
Abstract
Diabetic retinopathy (DR) is a retinal microvascular disease characterized by inflammatory and angiogenic pathways. In this study, we evaluated NLRP3 inflammasome in a double transgenic mouse model, Akimba (Ins2AkitaxVEGF+/−), which demonstrates hyperglycemia, vascular hyperpermeability and neovascularization seen in the proliferative DR. Retinal structural integrity, vascular leakage and function were examined by fundus photography, fluorescein angiography, optical coherence tomography, retinal flat mounts, laser speckle flowgraphy (LSFG), and electroretinography in Akimba and its parental strains, Akita (Ins2Akita) and Kimba (trVEGF029) mice. Inflammatory mechanisms involving NLRP3 inflammasome were investigated using real time-PCR, immunohistochemistry, ELISA and western blots. We observed an increased vascular leakage, reduced retinal thickness, and function in Akimba retina. Also, Akimba retina depicts decreased relative flow volume measured by LSFG. Most importantly, high levels of IL-1β along with increased NLRP3, ASC, and Caspase-1 at mRNA and protein levels were observed in Akimba retina. However, the in vivo functional role remains undefined. In conclusion, increased activation of macroglia (GFAP), microglia (Iba-1 and OX-42) and perivascular macrophages (F4/80 and CD14) together with pro-inflammatory (IL-1β and IL-6) and pro-angiogenic markers (PECAM-1, ICAM-1, VEGF, Flt-1, and Flk-1), suggested a critical role for NLRP3 inflammasome in the Akimba mouse model depicting advanced stages of DR pathogenesis.
Similar content being viewed by others


Introduction
Diabetic retinopathy (DR) is a common retinal microvascular complication of diabetes mellitus in working adults1,2. Clinically, DR is broadly categorized into an early stage non-proliferative DR (NPDR) and the advanced stage of proliferative-DR (PDR), with and without the development of diabetic macular edema (DME). DR involves multiple interlinked pathways that lead to cellular damage, vascular and neuronal abnormalities, and microvascular damage in the retina3,4. Chronic low-grade retinal inflammation caused by the activated microglia/macrophages have been described as a major contributor in the pathogenesis of DR and PDR responsible for the infiltration of leukocytes into the retina, release of inflammatory mediators, breakdown of the blood-retinal barrier (BRB), vascular leakage and neovascularisation5,6,7. In clinical studies, elevated serum and vitreous levels of pro-inflammatory cytokines such as C-reactive protein (CRP), Interleukin-1beta (IL-1β), Interleukin-6 (IL-6) and Tumour Necrosis Factor-alpha (TNF-α) have been demonstrated in DR and PDR8,9.
The immune system in the retina is evolutionally exempted from deleterious effects of exogenously triggered pathogen-associated molecular patterns (PAMPs), as well as endogenously generated damage-associated molecular patterns (DAMPs), sometimes referred to as alarmins10. These DAMPs are recognized by pattern recognition receptors (PRRs) such as toll-like receptors (TLRs), C-type lectin receptors (CLR), RIG-like receptors (RLR), and NOD-like receptors (NLR)11,12. Growing evidence suggest the involvement of DAMPs-bound PRRs in the induction of low-grade but chronic inflammatory responses seen in many ocular and non-ocular multifactorial diseases such as age-related macular degeneration, Alzheimer’s disease, Parkinson’s disease and multiple sclerosis13,14,15. The DAMPs such as oxidized lipoproteins, glycated proteins, uric acid, DNA and RNA from necrotic cells have been suggested to contribute to the pathogenesis of retinal diseases16,17. This may compromise the first line of defense system embodied by the innate immune system and protected by the blood-retina barrier (BRB) as seen in PDR and DME18.
DAMPs help to assemble a newly identified multimeric protein complex called inflammasome comprised of NLRP3 (NLR family pyrin domain containing 3), apoptosis associated speck-like protein (ASC) and Caspase-119. The NLRP3 component recognizes danger signals and assembles the protein complex ASC to activate caspase-1, which leads to the proteolytic secretion of the pro-inflammatory cytokine, IL-1β. A recent study on metabolic disorders such as obesity and type 2 diabetes have also shown to involve activated NLRP3 inflammsome20. Also, mice deficient in NLRP3 components showed attenuated diabetic phenotype21. Moreover, a recent report found traces of NLRP3 expression in the vitreous of PDR patients22. Although inflammatory pathways play a substantial role in DR pathogenesis, especially in the proliferative and angiogenic stages, the contribution of the NLRP3 inflammasome has not been described.
One of the major limitations to study late stage DR pathogenesis is the unavailability of a suitable animal model. To date, several DR rodent models have been reported, but they mostly represent early stages of DR and do not depict the pathology seen in the advanced stages of DR (PDR and DME). This could be probably due to the short lifespan and duration of diabetes. Various animal models with the detailed description of pros and cons have been summarized in the literature23,24. The most common rodent model described is streptozotocin (STZ)-induced type-I diabetes, which develops early symptoms of DR as early as 2–3 months characterized by pericyte loss, thickening of basement membrane and increased vascular permeability. However, advanced proliferative and neovascular changes were not observed in these rodents eyes even after 18–24 months of diabetes25,26.
Recently, mouse models such as Akita (Ins2Akita) and Kimba (trVEGF029) have been described in DR studies. The Akita mouse is a spontaneous type-I diabetes model that carries a dominant point mutation in the insulin-2 gene27. It develops hyperglycemia and exhibits early pathophysiological changes of DR but fails to progress to the advanced stage. The Kimba mouse, which has transient photoreceptor (PhR)-specific overexpression of human VEGF165 (hVEGF), depicts a less destructive and slower form of retinal neovascularization present in the advanced stages of DR28. Although this model has several vascular abnormalities seen in DR, it lacks hyperglycemic background. Due to the limitations of these mouse models, Akimba (Ins2AkitaxVEGF+/−) mice were generated by crossing Kimba and Akita29 described with advanced retinal lesions, increased leaky capillaries, venous beading, tortuous vessels and attenuation of vessels, characteristic features seen in human DR, providing a significant advancement in DR research24,30. Since inflammatory pathways contribute significantly to DR, we designed this study to evaluate the role of the NLRP3 inflammasome in Akimba retina to better understand the disease pathogenesis in the advanced stages of DR.
Results
Akimba mouse displays characteristic features of advanced stages of DR
The fundus photographs and fluorescein angiograms from the Akimba mouse display increased retinal vascular leakage and vascular changes observed in DR compared to their parental strains. In the present study, we describe central, superior, inferior, temporal and nasal retina photographs in 20–24 weeks old mouse retina to further describe the abnormalities in the retinal microvascular (Fig. 1A–X). The Akimba mouse (Fig. 1S–X) was characterized by capillary dropout, severe hemorrhage, neovascularization, venous beading and vessel tortuosity compared to the WT or parental strains (Fig. 1A–F). Kimba mouse showed less severe but similar vascular phenotype (Fig. 1G–L). Akita showed no leaky vessels and relatively mild phenotype compared to the other animal models studied (Fig. 1M–R). We measured relative fluorescein intensity using a mean grey area measurement of FFA images and found a significant increase in fluorescein leakage in Akimba mice across all areas of the retina (Fig. 1 AA–AE).

Color fundus photographs and Fundus fluorescein angiograms (FFA) of wild -type (WT), Kimba, Akita and Akimba mice. WT mouse showed regular arterial and venular caliber, branching, and tortuosity in the retina (A–F). Kimba mice had multiple spots of hyperfluorescein (bright spots) throughout the retina (G–L). Akita mice displayed a slight increase in tortuosity with no signs of leakage (M–R). Double transgenic Akimba mice showed vasculature abnormalities inherited from parental strains, with high severity of leakages accompanied with tortuosity and significant loss of capillary density (S–X). Relative fluorescein intensity calculated using mean grey area measurement of FFA images showed a significant increase in fluorescein leakage in Akimba mice across all photographed areas of the retina (AA–AE). n = 8 animals in each group. *p < 0.05; **p < 0.01 and ***p < 0.001 compared to WT group. Data represent mean ± SD. Scale bar, 500 µm.
Akimba mouse has neurodegenerative retina and aberrant retinal function
OCT imaging showed no significant difference in retinal thickness (Fig. 2E) between Akita (Fig. 2C) and WT (Fig. 2A) mice, but there was significant reduction in the retinal thickness in Akimba (Fig. 2D; p < 0.001) and Kimba mice (Fig. 2C; p < 0.001) compared to the WT controls. We next measured retinal function using ERG (Fig. 2F) and found the average scotopic a- and b-wave amplitudes (Fig. 2G, H, respectively) obtained at the flash intensity of 10 cd·s/m2 were significantly reduced in all the experimental groups studied compared to the WT (p < 0.001 for both a- and b-wave amplitude). Akimba showed the maximal decrease in the a- and b-wave amplitudes compared to their parental strains (Fig. 2F).

Akimba retina displayed reduced retinal thickness and disrupted retinal function. Optical coherence tomography (OCT) analysis showed disruption of retinal architecture and appearance of subretinal hemorrhage in (C) Kimba and (D) Akimba retina. This was accompanied by a significant reduction in retinal thickness in these mice (E). Abnormal ERG was observed in all the mice models (F–H), with significantly decreased scotopic a-wave (G) and b-wave (H) amplitudes. Akimba mice showed the most severe disruption of retinal function, corresponding with the loss of retinal thickness and subretinal hemorrhages. n = 6 animals in each group. ***p < 0.001 compared to WT. Data represent mean \(\pm \) SD. Scale bar, 50 µm.
Akimba retina shows decreased relative blood flow volume
Retinal glial cells are known to influence the activity-dependent regulation of blood flow in the pathogenesis of many ocular diseases, including DR31. Recently, a novel non-invasive method has been defined to calculate the relative blood flow volume (RFV) in the human retina using laser speckle flowgraphy (LSFG)32. RFV is influenced by vessel diameter and retinal flow velocity and derived by filtering the background choroidal flow from the overall blood flow value in a region of interest centered on a retinal vessel (Fig. 3A–D). Akimba retina showed a significant decrease in RFV compared to its parental strains (Fig. 3D, E, P < 0.001). However, there were no differences in the retinal vessel diameter measured among the animal models studied (Fig. 3F).

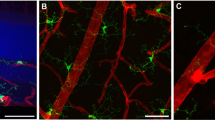
Reduced retinal flow volume and Müller cell gliosis in Akimba mouse. Laser speckle flowgraphy (LSFG) heat map of retinal blood flow in the animal models (A–D). Red indicates faster blood flow while blue indicates slower blood flow. Numbers 1–4 denotes four quadrants of the retina used for the analysis. Assessment of relative flow volume in the all four quadrants depicted significant attenuation of blood flow in Akimba (D, E). No differences were seen between the Kimba and Akita mice (E). Measurement of vessel diameter showed no difference in caliber amongst all four groups (F). Data represent mean \({\rm{\pm }}\) SD. n = 6 animals in each group. ***p < 0.001 compared to the WT. Akimba mouse showed maximal GFAP (J) stained cells compared with WT (G) or the parental strains (H, I). Similarly, GS immunostaining was observed throughout the entire inner retina and few cells migrating to the outer retina in the Akimba mice (N). Kimba retina showed similar staining patter but less intense (L) followed by Akita, where we found staining restricted to the inner retina (M) compared to the WT (K). n = 4 retina per group for immunohistochemistry, repeated twice. Arrowheads indicate positively stained cells. Scale bar, 50 µm.
Akimba retina exhibits hyperactive micro- and macroglial cells and increased mobilization of perivascular macrophages
PDR is primarily considered microangiopathy and neovascularization disease, but several reactive changes in Müller cells and activation of the resident or migrating macrophages/microglial cells have been reported in the disease pathogenesis33. We found a massive upregulation of GFAP stained macroglial cells throughout the Akimba retina (Fig. 3J) and relatively less expression in Kimba (Fig. 3H) followed by Akita (Fig. 3I) and further fewer cells in the inner retina of the WT (Fig. 3G) mice. A similar trend was observed with GS staining (Fig. 3K–N).
The presence of activated microglial cells (Iba-1 and OX-42) and resident or infiltrating macrophages (F4/80 and CD14) were confirmed by double immunostaining. In the normal retina, several ramified Iba-1 (Fig. 4A) and OX-42 (Fig. 4E) positive cells were observed in the outer plexiform layer (OPL), ganglion cell layer (GCL) or clustered around the perivascular region in the retina. In Kimba (Fig. 4B, F) and Akimba (Fig. 4D, H), these cells appeared hypertrophic or amoeboid and scattered in the entire retina. The most remarkable observation was the infiltration of these cells into the outer retina in Akimba mouse (Fig. 4L). In contrast, Akita (Fig. 4C, G) showed a marginal increase in activated microglial cells but none infiltrating into the outer retina.

Elevated expression of macrophage/microglia markers in Akimba mouse. Double immunostaining of Iba-1 (A–D) with OX-42 (E–H), and CD14 (M–P) with F4/80 (Q–T). Iba-1 staining (arrowheads) in WT (A) mice showed resident microglial cells with normal ramified morphology in the inner retina. Akita (C) mice showed minimal activation of microglia, however, had displaced microglia in the outer nuclear layer (ONL). Iba-1+ cells were both increased and amoeboid shaped in the outer retina of Kimba (B), and maximal activation was seen in Akimba (D) retina. OX-42 (block arrowheads) stains a cell surface antigen expressed by microglia, hence co-localized (arrows) with Iba-1+ microglia in WT (I). Majority of Iba-1+ microglia in Kimba (J), Akita (K) and Akimba (L) mice retina also stained positive for OX-42 (arrows). CD14 (arrowheads) is expressed by perivascular macrophages, which were minimal and located only around the vessels in the ganglion cell layer of the WT (M) mice. Increased expression was seen in the Kimba (N) and Akita (O) mice, and was drastically elevated in the outer retina of Akimba (P) mice, indicating migratory perivascular cells around new vessel sprouting from choroid into photoreceptor (PhR) region. F4/80 (block arrowheads) staining for macrophage showed predominant localization in the inner retina of the WT (Q) mice, which increased in Kimba (R) and Akita (S) mice, and was significantly elevated in GCL and ONL of Akimba (T) mice. While naïve CD14+ cell was not positive for F4/80 in WT mice (U, asterisk), co-localization (arrows) was seen in the GCL of Akita (W) mice, and in ONL/PhR of the Kimba (V) and Akimba (X) mice retina where sprouting vessels were located. n = 4 retina per group; repeated twice. Scale bar, 50 µm.
The second category of microglial precursors, also called perivascular macrophages, appear around the blood vessels and upon activation gradually lose dendritic processes and transform typically into phagocytic macrophages. In fact, activated macrophages have been reported earlier in the proliferative vitreoretinal disorders34. In Akita, we observed scant staining of F4/80 in the OPL (Fig. 4S). In both Kimba (Fig. 4R) and Akimba (Fig. 4T), F4/80 positive cells were seen in the photoreceptor layers. In contrast, CD14 was primarily present in the perivascular zones around the vessels in the ganglion cell layer of the WT (Fig. 4M) and Akita mice (Fig. 4O). Increased expression was seen in the Kimba (Fig. 4N), which was highly elevated in the outer retina of Akimba mice (Fig. 4P), indicating migratory perivascular cells around new vessel sprouting from choroid into photoreceptor (PhR) region. We observed that only a few F4/80 positive cells were co-localized with CD14 stained cells in Akita and Kimba mice (Fig. 4W, V, respectively), which amplified immensely in in Akimba (Fig. 4X) and none in WT retina (Fig. 4U).
Whole retina flat-mounts stained using Alexa Fluor 594-conjugated Griffonia simplicifolia (Bandeiraea) isolectin B4 indicate varying degrees of vascular permeability/leakage and altered retinal arteriolar microvasculature in all the animal models studied (Fig. 5D–L). BRBs were severely compromised in Akimba (Fig. 5J–L) retinas compared to Akita (Fig. 5G-I) and Kimba (Fig. 5D–F) as evidenced by the significantly increased capillary dropouts and severe vascular degeneration. Akimba mouse also exhibits retinal lesions accompanied with several instances of retinal neovascularization depicting advanced stages of DR (Fig. 5J–L).

Loss of retinal capillary density and vessel leakage in Akimba mice. Isolectin-GS IB4 staining on retinal flat mount showed normal vascular bed and uniform capillary distribution in WT (A–C) mouse, which was reduced markedly in Kimba (D–F), Akita (G–I) and significantly higher in Akimba (J–L) mice. Disruption of the capillary network was noted especially around the optic disk of Akimba (K) retina. Diffused staining (asterisk) around the optic head of Kimba (F) and Akimba (L) mice indicate loss of vessel integrity and leakage. n = 4 retina per group. Each retina flatmount image (A, D, G, and J) was stitched from at least 8 images were taken at 50× magnification; scale bar, 1 mm. Insets shown in middle row (B, E, H, and I) were taken at 100×; scale bar, 200 µm. Insets are shown in bottom row (C, F, I, and L) were taken at 200×; scale bar, 100 µm.
Akimba retina is characterized by increased IL-1β levels and high inflammatory index
One of the key factors in the pathogenesis of DR is the low-grade chronic inflammation driven by pro-inflammatory mediators including IL-1β and IL-6, which are thought to be released by activated macrophages and glia cells in the retina35. In both WT (Fig. 6A) and Akita (Fig. 6C) mice, specks of IL-1β expression were seen in the GCL, inner nuclear layer (INL) and plexiform layers. In Kimba (Fig. 6B), high expression of IL-1β was observed only in the GCL and minor staining was seen in the INL or plexiform layers. In Akimba (Fig. 6D), extensive damage was seen in all the retinal layers. In particular, the GCL was heavily stained. The INL and ONL have merged, and the entire area showed IL-1β expression. As shown in Fig. 6I, the relative mRNA expression of IL-1β increased by more than 4-fold in Akimba mouse (p < 0.001) compared with the age-matched Kimba or Akita. This increase was very well represented in the western blot (Fig. 6J), where IL-1β levels were significantly higher (p < 0.01) compared to the WT in the Akimba retina. This is further supported by the ELISA, which showed similar increases in IL-1β concentrations (p < 0.01) in the Akimba retina (Fig. 6K). Kimba or Akita retina showed an increasing trend but was not significant compared to the WT controls (Fig. 6I, J). On the contrary, we observed no increase in the IL-1α mRNA expression between WT and Akimba retina (Fig. 6I).

Akimba mice showed significantly high levels of the inflammatory milieu. Immunostaining with IL-1β (A–D) and IL-6 (E–H) showed basal levels of these cytokines in the WT (A, E, respectively), which were markedly increased in all the mice models studied. Akita showed diffused expression of IL-1β from the GCL to the OPL (C), which gradually increased in Kimba (B) followed by maximal expression in the Akimba (D) retina where we found its expression to be localized in the entire retina. We observed few specks of IL-6 staining in the GCL of WT (E) mice, with increasing expression levels from Akita (G), Kimba (F) with maximal expression in Akimba (H). Real-time PCR (I) followed the similar expression trend seen in immunostaining. The most remarkable findings were the significant high levels of IL-1β expression exclusively in Akimba retina found in western blots (J) and recapitulated with ELISA (K). n = 4 retina per group for immunohistochemistry, qPCR, western blot and ELISA; repeated twice. Arrowheads in A–H indicate IL-1β and IL6-positive cells. **p < 0.01; ***p < 0.001 compared to WT group. Data represent mean ± SD. Scale bar, 50 µm.
The WT-naïve retina (Fig. 6E) showed IL-6 immunoreactivity specifically in the nerve fiber layer (NFL) and specks of staining were also seen in the inner plexiform layer (IPL). A similar staining pattern was seen in the Akita (Fig. 6G). Kimba (Fig. 6F) showed similar staining to Akita, and few IL-6 positive cells were seen in the subretinal space. In Akimba mouse retina (Fig. 6H), IL-6 expression in NFL was even higher compared to its parental strains. More specks of staining were seen in the IPL. In addition, OPL and IS were also heavily stained. Quantitative IL-6 transcript levels followed the immunostaining trend and showed significantly higher expression (p < 0.001) exclusively in the Akimba mouse (Fig. 6I).
NLRP3 inflammasome was activated in Akimba mouse retina
Immunohistochemistry findings in the WT (Fig. 7A) revealed few NLRP3 positive cells in the GCL, and NFL layers. Akita showed similar staining pattern (Fig. 7C). Kimba (Fig. 7B) and Akimba (Fig. 7D) showed similar staining to Akita except that the OP was much more heavily stained and few specks in Akimba outer retina. With regard to ASC immunoreactivity, WT retinas showed expression mostly in the GCL (Fig. 7E). In Akita, the diffused staining of ASC was present throughout the retinal layers (Fig. 7G). In Kimba, there was slight staining in the GCL (Fig. 7F). Akimba showed significantly high diffused staining in the entire retina, mostly seen around the damaged retinal layers (Fig. 7H). WT retinas stained with Caspase-1 antibody exhibit diffused staining in the IPL (Fig. 7I). In Akita, NFL was heavily stained including the plexiform layers (Fig. 7K). In Kimba, the GCL and OPL were stained heavily (Fig. 7J). In Akimba, the staining was similar to Kimba but heavily stained cells were found in the outer retina (Fig. 7L).

Activated retinal NLRP3 inflammasome components in Akimba mice. Retinal sections were immunostained with NLRP3 (A–D), ASC (E–H) and Caspase-1 (I–L) antibodies. Full-length blots for NLRP3 and ASC are provided in supplementary figure 1. NLRP3 was constitutively expressed in the GCL and inner plexiform layer (IPL) in the WT retina (A). A gradual increase in NLRP3 immunofluorescence was seen from Akita (C), Kimba (B) to Akimba (D). Some staining was also seen in the outer plexiform layer (OPL) indicative of the highest level of inflammatory status in the Akimba retina. Similarly, ASC and Caspase-1 were localized in the GCL and IPL in all mice models (E–H and I–L, respectively). ASC was also observed in the outer retina with vessel intrusion in Akimba (H). Caspase-1 was also seen in Akimba subretinal regions (L) where retinal hemorrhage was observed. Expression levels of NLRP3 components were further confirmed with real-time PCR (M) and western blots (N–Q). The most significant increase of NLRP3, ASC and Caspase-1 were seen in the Akimba compared to Kimba or Akita or WT mice. NLRP1 mRNA expression showed no differences in any of the model studied compared to the WT expression (M). n = 4 retina per group for immunohistochemistry, qPCR and western blot. Arrowheads in A–L indicate stained cells. Scale bar, 50 µm. *p < 0.05; **p < 0.01 and ***p < 0.001 compared to WT. Data represents mean ± SD.
The NLRP3 transcript levels measured using qPCR indicated progressive increase in all the animal models compared to their age-matched controls (Fig. 7M). Akimba retina showed maximal levels (~6-fold, p < 0.001) followed by Akita (p < 0.001) and Kimba (p < 0.05). In contrast, we found no differences in the NLRP1 mRNA in any of the animal model studied indicating the specificity of NLRP3 activation in these mice models. Also, we did not find any increase in inflammasome components such as ASC in Kimba or Akita retinas and their transcript levels were similar to WT controls (Fig. 7M). We found a significant increase (p < 0.05) in Caspase-1 mRNA in Kimba mice but not in Akita compared to the WT mice. On the contrary, Akimba mouse retina showed a 4-fold increase (p < 0.001) in ASC and ~12-fold increase (p < 0.001) in the Caspase-1 mRNA levels (Fig. 7M). These results corroborate with increased levels of IL-1β found only in the Akimba retina but not in Akita or Kimba mice (Fig. 6I–K). Western blots validated our findings from qPCR and immunohistochemistry (Fig. 7N–Q). Akimba retina showed a 5-fold increase in the NLRP3 (p < 0.001, Fig. 7O), a 13-fold increase in ASC expression (p < 0.001, Fig. 7P) and a 12-fold increase in the Caspase-1 (p < 0.001, Fig. 7Q) levels. Similarly, Akita showed increased NLRP3 (p < 0.05) and Caspase-1 (p < 0.001), but no significant change in ASC, while Kimba showed significantly higher levels of NLRP3 (p < 0.001), ASC (p < 0.01) and caspase-1 (p < 0.001).
Akimba mouse retina shows increased angiogenic mediators
Since PDR involves neovascularization as one of the key components in the disease pathogenesis, we studied the angiogenic mediators involved in the Akita, Kimba and Akimba mice models. We first looked at the retinal VEGF and its receptors, Flt-1 and Flk-1 that are described as the major vascular permeability factors to cause vascular leakage and pathological neovascularization36. We found significantly increased transcript levels of VEGF, Flt-1 and Flk-1 in all the models studied with maximal expression seen in Akimba mouse retina (Fig. 8A). Western blots showed similar results with maximum expression of VEGF detected in Akimba retina except in Akita where we found no change in VEGF protein levels compared to the WT controls (Fig. 8B–E). These results validate our earlier findings with loss of vascular permeability in Kimba and Akimba mice models but not in Akita retina (Figs 1 and 5).

Pro-angiogenic factors were upregulated in Akimba retina. mRNA expression of VEGF showed a 3-fold increase in Akimba mouse (A), corresponding with a >2-fold increase in protein expression seen in Western blot (B, E). Similarly, ICAM-1 was elevated (B, D) up to 15-fold in Kimba and 25-fold in Akimba mice. PECAM-1 showed an upward trend but was not statistically significant in any of the mouse models studied (B, C). n = 4 retina per group for qPCR and western blot; repeated twice. *p < 0.05; **p < 0.01 and ***p < 0.001 compared to WT. Data represent mean \(\pm \) SD.
We also studied other angiogenic mediators such as platelet endothelial cell adhesion molecular (PECAM)-1 and intracellular adhesion molecule (ICAM)-1, both known to play a critical role in pathological retinal neovascularization. ICAM-1 protein concentrations were significantly increased in neural retina in all animal models studied compared to the WT controls (Fig. 8D). In contrast, we found an increasing trend in PECAM-1 expression (Fig. 8C) but was not significant.
Discussion
The inflammatory milieu in the advanced stages of DR results in the vascular leakage in DME and vaso-occlusion and retinal neovascularization in PDR37,38. The most challenging aspects were to find an appropriate animal model to recapitulate the cellular and pathological changes in the progression of advanced stages of human DR. Recently a double transgenic Akimba mouse model to study the advanced stages of DR has been characterized. Akimba mouse exhibit progressive retinopathic changes characterized by the photoreceptor cell death, thinning of the retina, BRB loss, vessel constriction, neovascularization, fibrosis, and edema. With aging, these mice show many clinical signs of DR including microaneurysms, retinal neuronal loss, leaky capillaries, venous beading, tortuous vessels, capillary dropout, and neovascularization. These findings suggest that Akimba mouse model represent several characteristic features of the advanced stages of human DR29,30. On the contrary, Kimba mouse shows several of these vascular changes but lacks diabetic background. It is noteworthy that the neovascular changes observed in the Akimba retina are not due to long-term hyperglycemia, as seen in human DR, but are due to the presence of the human VEGF165 transgene in the photoreceptors. Nevertheless, improvements in DR are mostly associated with the hyperglycemic control in patients only during the early stages of disease39,40. This is due to the fact that chronic hyperglycemic insult (>8–10 years in the human patients) causes irreversible neuronal and vascular damage in the retina, which cannot be improved merely by regulating hyperglycemia in the advanced stages of the DR.
Retinal glial cells have been implicated in the pathogenesis of DR41. In the early stages of diabetic retinopathy, Müller glial cells become hyperactive by releasing angiogenic and neurotrophic factors to protect the retina from hyperglycemia-induced stress. Over time, the chronic hyperglycemic insult provokes massive inflammatory milieu followed by the onset of overt neovascularization seen in the late stages of DR41. We found increased GFAP and GS immunoreactivity across all the animal models with maximal increase in Akimba retina accompanied by severe loss of retinal architecture and Müller cell gliosis. This was supported by the ERG data showing loss of retinal function observed in Akimba mouse. In fact, retinal neuron-to-glia cell communications are necessary intermediaries for the maintenance of blood flow dynamics42. This is controlled by glial cell-mediated cross-talk between neurons and endothelial cells, also referred to as neurovascular coupling. The recent discovery of a novel laser speckle flowgraphy (LSFG) imaging technique for the evaluation of the microvasculature had led to the development of a rapid and non-invasive method for the determination of basal blood flow and neuronal activity-dependent alterations in ocular diseases32. LSFG has been suggested to serve as a useful biomarker not only for detection but also for monitoring postoperative visual outcomes in the patients with retinal and choroidal vascular diseases43,44. In type 1 and type 2 diabetic patients, a substantial decrease in the flicker-evoked vasodilation was observed45,46,47, which suggests reduced neurovascular coupling causing hypoxia and eventually retinal neovascularization. We found a significantly reduced blood flow volume in the Akimba retina compared to their parental strains depicting a dysregulated retinal microvasculature.
Next, we looked at the resident and migratory retinal microglial cells using multiple rodent microglia/macrophage cell markers. Isolectin-B4-stained Akimba retinal flat mounts showed a significantly high degree of leaky and degenerated retinal vasculature compared to the WT or parental strains. Whether or not the retina contains antigen-presenting dendritic cells has been a topic of controversy for decades. Zhang and colleagues reported a small population of MHC-II+ cells in the rat retina48. A recent study by Rakoczy and group showed a dramatic increase in MHC class II+ cells throughout the retina in Akimba mice29. In the normal retina, microglial cells are long slender processes distributed in the inner and outer plexiform layers (IPL & OPL), GCL and nerve fiber layer (NFL), showing highly motile and ramified protrusions. In DR, the microglia increase in number and become hypertrophic (amoeboid) with shorter dendrites and larger somas at different stages of DR. Often, they resemble perivascular macrophages in the area of retinal injury and cluster around the vasculature49. We used Iba-1 and OX-42 as well as F4/80 and CD14 double immunostaining to distinguish the resident or migratory microglia and perivascular macrophages in the retina. Although both perivascular macrophages and microglia express Iba-1, OX-42 and F4/80, CD14 is highly expressed by infiltrating macrophages and not by microglial cells10,49. We found a progressive increase in the Iba-1 expressing cells co-expressed with OX-42 in the WT to Akita to Kimba retina to maximal expression was observed in Akimba, where Iba-1/OX-42 positive cells were seen in the outer retina. This could be due to the loss of BRB in the Akimba mouse. The expression of F4/80 in WT was meager but found upregulated in the inner retinal layers of the Akita mouse presumably because of the damaged inner BRB (iBRB)18. In Akimba mice retina, significantly increased F4/80 and CD14 positive cells were detected in the outer retinal layers which could be either due to the migration of activated resident macrophages or from the infiltration of activated circulating macrophages from the choroid into the retina through the compromised oBRB. It is noteworthy that we observed only a few cells co-stained with CD14 and F4/80, which could be due to the fact that CD14 expressing perivascular macrophages might have migrated to the outer retina50 and represent a different subset of retinal macrophages as reported earlier10. In addition, in DME, microglia can escape outer limiting membrane and migrate to the outer retina and subretinal space. In fact, the activation and proliferation of microglia have been reported in both the inner and outer retina in the Goto-Kakizaki rat model (GK) of DR51. However, the relative importance and the role of resident and migrating microglia/macrophages in the pathogenesis of DR is yet to be fully determined.
During the advanced stages of DR, chronic neuroinflammation released several pro-inflammatory mediators and increased oxidative and nitrosative stress that may compromise the BRB and contribute towards neuronal and vascular damage in the retina. Increased levels of IL-1β and IL-6 have been found in the serum and vitreous of patients with DR8,9. Uncontrolled release of IL-1β has also been suggested to play a crucial role in the formation of acellular capillaries and retinal capillary cell death35. Consistent with the previous report, we found a significant increase in IL-1β and IL-6 expression in the inner retinal layers of Akita including the plexiform layers, GCL and NFL. The most deleterious effects were seen in the Akimba neural retina with significantly high levels of IL-6 and IL-1β transcripts and protein levels. Indeed, a previous study has shown IL-1β expression in microglial and macroglial cells (astrocytes and Müller cells) localized adjacent to RGCs52. Hence, we propose that microglial cells are the major producers of secreted IL-1β and IL-6 further elevating an inflammatory response supported by a previous finding that knocking out IL-1β receptor protected the retinal capillaries from degeneration in diabetic animals53.
Since IL-1β can amplify the initial inflammatory response by activating Caspase-1 in an autocrine manner, we next looked at the regulation of IL-1β activation in our transgenic mouse models. It has been recently reported that both IL-1β maturation and its release are tightly regulated by NLRP3 inflammasome, which has been involved in several systemic inflammatory diseases such as gout, atherosclerosis, pulmonary fibrosis and contact hypersensitivity54. In addition, NLRP3 inflammasome has been reported to be a critical component in the pathogenesis of metabolic disorders such as obesity and type 2 diabetes. Very recently, a clinical study suggested the association of inflammasome in the vitreous of the PDR patients22. The NLRP3 inflammasome is best characterized as a protein scaffold upon which an ASC adaptor assembles and acts to recruit pro-Caspase-1.
Clinical studies have demonstrated NLRP3 inflammasome-mediated release of IL-1β in diabetic conditions can be therapeutically treated with IL-1β antagonist55,56. In addition, mice deficient in the components of NLRP3 have been shown to have reduced diabetic symptoms. A previous study has reported an increase in Caspase-1 activity in retinas of diabetic mice, galactose-fed mice, diabetic humans, and in retinal Müller cells treated with high glucose57. In the present study, NLRP3 was upregulated in the inner retina along with Caspase-1 and ASC expression compared to the WT retina. Although we found increased transcripts of NLRP3 components in all the animal models studied significantly higher levels of IL-1β protein were exclusively seen in the Akimba mouse retina using western blot and ELISA. On the contrary, NLRP1 inflammasome remained unchanged in any of the models studied, thus indicating the specificity of NLRP3 inflammasome activation in the retina. This suggests that NLRP3 inflammasome plays a key role in the Akimba retina to release pro-inflammatory mediators eliciting a prolonged inflammatory response involved in the pathogenesis of DR. However, the in vivo functional role of NLRP3 in PDR needs further studies.
Another predominant feature seen in late stages of DR is retinal neovascularization caused by increased vascular permeability. This leads to VEGF upregulation and activates Flk-1 on endothelial cells (ECs) to promote the aberrant formation of new vessels18. In addition, the VEGF/Flk-1 signaling axis facilitates the degradation of adhesion junctions and loosens the intercellular junctions between neighboring ECs58. We found a significant increase in the VEGF levels in Akimba retina (and a mild increase in Kimba) contrary to the increased VEGF, or its receptor transcripts in all the mice models studied. A previous study suggests that VEGF injected in naïve eyes triggered many of these vascular changes observed during diabetes, such as ICAM-1 upregulation and vascular permeability59. Similarly, we found an increased expression of intercellular adhesion molecule (ICAM)-1 in the retina at transcript levels as well as in protein levels in Akimba mice.
In summary, our study suggests that the dysregulated activation of the NLRP3 inflammasome in the Akimba mouse compromises retinal immune response resulting in the loss of BRB. These changes led micro- and macroglia cells to a continuous activated state causing vascular leakage and retinal neovascularization observed in the advanced stages of DR.
Materials and Methods
Animals
Akita, Kimba, and Akimba mice were procured from Centre of Ophthalmology and Visual Science, The University of Western Australia, Crawley, Australia and inbred for at least 10 generations. Akita mice have a dominant point mutation in the insulin-2 gene, making it a natural occurring type-I diabetic mouse model. Kimba mice models transiently overexpress human VEGF165 in the photoreceptors, hence depicts advanced stage neovascularization in the retina. The Akimba mice developed by Rakoczy et al.29 were generated by crossing homozygous Kimba with heterozygous Akita mice. All mice are on C57BL/6 J background and inbred for at least 10 generations before used for experiments. Six-months-old mice (20–24 weeks) were used in this study. There were a minimum of 6 animals in each experiment unless otherwise stated in the figure legends. Animals were housed in 12 h light/12 h dark cycle with food and water provided ad libitum.
Ethics Statement
All mice experimental procedures was approved by the Singhealth Institutional Animal Care and Use Committee (IACUC), Singapore and were conducted in accordance with the ARVO recommendations for animal experimentation, and data presented in accordance with the ARRIVE guidelines.
Fundus and Fluorescein Angiography (FFA)
Digital color fundus photographs were taken using MICRON IV (Phoenix Research Laboratories, Pleasanton, CA, USA). Mice were anesthetized with a combination of 20 mg/kg ketamine hydrochloride (Ketamine, Parnell Laboratories, NSW, Australia) and 2 mg/kg xylazine hydrochloride (Ilium Xylazil-20, Troy Laboratories, NSW, Australia). The eyes were dilated with 1% tropicamide (Alcon Laboratories, Inc., Fort Worth, TX, USA), followed by 2.5% phenylephrine (Bausch and Lomb Pharmaceuticals, Inc., Tampa, FL, USA). A small drop of coupling gel (Genteal, Novartis Pharmaceuticals, East Hanover, NJ, USA) was placed on each eye to prevent corneal dryness. For FFA, mice were intraperitoneally injected with 10% sodium fluorescein dye (AKORN, Lake Forest, IL, USA) diluted by 10× in sterile saline, and given at a dose of 0.1 ml/10 g body weight. For accurate comparison of fluorescein intensity in the retina, FFA images were taken 30 s after injection, in the sequence as follows: central, temporal, nasal, superior, inferior. One image was taken per retinal area, to avoid fluorescein saturation. Semi-quantitative analysis of fluorescein intensity was performed by measuring the mean gray value of FFA image from each area of the retina, and values obtained from Kimba, Akita and Akimba mice were normalized to the WT mice for relative fold intensity.
OCT
Image-guided spectral-domain OCT images were taken using inSight software (Phoenix Research Labs). Genteal was applied to the eyes before imaging to keep cornea moist. Trans-retinal images were taken from regions around the optic nerve. Retinal thickness was calculated in the software.
Electroretinography (ERG)
Mice were dark-adapted for a minimum of 12 hours. All ERG recordings were performed using an Espion system (Diagnosys LLC, Lowell, MA, USA) in a dark room under dim red light illumination. Mice were anesthetized, and the eyes were dilated as described above. Body temperature was maintained at 37 °C using pumped-water heat pad fixed on the ERG stage. Coupling gel was applied to each eye, before placing a monopolar electrode on the cornea. A gold-cup electrode was placed in the mouth to serve as the reference electrode, and a silver-silver chloride electrode was inserted into the tail as the ground electrode. The mouse head was positioned in front of a Ganzfeld stimulus. Scotopic ERG responses were recorded across increasing light intensities from −3.3 to 1.5 log cd.s.m−2 in 0.3-log-unit increments. Full-field ERG was recorded, and each response was the average of 3–5 trials. Signals were acquired at 2 kHz with high- and low-pass filtering at 0.1 and 100 Hz respectively.
Laser Speckle Flowgraphy (LSFG)
LSFG uses a laser speckle phenomenon to measure the real-time 2-dimensional fundus blood flow changes without pupillary dilations or contrast media providing new insights into the pathophysiology of retinal diseases. Movement of blood cells in the tissue disrupts the speckle pattern of laser irradiation on the retina, which can be expressed as flow rate. Mice were anesthetized, and eyes dilated as described in FFA section. They were placed into the stand of the LSFG with one eye facing downward. Coupling gel was applied to the top eye before a cover glass was placed onto the cornea. LSFG (Softcare Co. Ltd., Fukuoka, Japan) was performed at the optic nerve head of the eye. Images were acquired continuously for 4 s to produce a composite map of ocular blood flow. The procedure was repeated with the contralateral eye. Calculation of retinal flow volume was produced by first subtracting the background choroidal flow from the overall blood flow value of a region of interest centered on a retinal vessel, reflecting retinal flow velocity and vascular diameter. Up to 6 areas of a blood flow map were analyzed. All calculations and analysis were performed by a single masked observer.
Tissue Harvesting and Analysis
For analysis by real-time PCR, ELISA and western blot, enucleated eyes were dissected immediately on chilled PBS and processed as described previously60. For immunohistochemistry, enucleated eyes were first fixed in 4% paraformaldehyde (PFA) in 1× PBS. Eyes were treated to sucrose gradient (15% and 30%) cryoprotection before embedding in Tissue Freezing Medium® (Leica Biosystems Inc., Buffalo Grove, IL, USA) on dry ice. Retina sections of 6–8 µm thickness were cut using HM525 HX Cryostat (Thermo Fisher Scientific, Waltham, MA, USA) and collected on Polysine™-coated glass slides (Thermo Fisher Scientific). Glass slides were kept frozen at −80 °C until staining was performed. For flat mount staining, eyes fixed in 4% PFA for 10 min was hemisected ora ciliaris to isolate the neural retina. Flat mount staining was performed immediately.
Real-Time PCR
Neural retina tissue was homogenized using a handheld homogenizer (Sigma-Aldrich, St. Louis, MO, USA) in RLT buffer +1% β-mercaptoethanol. RNA was isolated and purified using RNeasy Mini Kit (Qiagen, Valencia, CA, USA) according to manufacturer’s instructions. cDNA was generated using GoScript reverse transcriptase (Promega, Madison, WI, USA). 250 ng of RNA was used as the starting materials. Real-time PCR with SYBR Green dye (Thermo Fisher Scientific) was conducted on the LightCycler 480 II System (Roche, Indianapolis, IN). The PCR run starts with preincubation at 95 °C for 5 min, followed by 45 amplification cycles at 95 °C for 10 s, 60 °C for 15 s, and 72 °C for 30 s. Melt curve analysis was conducted to verify amplification reaction. GAPDH was used as housekeeping gene. Primer sequences are listed in Supplementary Table 1.
Immunohistochemistry
Immunofluorescence staining was performed according to standardized protocol described earlier60. In brief, frozen slides were kept at room temperature for 15 min, then soaked in 1× PBS for 10 min. Tissue sections were post-fixed with 4% PFA for 10 minutes followed by two washes in 1× PBS for 5 min each. Blocking was done with 1% BSA for 20 minutes at room temperature. Primary antibody was incubated overnight at 4 °C, and secondary antibody incubated for 1 h at room temperature the following day. Double immunostaining was performed by sequential overnight incubations with antibodies directed for two different species. Slides were mounted with Vectashield® Mounting Medium with DAPI (Vector Laboratories, Burlingame, CA, USA). Retinal sections without primary antibody served as staining control. The list of antibodies used in this study is listed in Supplementary Table 2.
For flat mount with isolectin GS-IB4, Alexa Fluor® 594 conjugate (Thermo Fisher Scientific), flat mounts were washed in 1× PBS, blocked with PBS + 1% Triton X-100 (PBSTx), and left overnight with 1:100 antibody dilution in PBSTx. Flat mounts were cut into four quadrants and mounted with ProLong® Diamond antifade mountant (Thermo Fisher Scientific). Slides were kept at 4 °C for at least 12 hours before viewing. All sections were viewed and imaged on the Confocal Micros C2 (Nikon Instruments Inc, Melville, NY, USA).
Western Blotting
Total protein from each neural retina was extracted in 150 µl of RIPA buffer + protease and phosphatase inhibitor (Sigma-Aldrich) using handheld homogenizer. Protein quantification was performed using BCA protein assay kit (Pierce, Thermo Fisher Scientific). 15 µg of protein was loaded onto 4–20% pre-cast gels (Bio-Rad, Hercules, CA, USA). Electrophoresis was run at 200 V for 30 min. The transfer was made onto PVDF membrane overnight at 40 V in 4 °C. Immunoblotting was performed using 5% milk. All primary antibodies were incubated overnight at 4 °C, and secondary antibody was kept for 1 h at room temperature. Protein bands were visualized using SuperSignal Chemiluminescence (Pierce). Antibodies are listed in Supplementary Table 2. GAPDH was used as loading control.
ELISA
To further validate the expression of IL-1β, total expressed and secreted IL-1β was measured using mouse-specific IL-1 beta Quantikine ELISA kit (R&D Systems, Minneapolis, MN, USA) according to manufacturer’s instructions. Retina protein lysate was taken from homogenate made for western blot. Colorimetric readings were taken at 540 nm. The correction was made by subtracting readings from 450 nm.
Statistical Analysis
Statistical analysis was performed using Student t-test for comparison between 2 groups (GraphPad Prism 6.0, GraphPad Software, Inc., La Jolla, CA, USA). Comparison between multiple groups was done using one-way ANOVA followed by post-hoc analysis using Newman-Keuls test. Significance was taken when p < 0.05. Data were presented as mean ± SD. The number of animals or tissues used per analysis and repeats of each experiment are described in the figure legends.
References
Wong, T. Y., Cheung, C. M., Larsen, M., Sharma, S. & Simó, R. Diabetic retinopathy. Nat. Rev. Dis. Primers 2, 16012 (2016).
Yau, J. W. et al. Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care 35, 556–64 (2012).
Stitt, A. W. et al. The progress in understanding and treatment of diabetic retinopathy. Prog. Retin. Eye Res. 51, 156–86 (2016).
Roy, S., Kern, T. S., Song, B. & Stuebe, C. Mechanistic Insights into Pathological Changes in the Diabetic Retina: Implications for Targeting Diabetic Retinopathy. Am. J. Pathol. 187, 9–19 (2017).
Joussen, A. M. et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 18, 1450–2 (2004).
Chibber, R., Ben-Mahmud, B. M., Chibber, S. & Kohner, E. M. Leukocytes in diabetic retinopathy. Curr Diabetes Rev 3, 3–14 (2007).
Tang, J. & Kern, T. S. Inflammation in diabetic retinopathy. Prog. Retin. Eye Res. 30, 343–58 (2011).
Ting, D. S. et al. Biomarkers of Diabetic Retinopathy. Curr. Diab. Rep. 16, 125 (2016).
Sasongko, M. B. et al. Circulating markers of inflammation and endothelial function, and their relationship to diabetic retinopathy. Diabet. Med. 32, 686–91 (2015).
Xu, H. & Chen, M. Diabetic retinopathy and dysregulated innate immunity. Vision Res. pii: S0042–6989, 30091–3 (2017).
Richardson, M. B. & Williams, S. J. MCL and mincle: C-type lectin receptors that sense damaged self and pathogen-associated molecular patterns. Front. Immunol. 5, 288 (2014).
Ramasamy, R. et al. Advanced glycation end products and RAGE: A common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology 15, 16R–28R (2005).
Halle, A. et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 9, 857–65 (2008).
Tarallo, V. et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell 149, 847–59 (2012).
Freeman, L. C. & Ting, J. P. The pathogenic role of the inflammasome in neurodegenerative diseases. J. Neurochem. 136(Suppl 1), 29–38 (2016).
Murakami, Y. et al. Programmed necrosis, not apoptosis, is a key mediator of cell loss and DAMP-mediated inflammation in dsRNA-induced retinal degeneration. Cell Death Differ. 21, 270–7 (2014).
Gnanaguru, G., Choi, A. R., Amarnani, D. & D’Amore, P. A. Oxidized Lipoprotein Uptake Through the CD36 Receptor Activates the NLRP3 Inflammasome in Human Retinal Pigment Epithelial Cells. Invest. Ophthalmol. Vis. Sci. 574, 704–712 (2016).
Klaassen, I., Van Noorden, C. J. & Schlingemann, R. O. Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog. Retin. Eye Res. 34, 19–48 (2013).
Jin, C. & Flavell, R. A. Molecular mechanism of NLRP3 inflammasome activation. J. Clin. Immunol. 30, 628–31 (2010).
Schroder, K., Zhou, R. & Tschopp, J. The NLRP3 inflammasome: a sensor for metabolic danger? Science 327, 296–300 (2010).
Hu, C. et al. NLRP3 deficiency protects from type 1 diabetes through the regulation of chemotaxis into the pancreatic islets. Proc. Natl. Acad. Sci. USA 12, 11318–23 (2015).
Loukovaara, S. et al. NLRP3 inflammasome activation is associated with proliferative diabetic retinopathy. Acta Ophthalmol, Mar 8 https://doi.org/10.1111/aos.13427. [Epub ahead of print] (2017).
Kern, T. S. In vivo models of diabetic retinopathy. In: Duh EJ (ed.). Diabetic Retinopathy. Humana Press, Totowa, New Jersey, 137–156 (2008).
Robinson, R., Barathi, V. A., Chaurasia, S. S., Wong, T. Y. & Kern, T. S. Update on animal models of diabetic retinopathy: from molecular approaches to mice and higher mammals. Disease Models Mechanisms 5, 444–456 (2012).
Cheţa, D. Animal models of type I (insulin-dependent) diabetes mellitus. J Pediatr. Endocrinol. Metab. 11, 11–19 (1998).
Kern, T. S. et al. Topical administration of nepafenac inhibits diabetes-induced retinal microvascular disease and underlying abnormalities of retinal metabolism and physiology. Diabetes 56, 373–9 (2007).
Oyadomari, S. et al. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Invest. 109, 525–532 (2002).
Lai, C. M. et al. Generation of transgenic mice with mild and severe retinal neovascularisation. Br. J. Ophthalmol. 89, 11–6 (2005).
Rakoczy, E. P. et al. Characterization of a mouse model of hyperglycemia and retinal neovascularization. Am. J. Pathol. 177, 2659–2670 (2010).
Wisniewska-Kruk, J. et al. Molecular analysis of blood-retinal barrier loss in the Akimba mouse, a model of advanced diabetic retinopathy. Exp. Eye Res. 122, 123–31 (2014).
Kur, J., Newman, E. A. & Chan-Ling, T. Cellular and physiological mechanisms underlying blood flow regulation in the retina and choroid in health and disease. Prog. Ret. Eye Res. 31, 377–406 (2012).
Shiga, Y. et al. Relative flow volume, a novel blood flow index in the human retina derived from laser speckle flowgraphy. Invest. Ophthalmol. Vis. Sci. 55, 3899–904 (2014).
Abcover, S. F. Muller cell-microglia cross talk drives neuroinflammation in diabetic retinopathy. Diabetes 66, 261–263 (2017).
Esser, P., Heimann, K. & Wiedemann, P. Macrophages in proliferative vitreoretinopathy and proliferative diabetic retinopathy: differentiation of subpopulations. Br. J. Ophthalmol. 77, 731–3 (1993).
Kowluru, R. A. & Odenbach, S. Role of interleukin-1beta in the development of retinopathy in rats: effect of antioxidants. Invest. Ophthalmol. Vis. Sci. 45, 4161–6 (2004).
Miller, J. W. et al. Vascular endothelial growth factor/vascular permeability factor is temporally and spatially correlated with ocular angiogenesis in a primate model. Am. J. Pathol. 145, 574–84 (1994).
Cheung, N., Mitchell, P. & Wong, T. Y. Diabetic retinopathy. Lancet 376, 124–136 (2017).
Tan, G. S., Cheung, N., Simo, R., Cheung, G. C. & Wong, T. Y. Diabetic macular oedema. Lancet Diabetes Endocrinol. 5, 143–155 (2011).
Chew, E. Y. et al. Action to Control Cardiovascular Risk in Diabetes Eye Study Research Group. The effects of medical management on the progression of diabetic retinopathy in persons with type 2 diabetes: the Action to Control Cardiovascular Risk in Diabetes (ACCORD) Eye Study. Ophthalmology 121, 2443–51 (2014).
Ogura, S. et al. Sustained inflammation after pericyte depletion induces irreversible blood-retina barrier breakdown. JCI Insight 2, e90905 (2017).
Verkhratsky, A., Parpura, V., Pekna, M., Pekny, M. & Sofroniew, M. Glia in the pathogenesis of neurodegenerative diseases. Biochem. Society Transactions 42, 1291–1301 (2014).
Metea, M. R. & Newman, E. A. Signalling within the neurovascular unit in the mammalian retina. Exp. Physiol. 92, 635–40 (2007).
Nitta, F. et al. The effect of intravitreal bevacizumab on ocular blood flow in diabetic retinopathy and branch retinal vein occlusion as measured by laser speckle flowgraphy. Clin. Ophthalmol. 8, 1119–27 (2014).
Saito, M. et al. Pulse Waveform Changes in Macular Choroidal Hemodynamics With Regression of Acute Central Serous Chorioretinopathy. Invest. Ophthalmol. Vis. Sci. 56, 6515–22 (2015).
Garhofer, G. et al. Reduced response of retinal vessel diameters to flicker stimulation in patients with diabetes. British J. Ophthalmol. 88, 887–891 (2004).
Mandecka, A. et al. Influence of flickering light on the retinal vessels in diabetic patients. Diabetes Care 30, 3048–3052 (2007).
Pemp, B. et al. Reduced retinal vessel response to flicker stimulation but not to exogenous nitric oxide in type 1 diabetes. Invest. Ophthalmol. Vis. Sci. 50, 4029–4032 (2009).
Zhang, J., Wu, G. S., Ishimoto, S., Pararajasegaram, G. & Rao, N. A. Expression of major histocompatibility complex molecules in rodent retina. Immunohistochemical study. Invest. Ophthalmol. Vis. Sci. 38, 1848–1857 (1997).
Dick, A. D., Ford, A. L., Forrester, J. V. & Sedgwick, J. D. Flow cytometric identification of a minority population of MHC class II positive cells in the normal rat retina distinct from CD45 low CD11b/c+CD4 low parenchymal microglia. British J. Ophthalmol. 79, 834–840 (1995).
Chen, M. & Xu, H. Parainflammation, chronic inflammation, and age-related macular degeneration. J. Leukocyte Biol. 98, 713–725 (2015).
Omri, S. et al. Microglia/macrophages migrate through retinal epithelium barrier by a transcellular route in diabetic retinopathy: role of PKCζ in the Goto Kakizaki rat model. Am. J. Pathol 179, 942–53 (2011).
Liu, Y., Biarnes Costa, M. & Gerhardinger, C. IL-1beta is upregulated in the diabetic retina and retinal vessels: cell-specific effect of high glucose and IL-1beta autostimulation. PLoS One 7, e36949 (2012).
Vincent, J. A. & Mohr, S. Inhibition of caspase-1/interleukin-1beta signaling prevents degeneration of retinal capillaries in diabetes and galactosemia. Diabetes 56, 224–30 (2007).
McDermott, M. F. & Tschopp, J. From inflammasomes to fevers, crystals and hypertension: how basic research explains inflammatory diseases. Trends Mol. Med. 13, 381–388 (2007).
So, A., Desmedt, T., Revaz, S. & Tschopp, J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res. Ther. 9, R28 (2007).
Larsen, C. M. et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 356, 1517–1526 (2007).
Mohr, S., Xi, X., Tang, J. & Kern, T. S. Caspase activation in retinas of diabetic and galactosemic mice and diabetic patients. Diabetes 51, 1172–9 (2002).
Dejana, E., Tournier-Lasserve, E. & Weinstein, B. M. The control of vascular integrity by endothelial cell junctions: molecular basis and pathological implications. Dev. Cell 16, 209–21 (2009).
Miyamot, K. et al. Vascular endothelial growth factor (VEGF)-induced retinal vascular permeability is mediated by intercellular adhesion molecule-1 (ICAM-1). Am. J. Pathol. 156, 1733–9 (2000).
Su, X. et al. Characterization of Fatty Acid Binding Protein 7 (FABP7) in the Murine Retina. Invest. Ophthalmol. Vis. Sci. 57, 3397–408 (2016).
Acknowledgements
We will like to acknowledge excellent technical support from Ms. Alison Tan and Milan Mehta. The authors would also like to acknowledge start-up grant from Veterinary Medicine and Surgery department to S.S.C. and grants from Veteran Health Affairs Merit (5I01BX000357–05) and National Eye Institute, NIH (2RO1EY017294-06) to R.R.M. This work was also supported partially by grants from the National Medical Research Council, Singapore [NMRC/CG/SERI/2013, VAB] and Biomedical Research Council, Singapore [A*STAR Strategic Position, SiPRAD (IMCB-SERI Program in Retinal Angiogenic Diseases) to WTY & VAB]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
S.S.C. and V.A.B. designed the experiments; E.R. and W.T.Y. provided the materials; R.R.L., B.H.P., Y.S.W., B.B.T., C.D.L., R.A., A.G., A.M., R.R.M., V.A.B. and S.S.C. collected the data; S.S.C., R.R.L. and V.A.B. analysed and interpreted the data; and S.S.C., R.R.L. and V.A.B. wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chaurasia, S.S., Lim, R.R., Parikh, B.H. et al. The NLRP3 Inflammasome May Contribute to Pathologic Neovascularization in the Advanced Stages of Diabetic Retinopathy. Sci Rep 8, 2847 (2018). https://doi.org/10.1038/s41598-018-21198-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-21198-z
This article is cited by
-
Salidroside protects RGC from pyroptosis in diabetes-induced retinopathy associated with NLRP3, NFEZL2 and NGKB1, revealed by network pharmacology analysis and experimental validation
European Journal of Medical Research (2024)
-
PD protects Müller cells through the SIRT1/NLRP3 inflammasome pathway
International Ophthalmology (2024)
-
Research progress on the pathogenesis of diabetic retinopathy
BMC Ophthalmology (2023)
-
Diabetic retinopathy: a comprehensive update on in vivo, in vitro and ex vivo experimental models
BMC Ophthalmology (2023)
-
Effects of diabetes on microglial physiology: a systematic review of in vitro, preclinical and clinical studies
Journal of Neuroinflammation (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
