessments of changes in resistance than phenotypic assessments6. Knowledge of the genetic mechanisms underlying metabolic cross-resistance, which is far more difficult to predict a priori than cross-resistance at shared target sites7, is vital to allow implementation of insecticide combinations which will slow resistance development or allow susceptibility to return. Thus, improved knowledge of pyrethroid resistance mechanisms and provision of DNA markers for rapid predictive diagnosis8 and their evolution is an important goal to aid future insecticide deployment strategies.
Insecticide resistance occurs through four general mechanisms: insecticide target site alterations; elevated insecticide metabolism/sequestration; which are the primary focus here, and, potentially important but currently less-well described, reduced cuticular penetration and behavioural avoidance9. Target site resistance typically occurs through amino acid-altering substitutions, with the same mutations often found across diverse insect taxa10,11, or through copy number variation of alleles harbouring resistance mutations12,13. In Anopheles mosquitoes, and insects generally, pyrethroids and the organochlorine DDT target the voltage-gated sodium channel (Vgsc), within which knockdown resistance (kdr) mutations occur that can interfere with insecticide binding, preventing the normal insecticidal effect of repetitive nerve firing, paralysis and death14,15. In the primary malaria mosquito Anopheles gambiae kdr mutations exhibit limited variation in flanking haplotypes1,16,17. This conforms to a model of insecticide resistance evolution via hard selective sweeps from rare origins18, further supported by observed rapid increases of kdr variants toward fixation over a few years16,19,20. Similarly pronounced genomic footprints at the other major insecticide target site genes, acetylcholinesterase, AChE13 and GABA21 suggest that hard sweeps may be the norm for major resistance mutations in these essential neurotransmission genes. Such genomic signatures can greatly aid detection of major target site variants in genome scans, though selection toward fixation progressively reduces minor allele frequency and, as a direct consequence, statistical power to detect phenotypic association6,22.
With evidence of strong selection of target site mutations, it is perhaps surprising that metabolic resistance is usually considered the greater threat to vector control23. Indeed, the only widely accepted case of pyrethroid failure in malaria control to date was attributed to a local resurgence of An. funestus24,25 resistant to insecticide as a result of elevated expression of cytochrome P450 enzymes that metabolize pyrethroids26,27. Transcriptomic experiments conducted in Anopheles spp., have repeatedly linked overexpression of CYP6 subfamily P450 genes to insecticide resistance and shown that some genes have the ability to metabolize pyrethroids or even multiple insecticides28,29,30. In contrast, there are few studies in Anopheles which have identified DNA variation associated with metabolic resistance. The exceptions have targeted a single candidate gene for DDT resistance31,32 or a pair of duplicated P450 genes33. A paucity of resistance-associated DNA variants might seem to indicate a relatively greater role for gene overexpression in metabolic insecticide resistance evolution. Yet no regulatory variants for insecticide resistance-linked differential expression have been identified in Anopheles populations. This implies that allelic variants involved in metabolic resistance, whether altering proteins or regulatory regions, remain to be discovered.
Significant methodological challenges are involved in the identification of metabolic SNPs, such as phenotyping and genotyping sufficient mosquitoes to attain statistical power; population substructure within samples22 and typically extremely short linkage disequilibrium in the An. gambiae genome34, which reduces correlations between functional and marker polymorphisms35. An additional problem for identification of markers for metabolic resistance may result from their flexibility of function and exceptionally high polymorphism, at least in the largest detoxification gene superfamilies36. Both of these properties contrast markedly with highly-conserved target site genes subject to purifying selection13, and suggest that selection at detoxification genes is more likely to act upon standing genetic variation and result in soft selective sweeps, making their detection far more problematic18.
The aim of our study was to apply a candidate gene-enriched single nucleotide polymorphism (SNP) genotyping array to identify putatively pyrethroid resistance associated polymorphisms segregating within a wild population of An. gambiae from the Uganda-Kenya border area, an important focus of East African pyrethroid resistance37,38,39, with association established by reproducibility in additional field populations.
The study proceeded via the following steps:
-
1.
Collection of wild blood-fed females and rearing of progeny as isofemale lines.
-
2.
Genotyping of adult females individually from the collections using the SNP array to investigate sources of within population structure
-
3.
Bioassays and genotyping (using the same SNP array) of the each isofemale lines as a pools and association analysis based on relationship between family resistance phenotype and SNP genotypes
-
4.
Identification of putative candidates with additional analysis first within a laboratory colony established from the same location and subsequently in several distinct wild populations
-
5.
Sequencing and functional modelling analyses to attempt to identify causal SNPs and interpret the nature of selection
Our results suggest an important role for metabolic gene variants in pyrethroid resistance, which may be partially diagnosed using the SNPs identified as resistance-associated, and also indicate association of environmental humidity with pyrethroid resistance.
Results
Resistance phenotype
Isofemale families of An. gambiae from Tororo, Uganda were bioassayed using WHO permethrin papers for 60 min for males (N = 76 families) and 75 min for females (N = 98 families), which provided a full spectrum of family mortality levels ranging from zero to 100%, with a median of 76% (Figure S1). Humidity and temperature varied during the testing period in the field insectary (Figure S2), and across all families, mortality exhibited a significant inverse correlation with humidity (r = −0.43, P = 4 × 10−6; Figure S3), which was subsequently treated as a covariate in association analysis. Within each family, male and female mortalities were strongly correlated (r = 0.65; P = 2 × 10−9), and remained so following correction for humidity (partial r = 0.61; P = 3 × 10−6), suggesting a strong familial basis to resistance. Females were used for subsequent DNA analysis taking the proportionate mortality of the family from the bioassays (Figure S1) as a quantitative resistance phenotype.
Within population structure
We first examined evidence for within-population structure using individual genotypes from adult female An. gambiae, which comprised a collection of the wild caught mothers of the isofemale lines and others which did not produce sufficient offspring for use. These females were not classified by phenotype and were used solely for assessment of possible sources of within population structure and relatedness, which might impact subsequent association analysis using the pooled isofemale lines. Clustering analysis suggested three partitions, differentiation between which showed a strong localisation to the large 2La inversion region, with very limited inter-cluster divergence elsewhere in the genome (Fig. 1). Levels of differentiation within the 2La region were consistent with comparison among two homokaryotypic clusters and a heterokaryotypic cluster, and linkage disequilibrium was also extreme (Figure S4). This suggests that 2La inversion polymorphism was the only major source of structure within the sampled population, and that the sample did not comprise of groups of closely-related individuals. Though clear that 2La polymorphism is a major source of genomically-localised subdivision, it was unclear how it might influence association analyses because these females were not phenotyped. Therefore this was evaluated using principal components analysis as part of the association analysis of isofemale lines.

Analysis of sources of within-population structure in individually-genotyped adult females. Plots show FST values for each SNP compared among each of the three clusters identified using BAPS. The upper bar shows the location of the 2La inversion region.
Association analysis using isofemale lines
For pooled genotyping data from the isofemale lines, estimated allele frequencies were highly correlated with those calculated from individual genotypes after correction for SNP-specific dye bias (R2 = 0.98; Figure S5). Principal components analysis was used to identify covariates indicative of stratification in the pooled data based on 195 control (non-candidate gene) SNPs. The first principal component (PC1) was significantly correlated with resistance phenotype (partial correlation controlling for humidity: r = −0.33, P = 0.001; Table S1). PC1 primarily reflected variation within the 2La inversion region. All control (non-candidate) SNPs correlating strongly with PC1 (r≥0.5, N = 13) were located therein, and the mean correlation for 2La SNPs (r = 0.41, N = 32, was significantly higher (Mann-Whitney U-test, Z = 5.30, P < 0.001) than for control SNPs distributed throughout the rest of the genome (r = 0.11, N = 163). A second principal component was also correlated with mortality (r = 0.22, P = 0.03; Table S1), but showed no localisation of SNPs within the genome, and no difference in correlation between SNPs in 2La and elsewhere (Z = 1.45, P = 0.15). Owing to the concordance with individual genotyping results (above), in which the 2La region appeared to be the major source of stratification within the pooled dataset, we opted to consider PC1 further in order to apply correction for 2La variation. Elevated probabilities within the 2La region were evident when plotting all SNPs, but were removed by correction for PC1 as a covariate (Figure S6A), whereas if SNPs from the 2La region were removed a priori, correction for PC1 was not required to align the vast majority of SNPs with their expectation (Figure S6B). However, because correction for 2La variation (via PC1) might obscure SNPs which contributed to, or acted in addition to, the resistance association of 2La, and also because correction is likely to be imperfect, we considered results both with and without correction for PC1.
For the association analysis the relatively limited number of families analysed limited study power, with calculations suggested only moderate-large effects might be detected (Materials and Methods). Consequently, we did not apply formal thresholds for significance in the analysis, but rather identified candidate SNPs for further investigation from those with high ranking -logP values, with demonstration of association based on subsequent replication of results. Most of the SNPs with lowest P-values were on chromosome 2L, of which the two showing strongest evidence for association were a synonymous variant in Cyp4j5 and a non-synonymous variant in Cyp4j10 located 2kb apart (Fig. 2A; Table S2). Systematic elevation of P-values was clear within the 2La inversion, especially toward the ends, and following correction for PC1, P-values of most SNPs located within 2La reduced substantially. SNPs with the lowest P-values post-correction were from chromosome 2L but outside the 2La region; within the Vgsc target site gene and a carboxylesterase gene, Coeae1d (Fig. 2B).

Family pool association analysis for permethrin resistance (A) corrected for humidity as a covariate; (B) corrected for humidity and PC1 (a proxy for 2La polymorphism; see Fig. 1). Test probabilities are shown for each SNP arranged on a physical scale across chromosomes, centromere positions are shown by solid vertical lines; chromosome breaks by dashed lines. The purple bar indicates the 2La inversion region.
The candidate P450 and carboxylesterase gene SNPs were at high minor allele frequency (MAF, 0.35–0.49). However, those in the Vgsc exhibited much lower MAF (≤0.12), possibly as a result of a selective sweep of the kdr 1014S mutation, which is close to fixation (frequency = 0.94). To address the issue of potentially poor detectability of association of low MAF SNPs that might be influenced by strong selection in the wild, we performed an additional (individual-mosquito) association study using the class II pyrethroid deltamethrin. This experiment used bioassay survivors and dead from a recently-founded colony from the same location, which had not been previously been exposed to insecticide (Figure S7A). In the colony, MAFs of Vgsc-L1014S (MAF = 0.37) and other Vgsc SNPs were much higher than in the wild population (Table S3) and L1014S exhibited the strongest resistance association of all the SNPs typed (Figure S7B). The Vgsc SNP exhibiting the lowest probability in the previous analysis (Fig. 2B) and the SNP in Coeae1d were also strongly associated with resistance phenotype in the colony.
Marker association repeatability
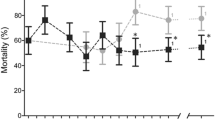
We chose four SNPs for further testing, the linked Cyp4j5 and Cyp4J10 variants, the Coeae1d SNP and the Vgsc-L1014S variant. When tested sequentially using stepwise regression, insectary humidity explained the highest proportion of the variance in permethrin resistance phenotype among the families, but each of the four SNPs explained significant additional variance, together totalling 22% among isofemale lines, with Cyp4j5 the strongest predictor (Table 1). Novel TaqMan assays were designed for the Cyp4j10, Coeae1d and Cyp4j5 SNPs. For the latter we used a non-synonymous SNP (not present on the array) in highest LD with the original marker, a leucine-phenylalanine change at codon 43 of the gene (L43F) (see Materials and Methods for details of source). These assays were combined with that for Vgsc-L1014F for screening in up to seven independent field samples phenotyped for susceptibility to three pyrethroid insecticides (Fig. 3). The known causal marker Vgsc-L1014S was significantly resistance-associated in two of the samples and the Coeae1d marker in three. The Cyp4j10 SNP failed to approach significance in three test samples and screening was discontinued, but the Cyp4j5-L43F marker was highly reproducible, with significant resistance association in five out of seven independent sample sets from both Uganda and Kenya (Fig. 3). Average effect sizes and their variability are shown in Table 2 and Table S4. For Vgsc-L1014S, odds ratios were highly variable, ranging from approximately 1 to over 10; were more consistent for Cyp4j5 (range 1.5–4.7) and lower but again quite consistent (range 0.7–2.7) for Coeae1d (Table S4). For Cyp4j5 we also investigated the predictive utility for a more extreme phenotype by comparing deltamethrin survivors of a 2 hour exposure with those killed by a one hour exposure; interestingly this yielded the highest odds ratio observed for this marker (OR = 6.9, P = 0.0001, Table S4).

Repeatability of SNP associations in independent field samples from Uganda and Kenya with three different pyrethroid insecticides (see Table S4 for full results). The dashed line indicates P = 0.05. Uga = Uganda; insecticides used for bioassays: λ-cy = lambda cyhalothrin; delta = deltamethrin; perm = permethrin. The year of collection is shown.
Temporal analysis of variation in marker frequencies in Tororo in 2013–2014 revealed relative stability of frequencies of each of the three metabolic gene markers (Figure S8). Interestingly, genotype frequencies of the Cyp4j5 and Cyp4j10 markers were strongly correlated (Table S5) as expected from their very close proximity in the genome, suggesting that the initial association of the Cyp4j10 marker for pyrethroid resistance may have in part arisen from co-variation with Cyp4j5. Neither Cyp4j5 nor Coeae1d marker genotype variation was correlated with 2La variation, despite apparent allele frequency covariation (Figure S5) highlighting their independent predictive utility (Table S5).
Investigation of possible causal SNPs
The resistance-associated variant at base 20288132 (VectorBase nucleotide numbering) in the Coeae1d gene is a synonymous variant. Therefore, to investigate whether any non-synonymous SNPs showed evidence of association which might be underpinning this, we sequenced a single individual from each of 24 families from the extremes of the mortality distribution (see Figure S1A; Table S6A). The Coeae1d gene is extremely polymorphic with a large number of missense variants (N = 116), many of which are at high frequency (31% with MAF > 0.1; 19% with MAF > 0.2). In this methodologically-independent replication of the initial association analysis, the 20288132 marker SNP exhibited a significant level of association (OR = 2.9, χ2 = 5.20, P = 0.022) but this was not exceeded by any function-altering polymorphism within the gene, and only marginally so by two other synonymous SNPs (Figure S9; Table S6B). We also genotyped the 2La inversion in the same samples, which showed significant association of the derived (2L+a) karyotype with permethrin resistance (OR = 4.0, χ2 = 4.85, P = 0.028).
Sequencing of the Cyp4j5 gene in Ugandan individuals revealed a split between resistant and susceptible haplotypes around the L43F variant, including similarity of resistant alleles from distinct locations (Jinja and Oyam) in southern and northern Uganda (Fig. 4). However, no marked difference in diversity, as would be expected with a strong selective sweep, was evident. We used structural modelling to evaluate the possible functional consequences of the L43F, and other non-synonymous substitutions in CYP4J5. Of the variants (Table S7) that could be modelled (noting that available templates bore only approximately 20% sequence identity to CYP4J5, limiting model quality), the substitution at L43F was not modelled reliably but seems unlikely to have an impact on protein stability. Of the other SNPs only one, a rare Alanine to Threonine substitution at codon 69, was predicted to potentially fall near enough to the active site for impact. However, this SNP was not detected as being in LD with the original marker, nor with the L43F variant; in fact the correlation with each was negative suggesting, if anything, weak linkage of the resistance associated allele at each marker with the wild type allele at A69T. Strong repeatability of the L43F mutation as a resistance predictor (above) suggests that either this is in strong LD with an, as yet unidentified, causal variant, or contributes directly to the resistance phenotype in a way that was not revealed by the current modelling approach.

Evolutionary relationships of the resistant (Phenylalanine) and susceptible (Leucine) alleles of CYP4J5 marker L43F in Ugandan samples. The optimal tree with sum of branch lengths = 0.062 is shown. Percentage bootstrap values (1000 replicates) are shown next to branches. The sample prefix indicates mosquito origin (Jinja or Oyam, Uganda).
Discussion
Using a candidate-GWAS approach we have identified two novel SNPs in genes from major metabolic superfamilies that are reliably and reproducibly associated with resistance to class I and II pyrethoids. We also detected a strong relationship between bioassay survival and humidity in our initial experiment and evidence for association of 2La inversion polymorphism with resistance. Moreover, although the genes containing the associated SNPs are located within and close to 2La, respectively, independent temporal dynamics of the SNP and 2La polymorphisms in Ugandan field samples suggests that additional resistance-associated variants remain to be discovered within 2La. The SNP markers identified are unlikely to be causal substitutions and we speculate that linkage disequilibrium between each marker SNP and one or more haplotypes, each likely to contain multiple non-synonymous changes in a resistance-conferring haplotype, may underpin the associations observed. Certainly the pattern of polymorphism deviates from that arising from selection on a single, relatively recent mutation13,16,17 and, particularly if haplotypes are driving the resistance association, selection from standing variation appears more likely40.
In the Eastern Ugandan Tororo field population strong association of the Vgsc-1014S (kdr) variant with both permethrin and DDT resistance has been demonstrated previously37 and we therefore suspected that the relatively weak association with permethrin-survival across families in our study might be attributed to limited statistical power arising from low polymorphism (1014S frequency ≈ 95%). Results from the Tororo colony, within which the frequency of the wild type 1014L allele was much higher, supported this hypothesis: L1014S was the SNP most strongly associated with survival following deltamethrin exposure. This is significant because: (i) the association of Vgsc-1014S has not previously been tested against a background of many other candidates; (ii) a significant link with class II resistance has proved difficult to establish37,41; (iii) it suggests that 1014S, and probably other kdr mutations continue to play a pivotal role in resistance in the presence of other resistance-linked mutations. Thus, it is important to recognise that even following rise toward fixation where kdr mutations explain relatively little of the variation in resistance within a population, they provide a strong baseline of resistance, which can be elevated further – rather than being superseded - by the presence of other resistance-linked variants6. An additional Vgsc mutation (N1575Y) detected in A. gambie and A. coluzzii across West Africa17 illustrates this point. The 1575Y allele occurs only on a Vgsc-1014F background and though the SNP itself exhibits a moderate odds ratio (≈2), this effectively doubles that of kdr1014F, and so translates into a large aggregate odds ratio (≈12)7,17.
Polymorphism of the 2La inversion has long been linked with important phenotypic variation in An. gambiae, primarily with adaptation to environmental aridity42,43, but also with refractoriness to Plasmodium falciparum44. Our data suggest that insecticide resistance can also be associated with the 2La inversion, with the major permethrin resistance-associated principal component (PC1) identified in our family pool study, correlating strongly with variation at SNPs within 2La. Owing to the known relationship between the 2La inversion and aridity, it is important to note that in the Tororo discovery dataset the association of PC1 remained significant following correction for humidity, i.e. both humidity and 2La polymorphism were determinants of resistance phenotype. In addition, when 2La polymorphism was typed directly, during our investigation of Coeae1d variation, the 2L+a karyotype was significantly associated with permethrin survival. Genomic stratification in the Tororo population was attributable to 2La polymorphism, therefore we opted to investigate further the most associated SNPs that emerged prior to, and post-correction using PC1 as a proxy for 2La variation. The resultant analyses showed that identification of top candidate SNPs solely post-correction for 2La would have been a mistake, because the strongest and most reproducible candidate SNP (Cyp4j5-L43F) would probably have been missed at the discovery phase. This is noteworthy and reflects a difference in genomewide stratification as expected in an admixed population, which would necessitate a priori correction to avoid false positives, and genomically-localised stratification, as seen here, which is a driver of the phenotype and unrelated to mating barriers.
Interestingly, when typed in a series of field samples collected from near Tororo between 2013 through 2014, neither Cyp4j5 (which was correlated with PC1) nor Coeae1d (uncorrelated with PC1) genotype frequencies were correlated within 2La karyotype variation, despite visual resemblance of summary data (see Figure S4). This is an important finding because it means that these two SNPs and 2La can serve as independent diagnostic markers, and perhaps most crucially because evolutionary dynamics of Coeae1d and especially Cyp4j5 (located within 2La) need not be constrained by dependence on 2La inversion polymorphism dynamics, which might be subject to multiple and potentially contrasting selection pressures. Whether such constraints are currently operative within the East African area we covered in our study is unclear, but recent indirect evidence suggests that insecticidal pressure might be the dominant selective factor for 2La variation. In A. gambiae s.s. samples from a 17-year time series collected in Western Kenya, Matoke-Muhia et al.45 detected a sharp decline in frequency of the previously numerically-dominant 2La karyotype in favour of the 2L+a karyotype, which correlated strongly (r = −0.96) with the changing coverage of pyrethroid-treated bednets in the area. To our knowledge our study is the first to provide direct association data directly linking pyrethroid resistance with 2La inversion polymorphism, adding corroborative evidence to the field survey results from Western Kenya.
An important objective is now to identify the genes/variants in 2La underpinning pyrethroid resistance association (noting that Cyp4j5 apparently is not the major cause). Contrasting with most of the An. gambiae genome22,34,46, LD within the 2La inversion region can be extreme, especially for a few megabases within the breakpoints around 22 and 42Mb22. Such strong LD creates the problem, commonly encountered in GWAS of human populations outside of Africa35, of how to isolate markers from large statistically-elevated haplotype blocks. Increasing sample size may be one answer, but will prove difficult in the near-term, because low LD throughout the majority of the genome argues strongly for the use of high density genotyping-by-sequencing approaches for which highly powered studies remain expensive. Instead, we suggest that investment of greater resources into post hoc investigation of repeatability of phenotype associations for smaller, filtered, sets of markers in wild populations could permit the signals from multiple linked markers to be teased apart. Polymorphic inversions are common in insects and low genomic LD in wild populations likely to be also, therefore such study design issues are of relevance to many taxa.
Our study demonstrates that pyrethroid resistance in eastern Uganda/western Kenya populations has a multivariate basis. In addition to the genetic polymorphism associations in the original discovery phase of the project, our data show a strong impact of humidity during bioassays on resistance, as reported previously in houseflies Musca domestica47. Although humidity varied naturally, rather than experimentally, in our study it explained over 17% of the variation among families in resistance and is very likely to be an important ecological determinant of insecticide resistance phenotypes under natural conditions. Of the genetic component of variation, both target site and metabolic variants play a role, with the two metabolic markers identified explaining a significant proportion (19%) of within-population variance. In a microarray study of the same Tororo population sampled the following year, relatively few significantly overexpressed genes were detected and only two detoxification genes48, though the design did not include a fully susceptible sample which will tend to accentuate discovery of overexpressed genes7. Nevertheless, available evidence suggests that the variants we identified are likely to link with mutations causing qualitative rather than quantitative protein variation, and we suggest that in Tororo this may be a more important determinant of pyrethroid resistant phenotypes. Owing to the paucity of agnostic DNA-based studies, which contrasts with the plethora of insecticide resistance-targeted gene expression experiments, whether this is population-specific, or the case more generally, remains to be determined.
Conclusion
With increased selective pressure arising from massive scale-up of long lasting insecticidal net (LLIN) distributions over the last decade, levels of pyrethroid resistance are likely to increase further and threat to control appears imminent. Identification of the genetic mechanisms leading to heightened resistance, and provision of simple assays for rapid diagnosis will aid attempts to manage resistance and retain pyrethroids as a viable option. Our study has highlighted the importance of allelic variants, outside the insecticide target site, as determinants of a significant fraction of variation in the resistance phenotype in a wild An. gambiae populations. Whole genome resequencing should permit identification of a spectrum of DNA polymorphisms underpinning resistance, and facilitate development and widespread application of DNA diagnostics as an accurate proxy for a priori assessment of phenotypic resistance.
Materials and Methods
Sample collections
Indoor-resting adult females were collected by aspiration from houses in Tororo, Uganda (00°40′41.6″N, 34°11′11.6″E) in November 2008 and transported to insectaries at the nearby National Livestock Resources Research Institute (NaLiRRI) facility. Morphological examination of almost 900 adult females revealed that the vast majority of samples (≈98%) were Anopheles gambiae s.l., with the remainder An. funestus, which were discarded. Adult female An. gambiae s.l. were held individually in plastic cups covered with fine netting, and contained moist cotton wool at the base covered by a disc of filter paper onto which eggs were laid. A cotton wool pad on top of each cup provided 10% sucrose solution ad libitum. Once eggs were laid, each filter paper disc was removed to a plastic bowl containing water, the adult female was sacrificed and preserved over silica gel. Following hatching, larvae were fed twice-daily with finely-ground Tetramin fish food. Pupae were transferred to netted cups containing a small volume of water, with one cup per isofemale family. On eclosion females and males were separated daily and isofemale line-specific bioassays were performed separately on 10–20 (median N = 15) 3–5 day old adult females (or males) in WHO tubes using standard 0.75% permethrin papers. Informed by bioassay data from preliminary tests on mixed F1 offspring from multiple families, we used an insecticide exposure time of 75 min (and 60 min for males), which aimed to produce an intermediate level of mortality (ca. 50%) for the population as a whole. Humidity and temperature were recorded during each assay. Approximately 24 h after exposure, all individuals were counted as alive or dead and preserved individually over silica gel.
For establishment and phenotype-classification of a colony for secondary association analysis, eggs were initially transported to the Liverpool School of Tropical Medicine from the Tororo collection site in December 2008; with a second supplementary collection added in May 2009. The colony was maintained without any insecticide exposure until September 2009 at which point 3–5 day old adult females were exposed to varying concentrations of the pyrethroid insecticide deltamethrin (Greyhound chemicals, UK) using WHO bioassay tubes and custom papers produced using the appropriate quantity of deltamethrin dissolved in acetone, added to silicone oil and spread over Whatman papers. Following the usual 24 h holding period, 20 adult females that died at low deltamethrin concentrations (<0.01%) and 36 surviving high deltamethrin concentrations (≥0.05%; note that 0.05% is the standard WHO resistance diagnostic concentration) were defined as susceptible and resistant classes, respectively and preserved individually over silica gel.
SNP array genotyping
DNA from An. gambiae s.l. individuals was extracted, quantified and typed to species using previously-described methods49. More than 96% of samples were An. gambiae (formerly An. gambiae s.s. S-form); the remainder were An. arabiensis, which were not considered further. The Illumina Goldengate 1536-SNP array used is described in detail elsewhere22,49 but briefly was enriched for SNPs at 266 candidate detoxification or target site genes, with approximately 20% of SNPs, located in intergenic regions or non-candidate genes. Although the SNP-array was populated by candidate genes, it should be noted that none of the SNPs present were known candidates in this geographical area. The genotype of the known resistance mutation, Vgsc (kdr) L1014S was also determined by direct sequencing of PCR products amplified using primers ex20+ F/R16. Mothers of isofemale lines and additional females collected but not producing eggs were genotyped individually, as were females from the laboratory colony. Female offspring from families were genotyped as single-family pools, using an equimolar amount of DNA from each individual to provide a final concentration per family pool of 50 ng/µl.
SNP array data analysis
Individuals
Genotyping arrays for 216 adult females (N = 98 mothers of the isofemale lines and N = 118 other females from house collections) and N = 56 Tororo colony specimens were scored using Beadstudio v3.2 (Illumina Inc). Haploview 4.150 was used to compute minor allele frequencies, linkage disequilibrium among SNPs as r2, observed and expected heterozygosities, and to test for deviation from Hardy-Weinberg expectations. Individual cluster analysis of genotypes at the control (intergenic and non-candidate) SNP loci was performed by BAPS 5.251,52, with multiple runs performed to obtain the optimum clustering solution.
Pools
Alleles labelled with different dyes seldom give a perfectly equal signal in heterozygotes and such dye bias may be assay-specific. Dye bias is typically not a major concern for individual genotyping but can greatly impact accurate estimation of allele frequencies from pooled DNA53. Therefore prior to analysis of pooled genotypes we computed the dye bias (k) for each SNP for which a minimum of two individual heterozygotes were available, as the mean cy3: cy5 dye ratio. For example, if heterozygotes exhibit a mean ratio of 0.5 (i.e. a twofold cy5 bias) then k = 0.5. We excluded SNPs exhibiting extreme or poorly estimated values of k, which can give rise to poor estimates of allele frequencies in pooled analysis53, based on two exclusion criteria: (1) k > upper 95th percentile of the range of k values; (2) standard deviation of k across individuals > upper 95th percentile of the range of k value standard deviations. Of the 1536 SNPs on the array 895 could be scored reliably, were polymorphic and exhibited k values meeting criteria 1 and 2: only these SNPs were used in the analyses. Raw X and Y (cy3 and cy5) signal data were extracted from Beadstudio v3.2 and used as the basis for all analyses of pooled DNA. Allele frequencies, p(X) and p(Y), were computed as:
Principal components analysis (PCA) using allele frequency data from 195 control SNPs was used to investigate possible within-sample stratification using the pooled genotype data. Association analysis was performed on rank-transformed data by computing correlations between allele frequency and proportionate bioassay mortality for each family, or partial correlations to correct for possible covariates (temperature, humidity and principal components; see Results).
Power calculation suggested that our analysis could detect a slope for mortality vs allele frequency in the analysis of 0.46 (as significantly different from zero), with 80% power and a Bonferroni-corrected alpha level (0.05/N SNPs), with the 894 SNPs successfully genotyped and our dataset of pooled genotypes from 98 families. This suggests that moderate-large effect sizes associated with any individual SNP would be detectable. Whilst this was compatible with our aim to discover markers with useful predictive power for phenotypes (i.e. those of larger effect) rather than a comprehensive set of variants, owing to the limited power we do not apply formal significance thresholds in the analysis but rather base assessment of association on reproducibility of candidates selected for further testing.
IBM SPSS 22 was used to perform subsequent stepwise regression analyses for the pooled genotype data and multiple logistic regression for the individual data from the reproducibility testing populations.
Associated gene sequencing
To attempt to identify the possible causal SNP and evaluate evidence of selection the Cyp4j5 gene was sequenced in several individuals from Jinja and Oyam, distinct locations from southern and northern Uganda, respectively (locations given in subsection below). Specimens were genotyped using the CYP4J5 marker (2L: 25635973) and six homozygotes for each allele Cyp4j5–43L and Cyp4j5-43F, the latter resistance associated, were selected for cloning and sequencing. The entire gene was amplified using primers 4J5F1 5′-AGCACCACGGTAAGGATGTC-3′ and 4J5R1 5′-GCGGAGAAACGTAACCCATA-3′ with a Phusion PCR kit (Thermo Scientific) prior to cloning in E.coli using a pJET1.2/blunt cloning vector. Due to the size of the product (~3.5 kb), two internal primers (4J5SEQ2 5′-ATTGCCGACTGTAGCTCGAT-3′ and CYP4J5-2 5′-GGCTTCTTTGGGACACACAT-3′) were used in addition to the pJET plasmid specific primers (pJETF and pJETR). Clones were sequenced by Macrogen, Korea and edited using CodonCode Aligner v4 (CodonCode Corporation). Gene trees were produced in MEGA 754 using the neighbor-joining method with 1000 bootstraps. Evolutionary distances were computed using the Maximum Composite Likelihood method using all codon positions. All positions containing gaps and missing data were eliminated yielding a total of 1596 positions in the final dataset.
The Coeae1d gene was sequenced in a single individual from 24 families from each end of the spectrum of mortalities, and each individual thus designated as resistant or susceptible. Initial PCR amplification of a 2047bp fragment used primers Agap5756his (5′ATCGTCAACGTGCTCAGTCA3′) and Agap5756exp (5′AGCACAGCGACTAACTCTTGC3′) in the following mixture 5 μl KapaTaq 10X buffer, 1 μl (10mM dNTPs), 10 picomoles of each primer, 1.5 U KapaTaq polymerase, 40.7 μl water & 1 μl DNA template. Cycling conditions were 95 °C for 5 min, followed by 40 cycles of 95 °C for 30 sec, 57 °C for 30 sec, 72 °C for 90 sec, with a final step of 72 °C for 10 min. Products were cleaned with QIAquick PCR Purification Kit (Qiagen) and sent for sequencing by Macrogen, Korea using the following overlapping sequencing primers covering 1951 bp within the amplicon, which included the whole gene (5756SEQ7i 5′AACTCCTCGAAACCTCACCTA3′; 5756SEQ1 5′ATGCAACCCTTCCTAGCG3′; 5756SEQ2i 5′ACCCCATTTGCTCAAACA3′; 5756SEQ3i 5′GGTTTCGAGGAGTTTTGTTGT3′; 5756SEQ5 5′TCACTCCGGCAGCTTCCTA3′; 5756SEQ4i 5′CAACTCAACTCTCTTACAGACATGC3′). Sequences were aligned to a reference sequence from VectorBase in CodonCode Aligner v4, edited and polymorphisms identified. Chi-square tests were conducted for each SNP identified to investigate association with permethrin resistance phenotype, by comparing allele frequencies between resistant and susceptible individuals. We also genotyped polymorphism of the 2La inversion in these samples using a PCR-based method55 for association analysis based on chi-square tests as before.
Functional modelling
Templates for modelling of Anopheles gambiae CYP4J5 were ranked by a search of the Protein Data Bank56 using HHsearch57. The best scoring 10 structures were used for comparative modelling using the recent high-resolution methods implemented in Rosetta58 to generate 30 models. The top model was used for interpreting the likely consequences of observed SNPs using structure-based methods FoldX59,60 and PoPMuSiC61. This analysis was confined to regions that comparison between models indicated were predicted reliably. SNPs positioned in regions not considered reliably modelled were interpreted using PolyPhen262. The structure of deltamethrin, obtained from the Toxin and Toxin Target Database63, was manually positioned using PyMOL (www.pymol.org) in the CYP4J5 model in a similar position to ritonavir bound to CYP3A464, one of the template structures. Its position relative to amino-acids altered by SNPs was assessed. Since the available templates bore only low (~20%) sequence identity with Anopheles gambiae CYP4J5 and apo-structures were among the templates, the binding cavity of the CYP4J5 model was not predicted accurately enough to allow for automated docking. Preliminary functional modelling of COEAE1D was performed but owing to a very large number of non-synonymous variants (see Results) was discontinued.
Replication study samples and assays
We designed novel TaqMan assays for two candidate CYP450 markers identified in the family pooled genotyping and colony individual genotyping experiments and the Coeae1d marker. For the Cyp4j5 marker, non-target polymorphism in the assay region prevented design for the associated SNP, therefore we sought to design an assay for a non-synonymous SNP within the gene in highest linkage disequilibrium (LD) with the marker. Data were obtained from whole genome sequences of 40 isofamily pools of females, collected in the same way as before from the same locations one year later48. LD was approximated by computing family-wise Pearson correlations for each of the variants identified with the original marker (Table S7). The SNP with the highest correlation coefficient (and similar minor allele frequency to the original marker) was a leucine to phenylalanine substitution at codon 43, for which an assay was successfully designed.
For independent replication of marker: phenotype associations, samples bioassayed for pyrethroid susceptibility were obtained from a number of spatially and/or temporally distinct collection locations in the East African region. Oyam (02°14′N 32°23′E), Apac (01°59′N 32°32′E) Jinja65, (00°46′N 34°01′E) Nagongera66 all in Uganda and the Busia/Bungoma area of Western Kenya38. All were collected as larvae with rearing as above and bioassays-screened using standard WHO methods. Samples were genotyped for SNPs using a TaqMan SNP genotyping assay for Vgsc L1014S67, and novel TaqMan assays for the: (1) CYP4J10 marker (2L: 25,636,722) primers were (forward 5′-ATCACGGTGCAGATCGT-3′) and (reverse 5′-GCTTCAAGAATCTGAATCGC-3′) and SNP specific probes (G: 5′-6FAM-CATGTCACACGTCCACA-3′ and A: 5′-VIC-CATGTCACACATCCACA-3′); (2) COEAE1D marker (2L: 20,288,132) primers were (forward 5′-GAGAGTGCAGGAGCTAAGGC-3′) and (reverse 5′-CTCCACTTTGACAGATCACTCGAT-3′) and SNP specific probes (G: 5′-6FAM-CCTATCTGCATTACCTTT-3′ and A: 5′-VIC-CCTATCTGCACTACCTTT-3′); (3) CYP4J5 marker (2L: 25635973) primers were (forward 5′-AGCCTGCGCGTGTGATA-3′) and (reverse 5′-CTTCTTCTCCTGTGGTTCGTTTG-3′) and SNP specific probes (G: 5′-6FAM-TTGCCGGAAGGCAGT-3′ and A: 5′-VIC-TTGCCGGAGGGCAGT-3′). Probes carried non-fluorescent quenchers at the 3′ end. Assays were performed in a 10 μl volume containing 1x Sensimix (Bioline), 1x primer/probe mix and 1μl template DNA with a temperature profile of 95 °C for 10 min followed by 40 cycles of 92 °C for 15 s and 60 °C for 1 min on an Agilent MX3005 real-time PCR machine. VIC and FAM fluorescence was captured at the end of each cycle and genotypes called from endpoint fluorescence using Agilent MXPro software. Genotype-phenotype associations were examined using χ2 tests and odds ratios.
References
Clarkson, C. S. et al. Adaptive introgression between Anopheles sibling species eliminates a major genomic island but not reproductive isolation. Nature Communications 5, e4248, https://doi.org/10.1038/ncomms5248 (2014).
Bhatt, S. et al. The effect of malaria control on Plasmodium falciparum in Africa between 2000 and 2015. Nature 526, 207-+, https://doi.org/10.1038/nature15535 (2015).
Ranson, H. & Lissenden, N. Insecticide resistance in African Anopheles mosquitoes: a worsening situation that needs urgent action to maintain malaria control. Trends Parasitol (2016).
Kleinschmidt, I. et al. Design of a study to determine the impact of insecticide resistance on malaria vector control: a multi-country investigation. Malar. J. 14, 13, https://doi.org/10.1186/s12936-015-0782-4 (2015).
WHO-GMP. Global plan for insecticide resistance management in malaria vectors (GPIRM). (World Health Organization, 2012).
Weetman, D. & Donnelly, M. J. Evolution of insecticide resistance diagnostics in malaria vectors. Transactions of The Royal Society of Tropical Medicine and Hygiene 109, 291–293, https://doi.org/10.1093/trstmh/trv017 (2015).
Donnelly, M. J., Isaacs, A. & Weetman, D. Identification, validation, and application of molecular diagnostics for insecticide resistance in malaria vectors. Trends in Parasitology 32, 197–206, https://doi.org/10.1016/j.pt.2015.12.001 (2016).
Donnelly, M. J., Isaacs, A. & Weetman, D. Identification, validation, and application of molecular diagnostics for insecticide resistance in malaria vectors. Trends Parasitol, https://doi.org/10.1016/j.pt.2015.12.001 (2016).
Hemingway, J., Hawkes, N. J., McCarroll, L. & Ranson, H. I. The molecular basis of insecticide resistance in mosquitoes. Insect Biochemistry and Molecular Biology 34, 653–665 (2004).
Weill, M. et al. Insecticide resistance: a silent base prediction. Current Biology 14, R552–R553 (2004).
Davies, T. G. E., Field, L. M., Usherwood, P. N. R. & Williamson, M. S. A comparative study of voltage-gated sodium channels in the Insecta: implications for pyrethroid resistance in Anopheline and other Neopteran species. Insect Molecular Biology 16, 361–375 (2007).
Remnant, E. J. et al. Gene duplication in the major insecticide target site, Rdl, in Drosophila melanogaster. Proceedings of the National Academy of Sciences of the United States of America 110, 14705–14710, https://doi.org/10.1073/pnas.1311341110 (2013).
Weetman, D. et al. Contemporary evolution of resistance at the major insecticide target site gene Ace-1 by mutation and copy number variation in the malaria mosquito Anopheles gambiae. Molecular ecology 24, 2656–2672, https://doi.org/10.1111/mec.13197 (2015).
Ranson, H. et al. Identification of a point mutation in the voltage-gated sodium channel gene of Kenyan Anopheles gambiae associated with resistance to DDT and pyrethroids. Insect Molecular Biology 9, 491–497 (2000).
Martinez-Torres, D. et al. Molecular characterization of pyrethroid knockdown resistance (kdr) in the major malaria vector Anopheles gambiae s.s. Insect Molecular Biology 7, 179–184 (1998).
Lynd, A. et al. Field, genetic and modelling approaches show strong positive selection acting upon an insecticide resistance mutation in Anopheles gambiae s.s. Molecular Biology and Evolution 27, 1117–1125 (2010).
Jones, C. et al. Footprints of positive selection associated with a novel mutation (N1575Y) in the voltage gated sodium channel of Anopheles gambiae. Proceedings of the National Academy of Sciences of the United States of America 109, 6614–6619 (2012).
Messer, P. W. & Petrov, D. A. Population genomics of rapid adaptation by soft selective sweeps. Trends Ecol. Evol. 28, 659–669, https://doi.org/10.1016/j.tree.2013.08.003 (2013).
Mathias, D. et al. Spatial and temporal variation in the kdr allele L1014S in Anopheles gambiae s.s. and phenotypic variability in susceptibility to insecticides in Western Kenya. Malaria Journal 10, e10 (2011).
Norris, L. C. et al. Adaptive introgression in an African malaria mosquito coincident with the increased usage of insecticide-treated bed nets. Proceedings of the National Academy of Sciences of the United States of America 112, 815–820, https://doi.org/10.1073/pnas.1418892112 (2015).
Lawniczak, M. K. N. et al. Widespread Divergence Between Incipient Anopheles gambiae Species Revealed by Whole Genome Sequences. Science 330, 512–514 (2010).
Weetman, D. et al. Association mapping of insecticide resistance in wild Anopheles gambiae populations: major variants identified in a low-linkage disequilbrium genome. PLoS ONE 5, e13140 (2010).
Ranson, H. et al. Pyrethroid resistance in African anopheline mosquitoes: what are the implications for malaria control? Trends Parasitol 27, 91–97 (2011).
Hargreaves, K. et al. Anopheles funestus resistant to pyrethroid insecticides in South Africa. Medical and Veterinary Entomology 14, 181–189, https://doi.org/10.1046/j.1365-2915.2000.00234.x (2000).
Maharaj, R., Mthembu, D. J. & Sharp, B. L. Impact of DDT re-introuction on malaria transmission in KwaZulu-Natal. Samj South African Medical Journal 95, 871–874 (2005).
Wondji, C. S. et al. Mapping a Quantitative Trait Locus (QTL) conferring pyrethroid resistance in the African malaria vector Anopheles funestus. BMC Genomics 34 (2007).
Wondji, C. S. et al. Two duplicated P450 genes are associated with pyrethroid resistance in Anopheles funestus, a major malaria vector. Genome Res. 19, 452–459, https://doi.org/10.1101/gr.087916.108 (2009).
Mitchell, S. et al. Identification and validation of a gene causing cross-resistance between insecticide classes in Anopheles gambiae from Ghana. Proceedings of the National Academy of Sciences of the United States of America 109, 6147–6152 (2012).
Müller, P. et al. Field-caught permethrin-resistant Anopheles gambiae overexpress CYP6P3, a P450 that metabolises pyrethroids. PLoS genetics 4, e1000286 (2008).
Edi, C. V. et al. CYP6 P450 enzymes and ACE-1 duplication produce extreme and multiple insecticide resistance in the malaria mosquito Anopheles gambiae. PLoS genetics 10, e1004236–e1004236, https://doi.org/10.1371/journal.pgen.1004236 (2014).
Mitchell, S. N. et al. Metabolic and Target-Site Mechanisms Combine to Confer Strong DDT Resistance in Anopheles gambiae. PLoS ONE 9, https://doi.org/10.1371/journal.pone.0092662 (2014).
Riveron, J. M. et al. A single mutation in the GSTe2 gene allows tracking of metabolically based insecticide resistance in a major malaria vector. Genome Biology 15, 20, https://doi.org/10.1186/gb-2014-15-2-r27 (2014).
Ibrahim, S. S. et al. Allelic Variation of Cytochrome P450s Drives Resistance to Bednet Insecticides in a Major Malaria Vector. PLoS genetics 11, e1005618, https://doi.org/10.1371/journal.pgen.1005618 (2015).
Neafsey, D. E. et al. SNP Genotyping Defines Complex Gene-Flow Boundaries Among African Malaria Vector Mosquitoes. Science 330, 514–517, https://doi.org/10.1126/science.1193036 (2010).
Teo, Y. Y., Small, K. S. & Kwiatkowski, D. P. Methodological challenges of genome-wide association analysis in Africa. Nature Reviews Genetics 11, 149–160, https://doi.org/10.1038/nrg2731 (2010).
Wilding, C. S., Weetman, D., Steen, K. & Donnelly, M. J. High, clustered, nucleotide diversity in the genome of Anopheles gambiae revealed by SNP discovery through pooled-template sequencing: implications for high-throughput genotyping protocols. BMC Genomics 10, e320 (2009).
Ramphul, U. et al. Insecticide resistance and its association with target-site mutations in natural populations of Anopheles gambiae from eastern Uganda. Transactions of the Royal Society of Tropical Medicine and Hygiene 103, 1121–1126, https://doi.org/10.1016/j.trstmh.2009.02.014 (2009).
Ochomo, E. et al. Pyrethroid susceptibility of malaria vectors in four districts of western Kenya. Parasites and Vectors 7, e310 (2014).
Verhaeghen, K. et al. Spatio-temporal patterns in kdr frequency in permethrin and DDT resistant Anopheles gambiae s.s. from Uganda. American Journal of Tropical Medicine and Hygiene 82, 566–573, https://doi.org/10.4269/ajtmh.2010.08-0668 (2010).
Karasov, T., Messer, P. W. & Petrov, D. A. Evidence that adaptation in Drosophila is not limited by mutation at single sites. PLoS Genet. 6, 10, https://doi.org/10.1371/journal.pgen.1000924 (2010).
Reimer, L. et al. Relationship between kdr mutation and resistance to pyrethroid and DDT insecticides in natural populations of Anopheles gambiae. Journal of Medical Entomology 45, 260–266 (2008).
Coluzzi, M., Sabatini, A., Petrarca, V. & Di Deco, M. A. Chromosomal differentiation and adaptation to human environments in the Anopheles gambiae complex. Transactions of the Royal Society of Tropical Medicine and Hygiene 73, 483–497 (1979).
Cheng, C. D. et al. Ecological genomics of Anopheles gambiae along a latitudinal cline: a population-resequencing approach. Genetics 190, 1417-+, https://doi.org/10.1534/genetics.111.137794 (2012).
Riehle, M. M. et al. Natural malaria infection in Anopheles gambiae is regulated by a single genomic control region. Science 312, 577–579 (2006).
Matoke-Muhia, D. et al. Decline in frequency of the 2La chromosomal inversion in Anopheles gambiae (s.s.) in Western Kenya: correlation with increase in ownership of insecticide-treated bed nets. Parasites & vectors 9, 334, https://doi.org/10.1186/s13071-016-1621-3 (2016).
Harris, C., Rousset, F., Morlais, I., Fontenille, D. & Cohuet, A. Low linkage disequilibrium in wild Anopheles gambiae s.l. populations. BMC Genetics 11, https://doi.org/10.1186/1471-2156-11-81 (2010).
Bong, L. J. & Zairi, J. Temporal fluctuations of insecticides resistance in Musca domestica Linn (Diptera: Muscidae) in Malaysia. Tropical Biomedicine 27, 317–325 (2010).
Wilding, C. S. et al. Parallel evolution or purifying selection, not introgression, explains similarity in the pyrethroid detoxification linked GSTE4 of Anopheles gambiae and An. arabiensis. Molecular Genetics and Genomics 290, 201–215, https://doi.org/10.1007/s00438-014-0910-9 (2015).
Weetman, D., Wilding, C. S., Steen, K., Pinto, J. & Donnelly, M. J. Gene flow-dependent genomic divergence between Anopheles gambiae M and S forms. Molecular Biology and Evolution 29, 279–291, https://doi.org/10.1093/molbev/msr199 (2012).
Barrett, J. C., Fry, B., Maller, J. & Daly, M. J. W. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265 (2005).
Corander, J., Marttinen, P., Siren, J. & Tang, J. Enhanced Bayesian modelling in BAPS software for learning genetic structures of populations. BMC Bioinformatics 9, 14, https://doi.org/10.1186/1471-2105-9-539 (2008).
Corander, J. & Marttinen, P. Bayesian identification of admixture events using multilocus molecular markers. Mol. Ecol. 15, 2833–2843, https://doi.org/10.1111/j.1365-294X.2006.02994.x (2006).
Visscher, P. M. & Le Hellard, S. Simple method to analyze SNP-based association studies using DNA pools. Genet. Epidemiol. 24, 291–296, https://doi.org/10.1002/gepi.10240 (2003).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 33, 1870–1874, https://doi.org/10.1093/molbev/msw054 (2016).
White, B. J. et al. Molecular karyotyping of the 2LA inversion in Anopheles gambiae. American Journal Of Tropical Medicine And Hygiene 76, 334–339 (2007).
Rose, P. W. et al. The RCSB Protein Data Bank: new resources for research and education. Nucleic Acids Res. 41, D475–D482, https://doi.org/10.1093/nar/gks1200 (2013).
Soding, J., Biegert, A. & Lupas, A. N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Research 33, W244–248, https://doi.org/10.1093/nar/gki408 (2005).
Song, Y. F. et al. High-Resolution Comparative Modeling with RosettaCM. Structure 21, 1735–1742, https://doi.org/10.1016/j.str.2013.08.005 (2013).
Schymkowitz, J. et al. The FoldX web server: an online force field. Nucleic Acids Res. 33, W382–W388, https://doi.org/10.1093/nar/gki387 (2005).
Guerois, R., Nielsen, J. E. & Serrano, L. Predicting changes in the stability of proteins and protein complexes: A study of more than 1000 mutations. J. Mol. Biol. 320, 369–387, https://doi.org/10.1016/s0022-2836(02)00442-4 (2002).
Dehouck, Y., Kwasigroch, J., Gilis, D. & Rooman, M. PoPMuSiC 2.1: a web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinformatics 12, e151 (2011).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249, https://doi.org/10.1038/nmeth0410-248 (2010).
Lim, E. et al. T3DB: a comprehensively annotated database of common toxins and their targets. Nucleic Acids Res. 38, D781–D786, https://doi.org/10.1093/nar/gkp934 (2010).
Sevrioukova, I. F. & Poulos, T. L. Structure and mechanism of the complex between cytochrome P4503A4 and ritonavir. Proc. Natl. Acad. Sci. USA 107, 18422–18427, https://doi.org/10.1073/pnas.1010693107 (2010).
Mawejje, H. D. et al. Insecticide resistance monitoring of field-collected Anopheles gambiae s.l. populations from Jinja, eastern Uganda, identifies high levels of pyrethroid resistance. Medical and Veterinary Entomology 27, 276–283, https://doi.org/10.1111/j.1365-2915.2012.01055.x (2013).
Kilama, M. et al. Estimating the annual entomological inoculation rate for Plasmodium falciparum transmitted by Anopheles gambiae s.l. using three sampling methods in three sites in Uganda. Malaria Journal 13, 111, https://doi.org/10.1186/1475-2875-13-111 (2014).
Bass, C. et al. Detection of knockdown resistance (kdr) mutations in Anopheles gambiae: a comparison of two new high-throughput assays with existing methods. Malaria Journal 6, e111 (2007).
Acknowledgements
The project described was supported by Award Numbers U19AI089674 and R01AI082734 from the National Institute of Allergy and Infectious Diseases (NIAID) and by the Innovative Vector Control Consortium. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIAID or NIH.
Author information
Authors and Affiliations
Contributions
D.W., C.S.W., P.M., L.M.O. and M.J.D. conceived/designed the research. D.W., C.S.W., P.M., E.O., A.T.I., K.S., E.J.R., J.C.M., H.D.M. conducted the experiments. D.E.N., D.J.R. and L.M.O. contributed new reagents and/or analytical tools. D.W., C.S.W., A.I.T., D.E.N., D.J.R., M.J.D. analyzed the data. D.W. and M.J.D. wrote the manuscript. All authors read, corrected and approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Weetman, D., Wilding, C.S., Neafsey, D.E. et al. Candidate-gene based GWAS identifies reproducible DNA markers for metabolic pyrethroid resistance from standing genetic variation in East African Anopheles gambiae. Sci Rep 8, 2920 (2018). https://doi.org/10.1038/s41598-018-21265-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-21265-5
This article is cited by
-
Signatures of adaptation at key insecticide resistance loci in Anopheles gambiae in Southern Ghana revealed by reduced-coverage WGS
Scientific Reports (2024)
-
LLIN Evaluation in Uganda Project (LLINEUP)–effects of a vector control trial on Plasmodium infection prevalence and genotypic markers of insecticide resistance in Anopheles vectors from 48 districts of Uganda
Scientific Reports (2024)
-
Molecular drivers of insecticide resistance in the Sahelo-Sudanian populations of a major malaria vector Anopheles coluzzii
BMC Biology (2023)
-
Modelling spatiotemporal trends in the frequency of genetic mutations conferring insecticide target-site resistance in African mosquito malaria vector species
BMC Biology (2022)
-
Genetic variation at the Cyp6m2 putative insecticide resistance locus in Anopheles gambiae and Anopheles coluzzii
Malaria Journal (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
