
Abstract
Bemisia tabaci whitefly species are some of the world’s most devastating agricultural pests and plant-virus disease vectors. Elucidation of the phylogenetic relationships in the group is the basis for understanding their evolution, biogeography, gene-functions and development of novel control technologies. We report here the discovery of five new Sub-Saharan Africa (SSA) B. tabaci putative species, using the partial mitochondrial cytochrome oxidase 1 gene: SSA9, SSA10, SSA11, SSA12 and SSA13. Two of them, SSA10 and SSA11 clustered with the New World species and shared 84.8‒86.5% sequence identities. SSA10 and SSA11 provide new evidence for a close evolutionary link between the Old and New World species. Re-analysis of the evolutionary history of B. tabaci species group indicates that the new African species (SSA10 and SSA11) diverged from the New World clade c. 25 million years ago. The new putative species enable us to: (i) re-evaluate current models of B. tabaci evolution, (ii) recognise increased diversity within this cryptic species group and (iii) re-estimate divergence dates in evolutionary time.
Similar content being viewed by others

Introduction
Bemisia tabaci species are phloem-feeding insects that damage a wide range of crops, including beans, cassava, cotton, cucurbits, potato, sunflower and tomato1,2. This group of cryptic species also vector more than 200 plant viruses3 that cause a wide range of plant diseases associated with devastating economic losses to many agricultural crops worldwide1,4,5. To date, 39 morphologically indistinguishable, but genetically diverse species have been reported and they differ greatly in their biological characteristics6,7,8,9 including: host-plant range10,11, inducement of phytotoxic disorders4,12, resistance to insecticides13,14, invasiveness4,15 and specificity of begomovirus transmission3,16. For example, estimated economic losses attributed to one putative species, the ‘Middle East-Asia Minor 1′ (MEAM1) alone are US$714 million annually17.
Bemisia tabaci species are present across the globe between 30 °N and 30 °S15. In Africa, the B. tabaci species Sub-Saharan Africa 1‒5 (SSA1‒5), Uganda 3 (herein named Sub-Saharan Africa 6), Africa (named Sub-Saharan Africa 7), East Africa 1, Mediterranean (MED), Middle-East Asia Minor 1 (MEAM1), and Indian Ocean (IO) colonise important agricultural crops such as cassava, beans, sweetpotato, as well as several weed species1,18,19,20,21,22. The B. tabaci species that colonise cassava, specifically SSA1 and SSA2 vector two economically important viruses: cassava mosaic begomoviruses (CMBs) and cassava brown streak ipomoviruses (CBSI)23,24. The two whitefly species have been associated with significantly high whitefly abundance; SSA2 in the 1990s1 and SSA1 more recently19,21.
Genetic diversity of the B. tabaci group has been studied previously using various molecular markers2,25,26 including 16 S rDNA25, mitochondrial cytochrome oxidase 1 (mtCO1)25 and ribosomal internal transcribed spacer 1 (ITS1) sequences27,28. The most commonly used marker is a partial fragment of the mtCO1 gene that has been used to establish the phylogeographic distribution of the group1,2,25,26. Amplification of the target mtCO1 region has generally used a primer set (MT10/C1-J-2195 and MT12/TL2-N-3014)29, but despite its widespread use, problems with this primer set for the amplification of some B. tabaci DNAs have been reported30.
The geographical origin and distribution of the different species within the B. tabaci group has been investigated2,25,26 and Sub-Saharan Africa was inferred to be the likely centre of origin26. Frohlich et al.25 analysed a representative collection of B. tabaci from all around the world and identified two main groups: Old World and New World. The Old World group was further separated into the Indian subcontinent, equatorial Africa and Sahel-region groups25. Subsequent studies showed a similar geographical distribution pattern of B. tabaci species1,28,31. For example, New World species were identified in the Americas4,32,33, while the Asian species (Asia I–IV, Japan, China 1‒3)7,34 and SSA1‒620,21,35 species were identified in Asia and Africa, respectively. Apart from MEAM1 and MED, B. tabaci species still occupy distinct geographical regions across the globe. The worldwide distribution of the MEAM1 and MED species has clearly resulted from recent introductions, through the movement of plant-material by humans, combined with the ability of these species to invade new regions and displace indigenous species4,15,31.
Since the report by Frohlich et al.25, additional B. tabaci genetic groups have been discovered in Sub-Saharan Africa that show the evolutionary importance of the region. These include a single specimen from Sudan (EU760727) that is probably a recent introduction from America, because it clusters within the New World clade (Supplementary Fig. S1), from Cameroon (EU760739; named Sub-Saharan Africa 7) that is ancestral to the Australia-Asia clade (Supplementary Fig. S1)22,36 and from Morocco (HE863764 and HE863760) that groups next to the Italy clade37. The single Sudanese specimen (EU760727) is not ancestral and so, prior to the discovery of the new species reported here, there was no strong evidence for an evolutionary link between African and New World species.
The discovery of a close link between African and New World B. tabaci requires a re-evaluation of the molecular dating evidence for the evolutionary divergence of B. tabaci. Previous analyses have suggested that the current bio-geographical distribution of B. tabaci species was due to the breakup of Gondwanaland and subsequent plate tectonic movements36. Western Gondwanaland was believed to have separated into South America and Africa from 120–84 mya38. About 95 ± 5 mya, Australia broke away from Antarctica, while India broke away from Madagascar and drifted north to collide with Asia38,39,40.
Evolutionary divergence dates for B. tabaci species remain controversial36,41, however, Campbell et al.42, Boykin et al.36 and Santos-Garcia et al.41 estimated, based on DNA sequences and fossil material43, that the genus Bemisia diverged from the other whiteflies approximately 90–87 million years ago (mya) when South America separated from Africa38,44. Boykin et al.36 estimated that the MEAM1 and MED species of the B. tabaci species complex diverged approximately 13 mya, while Santos-Garcia et al.41 estimated that MEAM1 and MED diverged approximately 2.9–0.4 mya. These estimates are inconsistent, but all agree that the evolutionary changes pre-date the advent of agriculture by millions of years. Here, we add the newly discovered species to published data and re-analyse the world-wide phylogenetic relationships and evolutionary history of the B. tabaci group of species.
Results
New primer design
Total DNAs prepared from some of the B. tabaci specimen collected in our study were not amplified with the primer pair that has been used most frequently for this purpose to generate B. tabaci mtCO1 partial sequences. This pair has a forward primer (MT10/C1-J-2195) designed to target insects in general, while the reverse primer (MT12/TL2-N-3014) targets gerrids, weevils, mosquitoes, flies and Lepidoptera29. To investigate why amplification was not occurring, these primer sequences were aligned with complete mtCO1 sequences of B. tabaci and B. afer present in Genbank. Figure 1 shows the alignment and reveals that the above primers have several mismatches. This necessitated the development of a new degenerate primer set (2195Bt and C012/Bt-sh2) with improved specificity for B. tabaci and B.afer (Figs 1 and 2). The new primer set (2195Bt and C012/Bt-sh2) amplified all the specimens, including those that failed with the old primer set (Fig. 2).

Alignment of partial mtCO1 gene sequences of four Bemisia tabaci species and Bemisia afer showing the positions of the new (2195Bt and C012/Bt-sh2) and old (MT10/C1-J-2195 and MT12/TL2-N-3014) primer sets.

A comparison of the performance of the old (a) [MT10/C1-J-2195:MT12/TL2-N-3014] - Lane 1, negative control (no DNA template); lane 2 and 3, positive (whitefly DNA) controls; lane 4–15, whiteflies collected from mint (lanes 4–7), sesame (lanes 8–11) and sunflower (lanes 12–15) and new (b) [2196Bt:CO12/Bt-sh2] primer set with successful amplification using the same order of specimens. M, 1Kb plus molecular weight marker (BIOLABS, UK).
To investigate the genetic diversity of B. tabaci species present in Uganda, adult whiteflies were collected from cassava and five weed species occurring commonly around cassava fields (Table 1 and Supplementary Dataset) in 26 districts (Fig. 3). The new degenerate primer set was used to generate a total of 121 partial (867 bp) mtCO1 sequences (GenBank accession numbers KX570749–KX570869) that were used for species determination against the reference B. tabaci species2,45.

The locations (red circles) in Uganda where whitefly specimens were collected during August–November 2013.
Phylogenetic analysis
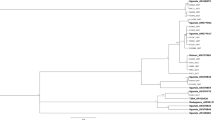
Based on the criterion that sequence divergence of more than 3.5‒4.0% indicates different B. tabaci species2,45, twelve B. tabaci species and one non-tabaci species were identified from the partial 121 mtCO1 sequences. Among them, five new previously undescribed putative species are reported for the first time here, which have been named Sub-Saharan Africa 9 to 13 (SSA9 – SSA13). Two of these putative new species, Sub-Saharan Africa 10 (SSA10) and Sub-Saharan Africa 11 (SSA11) clustered away from the other African species and next to species named New World 1 and New World 2 from the Americas (Fig. 4b), with which they shared 84.8‒86.5% sequence identity (Table 2). This provides new evidence for a close evolutionary link between African and American B. tabaci species, with the SSA10‒11 clade most closely related to the New World clade (Supplementary Fig. S1).

The newly identified putative species are shown in the MrBayes tree. Three sections (a–c) of the entire phylogenetic tree that are highlighted in grey are expanded in the sub-figures adjacent to them. The new putative species are highlighted in red (Sub-Saharan Africa 9, Sub-Saharan Africa 12, and Sub-Saharan Africa 13) and blue text (Sub-Saharan Africa 10 and Sub-Saharan Africa 11), respectively. The newly discovered link between African and New World B. tabaci are shown in sub-figure b. Reference sequences from GenBank used in the analysis appear in green text. Previously reported putative species also found during this study are highlighted in black text.
The global phylogeny of the 121 sequences, analysed with 570 reference sequences for known global B. tabaci species is shown in the Supplementary Figure S1. The topology showed that the B. Uganda1 species, together with B. atriplex and B. Japan2 were basal to the monophyletic group of B. tabaci species (Supplementary Fig. S1). B. Uganda1 was present on the five weed species, but not on cassava, confirming previous reports12,46,47 (Table 1) that cassava is not one of the hosts of this whitefly species. The Sub-Saharan Africa clade was basal to all other clades within the B. tabaci group of species (Supplementary Fig. S1) and was composed of SSA1, SSA2, SSA3, SSA4, SSA5, SSA6, SSA8 and SSA9 species. In our sample collection, SSA1, SSA2, SSA6 and SSA9 were identified.
In the previous literature, there have been two reports designating SSA5 to new putative B. tabaci species identified on cassava in Uganda and South Africa20,36. Our current study opted to rename the SSA5 (accession number: AM040598), identified by Boykin et al.36 on cassava in Uganda, as SSA8, while retaining the name SSA5 for the B. tabaci species identified on cassava and other weeds in South Africa by Esterhuizen et al.20 (accession number: JN104719). The species collected on wild mint previously called “Uganda 3”12 (accession number: AY903561) was named SSA6 here for consistency following the new naming system for B. tabaci species.
New mtCO1 sequences were obtained for previously recognised species in the SSA clade (SSA1, SSA2, SSA6), and these shared 98.4‒100% sequence identity with their closest relatives (AY903463, AY057173 and AY903561) in GenBank (Table 2). We identified SSA9, however, as a new species that clustered with SSA1–SSA6 in what has previously been referred to as the SSA clade2,48 (Fig. 4a) and it shared 92.8‒93.5% sequence identity with its closest relative, UgCsNm3 (AY903463) in GenBank (Table 2).
The Italy and Australia-Asia clades were basal to the New World and Africa-Middle East Asia Minor clades (Supplementary Fig. S1), the former being composed of Italy1, Morocco, SSA7 (from Cameroon), Japan 1, Chinese, Asian and Australian species, none of which were found in Uganda. Fourteen mtCO1 sequences obtained in this study clustered with sequences assigned previously to the New World clade. The New World clade was basal to the Africa-Middle East-Asia Minor clade (Supplementary Fig. S1), the latter being composed of IO, MED, MEAM1, MEAM2, SSA12 and SSA13 species, which were all found in Uganda. The IO, MED, MEAM1, MEAM2 sequences obtained from our samples shared 96.5–99.9% sequence identity with their closest relatives in GenBank (Table 2). The putative new species SSA12 and SSA13, however, shared only 93.9–94.7% sequence identity with their closest relatives in GenBank (Table 2). SSA12 and SSA13 grouped next to each other and were in the Africa-Middle East-Asia Minor clade (Fig. 4c).
Estimated global divergence of B. tabaci species
To test whether or not the discovery of the new species altered our view of the evolutionary history of B. tabaci, the global partial mtCO1 data-set was re-analysed together with the sequences generated in this study. We considered that the discovery of SSA10 and SSA11 and their close link to the New World species could provide a good test of the hypothesis that the separation of South America from Africa, which occurred between 120 to 84 mya38, was responsible for this evolutionary split. For this hypothesis to be correct, we would predict that divergence of Old World from the New World B. tabaci species should occur over a similar time-frame.
Our molecular clock analysis showed that B. tabaci diverged from the rest of Bemisia species c. 40 mya. (Fig. 5), where it split into two major groups: (i) the SSA clade and (ii) Africa-Middle East-Asia Minor, New World, Italy and Australia-Asia clades (Fig. 5). Basal to the split of B. tabaci species was the SSA clade whose members split into two groups about 15 mya. The first group composed of the SSA2, SSA3 and SSA9 species split c. 10 mya and diversified c. 8‒1 mya, while the second group was composed of the SSA1, SSA4, SSA5, SSA6, and SSA8 species split c. 12 mya and diversification occurred 6–1 mya.

Time-calibrated phylogenetic tree of B. tabaci based on partial mtCO1 sequences. Divergence estimates expressed in million years ago (mya) are shown above the branches with 95% confidence intervals (red bars).
The Africa-Middle East-Asia Minor clade (composed of IO, MED, MEAM1, MEAM2, SSA12 and SSA13) split from the New World clade (composed of New World 1, New World 2, SSA10 and SSA11 species) c. 29 mya. About 25 mya, the SSA10 and SSA11 species split from the New World species and diversified about 2‒1 mya. Diversification within the New World species occurred c. 7–1 mya. About 13 mya, the IO species split from MED, MEAM1/MEAM2, SSA12 and SSA13 species. Diversification within the IO species occurred c. 2 mya. The SSA12 and SSA13 species split from MED, MEAM1 and MEAM2 species c. 10 mya. Diversification within SSA12 and SSA13 species occurred 2 mya and for the MED, MEAM1 and MEAM2 species, c. 6–5 mya.
Approximately 35 mya, the Italy and Australia-Asia clade split from the Africa-Middle East-Asia Minor and New World clades. The Italy clade (Italy 1 and Morocco) then diverged from the Australia-Asia clade (SSA7, Japan1, China, Australia and Asian species) about 30 mya and diversified about 1 mya. Within the Australia-Asia clade, the SSA7 split from Japan1, China, Australia and Asian species at c. 27 mya. The AsiaII split from Australia, Asia1, AsiaIII, Asia IV, China and Japan 1 species c. 26 mya and the former species diversified 18–1 mya. Diversification within the AsiaII occurred 15–4 mya.
Discussion
This study is the first to report mtCO1 sequences from B. tabaci species in the Old World (Africa) clustering close to the New World species and thus provides the first strong evidence for a close evolutionary link between the Old (African) and New World species. The development of a new primer set for amplification of the mtCO1 ‘barcode’ of B. tabaci species enabled 12 putative B. tabaci species to be identified from the Ugandan whitefly samples, of which five were new including the two (SSA10 and SSA11) most closely related to the New World species (Fig. 4).
Gueguen et al.22 reported a single specimen from the Sudan (EU760727) that was apparently a New World species (according to the 3.5% mtCO1 divergence criterion), most probably a recent introduction from the New World into Africa by human trade. The mtCO1 sequence of the Sudanese sample is quite different from the SSA10 and SSA11 sequences that were amplified from samples collected from multiple locations in our study (Table 1).
In addition to the SSA10 and SSA11 species, 10 other putative species of B. tabaci and a non-tabaci species were identified amongst the whitefly samples collected from weeds. Of the 10, three were new putative species and were named: SSA9, SSA12 and SSA13. The three new putative species, SSA9 and SSA12 and SSA13, grouped with the SSA and Africa-Middle East-Asia Minor higher-level genetic groups (11% level or greater mtCO1 sequence divergence), respectively, identified by Dinsdale et al.2. The rest of the seven species (IO, MED, MEAM1, MEAM2, SSA1, SSA2 and SSA6) were reported previously in Uganda12,19,21 and elsewhere49. This high genetic diversity present in the study area further supports SSA and equatorial Africa, in particular, as the centre of origin of B. tabaci26,50.
MEAM2 B. tabaci species identified in this study shared 97.1% sequence similarity with the closest published partial mtCO1 DNA of a whitefly individual from Reunion (AJ550177). Similar to our study, previous studies carried out in Reunion51, Japan49 and Turkey52 identified this whitefly as a distinct species within the B. tabaci species complex. However, analysis of mitogenomes of three Peruvian individuals expected to be MEAM2 based on the Sanger sequence derived partial mtCO1 DNA revealed that they were MEAM153. The misidentification of MEAM2 sequences based on the partial mtCO1 DNA was attributed to the amplification of nuclear mitochondrial DNA (NUMTS) or PCR artefacts such as DNA polymerase-introduced errors. Although partial mtCO1 sequences of MEAM2 species are reported in this study, a full mitogenome of this species has been generated from a single whitefly collected in Uganda. This confirms that the MEAM2 species generated in this study and currently occurring in Uganda is genuine and was not as result of NUMTS or PCR artefacts as reported by Tay et al.53.
Bemisia Uganda1 was also found by our study to occur in Uganda and this has been previously included in the B. tabaci group12,54. Our new analysis, however, shows that B. Uganda1 and Bemisia Japan2 (AB308110)7 clearly group outside the B. tabaci complex (Supplementary Fig. S1), although the adults of the former are morphologically indistinguishable (H. Mugerwa, personal observation). The mtCO1 marker provides only limited phylogenetic information and so further genetic information, such as that provided by single-copy nuclear genes, is required to conclude with more certainty whether or not Bemisia Uganda1 is a member of the B. tabaci species complex.
The estimated date of divergence of SSA10 and SSA11 species from the New World clade was c. 25 mya (Fig. 5) and the separation of South America from Africa occurred over a prolonged period of 120–84 mya38. Our new data and analyses, therefore are clearly inconsistent with the hypothesis that divergence of the New and Old World species corresponded to the geological separation of South America from Africa. In addition, New World 1 is ancestral to New World 2 and this could be explained by the invasion of New World 1 into South America when the two land masses became joined c. 3 mya55,56. This hypothesis can only be tested more rigorously when, and if, more whitefly fossils, molecular data and further key species that cluster within the B. tabaci group are discovered.
As an alternative hypothesis, our estimated date of divergence of SSA10 and SSA11 species from the New World species corresponds with the most plausible date for the Old World/New World split in begomoviruses, which was estimated to be c. 20‒30 mya50. The presence of the Beringian land bridge connecting Asia and Western North America and a warm temperate climate between c. 65‒35 mya, enabled considerable exchange of terrestrial fauna and flora57 between these land masses. Movement of early whitefly-transmitted begomoviruses between Asia and North America via the Beringian land bridge would therefore have been possible up to c. 35 mya50. Our new data and the intimate relationship between begomoviruses and their B. tabaci vector species, therefore, strongly support the hypothesis that invasion of the New World occurred into North America through the Beringian land bridge and subsequently into Latin America. In addition, the SSA and Africa-Middle East-Asia Minor clade diverged from Italy and Australia-Asia clades between c. 35 mya. The warm temperate climate across the globe during that period would have enabled further movement and speciation amongst the B. tabaci major clades.
Our new estimates of when the members of the B. tabaci species diverged also differed from those of other researchers. Santos-Garcia et al.41, for example, reported that diversification within MEAM1 and MED species took place 0.63–0.16 mya (estimated using runAB and BEAST2) and 2.88–0.44 mya (estimated using PhyloBayes3). Boykin et al.36 estimated diversification of MEAM1 and MED species at about 13 mya. We estimate the diversification of MEAM1 and MED occurred c. 6–5 mya. Different outgroup calibration dates ranging from 263‒125 mya and full (1,341 bp) vs partial (657 bp) mtCO1 gene sequences were used in these different analyses. We conclude that more precise divergence estimates for B. tabaci species are only likely to be attained when additional fossil specimens within Bemisia are discovered and used to set accurate calibration points in geological time. We also consider that nuclear markers shall also be required, because they may reveal different evolutionary histories to the mitochondrial genome58.
Sub-Saharan Africa 7 that links Asia to Africa22, the Morocco species that links Italy to Africa37, as well as SSA10 and SSA11 that link the New World to Africa, all provide further evidence that members of the B. tabaci complex have a common ancestor that originated in Sub-Saharan Africa. Since SSA7 is represented by only one sequence in GenBank, we suggest that further collections be made in the region where SSA7 was found, to strengthen the weight of evidence for this theory.
The majority of studies on East African cassava whiteflies in the previous decade have focussed mainly on cassava and the two known B. tabaci species, SSA1 and SSA2 that colonise it1,12,19,21,35. The new species reported here were not found in these earlier studies and this is particularly surprising for SSA10, because its preferred host-plant appears to be cassava (Fig. 3b) and it was detected in six districts in our study. The most probable reason for this is that the primer set used by whitefly researchers globally has generally been the MT10/C1-J-2195/MT12/TL2-N-3014 pair. Figure 1 shows that there are six annealing mismatches in the MT12/TL2-N-3014 primer and the B. tabaci mitogenome sequences obtained by independent means59,60,61,62. Given the clear problems with the MT10/C1-J-2195/MT12/TL2-N-3014 primer set, it is surprising that they have been used so widely in the past. The modifications described here, however, will help reveal the true complexity and diversity within the B. tabaci groups of species.
The 2195Bt and C012/Bt-sh2 primer set designed in this study produced PCR amplicons of the same size as the MT10/C1-J-2195 and MT12/TL2-N-3014 primer set because both primer sets were designed from the same positions (Fig. 1). In contrast to Shatters et al.30 study, a modified primer set designed efficiently amplified partial mtCO1 gene for Bemisia and some related Aleyrodidae, however, it produced relatively short PCR amplicons (~748 bp) compared to the MT10/C1-J-2195 and MT12/TL2-N-3014 primer set. The relatively short PCR amplicon was due to Shatters et al.30 forward primer (Btab-uni-PrimerR) designed 52 base pairs downstream from the MT10/C1-J-2195 forward primer. In an era when more than 99% of species that ever lived on earth are now extinct63,64, our study reports the discovery of five new species within the B. tabaci species complex and suggests that with more sampling in Sub-Saharan Africa and the use of more efficient primers, it is probable that many more species will be discovered. In addition to the partial mtCO1 marker used to identify new putative species in this study, we recommend that biological studies such as mating crosses be carried out between the new identified putative species and other SSA species. These findings (new putative species) also require us to re-evaluate current models and the time-frame of whitefly evolution. As low-cost genome sequencing becomes increasingly available, the proposed model for evolution should be expanded and validated by including a range of phylogenetically informative nuclear genes. Comparative genomic studies will then become a resource to catalyse the development of novel tools and technologies to manage these economically devastating pest species.
Materials and Methods
Whitefly field collections
Adult whiteflies were collected from cassava (Manihot esculenta Crantz) (n = 9) and five weed species: Wandering Jew (Commelina benghalensis L.) (n = 37), Muwugula (Pavonia urens Cav.) (n = 27), Mexican fire plant (Euphorbia heterophylla L.) (n = 16), Joobyo (Cleome gynandra L.) (n = 14), Mululuza (Vernonia amygdalina Delie) (n = 18), which occur frequently within and/or the surroundings of cassava fields across 26 districts in Uganda between July and August 2013 (Table 1 and Supplementary Dataset). Collections were made using an aspirator and the whitefly samples stored in 1.5-ml Eppendorf tubes containing 90% ethanol. Samples from the same host plant species at each site were stored in the same Eppendorf tube. For each collection site, the geo-coordinates (latitude and longitude) were recorded using a Geographical Positioning System (GPS, Garmin eTrex Vista Cx) together with the village and district name. The samples were maintained at room temperature during field collection in Uganda, but stored at −20 °C upon arrival at Natural Resources Institute (NRI), UK until molecular analysis.
Generation of a map
The geo-coordinates for each collection site were used to generate a map (Fig. 3) using the ArcMap version 10.2.2 software program (http://www.arcgis.com/features/).
Global B. tabaci samples and outgroups
The data set of B. tabaci and outgroup sequences representing global diversity was obtained from GenBank. Five hundred seventy individual 657 bp mtCO1 sequences’ 3′-end region were selected and used in the analysis. Published host data were obtained for each sample. Outgroup taxa were selected based on their morphological similarities to the fossil Aleurodicus burmiticus43,65. Of the selected 570 sequences, 31 individual outgroup sequences representing 19 other whitefly species were used in the analysis. Uganda and Japan2 species grouped most closely to B. emiliae and B. atriplex. To avoid the confusion caused by their unusual names, we henceforth refer to Uganda and Japan2 species as, Bemisia Uganda1 and Bemisia Japan2, respectively.
Whitefly DNA extraction
Three adult whiteflies were selected randomly from those collect for each sampled plant species and location. Genomic DNA was extracted from single whiteflies by crushing each in 50 µl of 10% (w/v) Chelex 100 sodium form solution (Sigma Aldrich, St Louis, USA) in a 1.5-ml Eppendorf tube using a sterile plastic rod66. The extracts were incubated for 20 min at 56 °C and then for 5 min at 100 °C. Subsequently, extracts were centrifuged for 5 min at 15,900 g in a 5424 R Eppendorf centrifuge (Eppendorf UK Limited) and placed immediately on ice prior to proceeding to using the individual whitefly extracts as templates for PCR amplification.
Mitochondrial DNA amplification and sequencing
Amplification of a partial fragment (867 bp) of the mtCO1 gene was performed using the forward primer MT10/C1-J-2195 (5′-TTGATTTTTTGGTCATCCAGAAGT-3′) in combination with a reverse primer MT12/TL2-N-3014 (5′-TCCAATGCACTAATCTGCCATATTA-3′)29. The PCR reaction mixture (20 µl) contained 10 µl of 2× reSource™ Taq Mix (Source BioSciences, UK), 1 µl of each primer stock (10 µM), 6 µl of molecular biology grade water (Sigma Aldrich, St Louis, USA) and 2 µl of DNA template. Initial denaturation of template DNA was conducted at 94 °C for 2 min followed by 35 cycles of denaturation at 94 °C for 20 s, primer annealing at 52 °C for 30 s and extension at 72 °C for 1 min. The final extension of 10 min was run at 72 °C and the reaction held at 4 °C in a 2720 Applied Biosystems thermal cycler (Applied Biosystems, UK). Electrophoresis of PCR products was performed on 2% (w/v) agarose gels in 0.5× TBE buffer stained using RedSafe™ according to manufacturer’s guidelines (iNtRON BIOTECHNOLOGY, Korea). PCR products were visualised under UV light (302 nm) and those of the expected (~867 bp) size were purified for sequencing and DNA cloning using a reSource™ PCR purification multipack kit (Source BioScience, UK) as per the manufacturer’s procedure. Purified PCR products were cloned in three replicates using the pGEM®-T easy vector kit (Promega, UK) as per the manufacturer’s instructions. Purified and cloned PCR-amplified products were sent for sequencing at Source BioScience, UK.
Modified mtCO1 primers
Due to mismatches observed in the MT10/C1-J-2195 and MT12/TL2-N-3014 primer pair to B. tabaci mitogenome sequences (Asia I: KJ778614, MED: JQ906700, New World I: AY521259 and B. afer: KF734668), a modified primer set that most matches the B. tabaci sequences was designed (Fig. 1). This was done through multiple sequence alignment of mtCO1 gene sequences (nucleotide positions: ~600 to 1700) available in GenBank for complete mitogenomes and unpublished sequences of various species of the B. tabaci complex (Asia I, New World, MED and SSA1) and B. afer using MAFFT v7.27367. The alignment block was edited and illustrated using TEXshade program68. Regions where the MT10/C1-J-2195 and MT12/TL2-N-3014 primer pair aligned were identified (Fig. 1). The modified primer set: 2195Bt (5′-TGRTTTTTTGGTCATCCRGAAGT-3′) and C012/Bt-sh2 (5′-TTTACTGCACTTTCTGCC-3′) was designed to anneal to the same region as the MT10/C1-J-2195 and MT12/TL2-N-3014 primer pair to ensure compatibility of sequences for phylogenetic analyses (Fig. 1). The PCR reaction mixture and conditions of the new primer set were the same as for the MT10 & MT12 primer set.
Phylogenetic analysis
Whitefly mtCO1 sequences were aligned together with reference whitefly sequences obtained from GenBank in Geneious v 9.1 with the MUSCLE69 alignment option set to 50 iterations, then visually inspected and manually adjusted where necessary. The final alignment was translated to ensure sequences were aligned within the correct reading frame.
The model of molecular evolution was determined using Modeltest 3.670. MrBayes 3.2.271 was used to partition data based on codon position in the alignment. Codon positions 1 and 2 were treated with the following commands in MrBayes: lsetapplyto = (1,2) nst = 2 rates = gamma and the third position: lsetapplyto = (3) nst = 6 rates = gamma. MrBayes 3.1.2 was run for 50 million generations and trees were sampled every 1000 generations. All runs reached a plateau in likelihood score (i.e. stationarity), which was indicated by the standard deviation of split frequencies (0.0015), and the potential scale reduction factor (PSRF) was close to one, indicating the MCMC chains converged. Convergence of the runs was also checked using Tracer v1.6 and the effective sample size (ESS) values were well above 200 for each run. Twelve thousand five hundred trees were suboptimal at the beginning of the runs and were therefore discarded.
Divergence estimates
BEAUti v1.8.272 was used to generate the xml file for the multiple BEAST runs. Four independent runs of BEAST were conducted following the model sets Boykin et al.36. The tree height prior was set to a normal distribution, with an initial value of 123, to represent the age of the fossil 130 mya43 with a standard deviation of 20. The MCMC were run for 50 million generations and sampled every 1000th generation. Convergence of the multiple runs was checked using Tracer v1.6.0 and the ESS values were well above 200 for each run. Two independent BEAST runs were completed and the two tree files were combined using Logcombiner. The trimmed output was combined to yield posterior estimates of mean.rate, coefficient variation in rates, ucld.mena, ucld.stdev and the mean and 95% highest posterior density. TreeAnnotator was used to generate a final tree, which was viewed in FigTree v1.4.2.
References
Legg, J. P., French, R., Rogan, D., Okao-Okuja, G. & Brown, J. K. A distinct Bemisia tabaci (Gennadius) (Hemiptera: Sternorrhyncha: Aleyrodidae) genotype cluster is associated with the epidemic of severe cassava mosaic virus disease in Uganda. Mol. Ecol. 11, 1219–1229 (2002).
Dinsdale, A., Cook, L., Riginos, C., Buckley, Y. M. & De Barro, P. Refined global analysis of Bemisia tabaci (Hemiptera: Sternorrhyncha: Aleyrodoidea: Aleyrodidae) mitochondrial cytochrome oxidase 1 to identify species level genetic boundaries. Ann. Entomol. Soc. Am. 103, 196–208 (2010).
Polston, J. E., De Barro, P. & Boykin, L. M. Transmission specificities of plant viruses with the newly identified species of the Bemisia tabaci species complex. Pest Manag. Sci. 70, 1547–1552 (2014).
Brown, J. K., Frohlich, D. R. & Rosell, R. C. The sweetpotato or silverleaf whiteflies: biotypes of Bemisia tabaci or species complex? Annu. Rev. Entomol. 40, 511–534 (1995).
Seal, S. E., VandenBosch, F. & Jeger, M. J. Factors influencing begomovirus evolution and their increasing global significance: implications for sustainable control. CRC. Crit. Rev. Plant Sci. 25, 23–46 (2006).
Liu, J., Zhao, H., Jiang, K., Zhou, X.-P. & Liu, S.-S. Differential indirect effects of two plant viruses on an invasive and an indigenous whitefly vector: implications for competitive displacement. Ann. Appl. Biol. 155, 439–448 (2009).
Firdaus, S. et al. The Bemisia tabaci species complex: additions from different parts of the world. Insect Sci. 20, 723–733 (2013).
Qin, L., Pan, L.-L. & Liu, S.-S. Further insight into reproductive incompatibility between putative cryptic species of the Bemisia tabaci whitefly complex. Insect Sci. 23, 215–224 (2016).
Alemandri, V. et al. Three members of the Bemisia tabaci (Hemiptera: Aleyrodidae) cryptic species complex occur sympatrically in Argentine horticultural crops. J. Econ. Entomol. 108, 405–413 (2015).
Zang, L.-S., Chen, W.-Q. & Liu, S.-S. Comparison of performance on different host plants between the B biotype and a non-B biotype of Bemisia tabaci from Zhejiang, China. Entomol. Exp. Appl. 121, 221–227 (2006).
Xu, J., Lin, K. K. & Liu, S. S. Performance on different host plants of an alien and an indigenous Bemisia tabaci from China. J. Appl. Entomol. 135, 771–779 (2011).
Sseruwagi, P. et al. Genetic diversity of Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae) populations and presence of the B biotype and a non-B biotype that can induce silverleaf symptoms in squash, in Uganda. Ann. Appl. Biol. 147, 253–265 (2005).
Wang, X.-W. et al. Analysis of a native whitefly transcriptome and its sequence divergence with two invasive whitefly species. BMC Genomics 13, 529 (2012).
Horowitz, A. R. & Ishaaya, I. Dynamics of biotypes B and Q of the whitefly Bemisia tabaci and its impact on insecticide resistance. Pest Manag. Sci. 70, 1568–1572 (2014).
Liu, S.-S. et al. Asymmetric mating interactions drive widespread invasion and displacement in a whitefly. Science (80-.). 318, 1769–1772 (2007).
Maruthi, M. N., Colvin, J., Seal, S., Gibson, G. & Cooper, J. Co-adaptation between cassava mosaic geminiviruses and their local vector populations. Virus Res. 86, 71–85 (2002).
Mascarin, G. M., Kobori, N. N., Quintela, E. D. & Delalibera, I. The virulence of entomopathogenic fungi against Bemisia tabaci biotype B (Hemiptera: Aleyrodidae) and their conidial production using solid substrate fermentation. Biol. Control 66, 209–218 (2013).
Berry, S. D. et al. Molecular evidence for five distinct Bemisia tabaci (Homoptera: Aleyrodidae) geographic haplotypes associated with cassava plants in sub-SaharanAfrica. Systematics 97, 852–859 (2004).
Mugerwa, H. et al. Genetic diversity and geographic distribution of Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae) genotypes associated with cassava in EastAfrica. Ecol. Evol 2, 2749–2762 (2012).
Esterhuizen, L. L. et al. Genetic identification of members of the Bemisia tabaci cryptic species complex from South Africa reveals native and introduced haplotypes. J. Appl. Entomol. 137, 1–14 (2013).
Legg, J. P. et al. Spatio-temporal patterns of genetic change amongst populations of cassava Bemisia tabaci whiteflies driving virus pandemics in East and Central Africa. Virus Res. 186, 61–75 (2014).
Gueguen, G. et al. Endosymbiont metacommunities, mtDNA diversity and the evolution of the Bemisia tabaci (Hemiptera: Aleyrodidae) species complex. Mol. Ecol. 19, 4365–4378 (2010).
Storey, H. H. & Nichols, R. F. W. Studies of the mosaic disease of cassava. Ann. Appl. Biol. 25, 790–806 (1938).
Maruthi, M. N. et al. Transmission of Cassava brown streak virus by Bemisia tabaci (Gennadius). J. Phytopathol. 312, 307–312 (2005).
Frohlich, D. R., Torres-Jerez, I. D., Bedford, D., Markham, G. P. & Brown, J. K. A phylogeographical analysis of the Bemisia tabaci species complex based on mitochondrial DNA markers. Mol. Ecol. 8, 1683–1691 (1999).
Boykin, L. M. et al. Global relationships of Bemisia tabaci (Hemiptera: Aleyrodidae) revealed using Bayesian analysis of mitochondrial COI DNA sequences. Mol. Phylogenet. Evol. 44, 1306–1319 (2007).
De Barro, P. J., Driver, F., Trueman, J. W. & Curran, J. Phylogenetic relationships of world populations of Bemisia tabaci (Gennadius) using ribosomal ITS1. Mol. Phylogenet. Evol. 16, 29–36 (2000).
De Barro, P. J. Genetic structure of the whitefly Bemisia tabaci in the Asia-Pacific region revealed using microsatellite markers. Mol. Ecol. 14, 3695–3718 (2005).
Simon, C. et al. Evolution,weighting and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 87, 651–701 (1994).
Shatters, R. G., Powell, C. A., Boykin, L. M., Liansheng, H. E. & Kenzie, C. L. M. C. Improved DNA barcoding method for Bemisia tabaci and related Aleyrodidae: development of universal Bemisia tabaci biotype-specific mitochondrial cytochrome c oxidase I polymerase chain reaction primers. Mol. Entomol. 102, 750–758 (2009).
McKenzie, C. L., Anderson, P. K. & Villarreal, N. An extensive survey of Bemisia tabaci (Homoptera: Aleyrodidae) in agricultural ecosystems in Florida. Florida Entomol. 87, 403–407 (2004).
Marubayashi, J. M. et al. At least two indigenous species of the Bemisia tabaci complex are present in Brazil. J. Appl. Entomol. 137, 113–121 (2013).
Barbosa, L. et al. Indigenous American species of the Bemisia tabaci complex are still widespread in the Americas. Pest Manag. Sci. 70, 1440–1445 (2014).
Ellango, R. et al. Distribution of Bemisia tabaci genetic groups in India. Environ. Entomol. 44, 1258–1264 (2015).
Ghosh, S., Bouvaine, S. & Maruthi, M. Prevalence and genetic diversity of endosymbiotic bacteria infecting cassava whiteflies in Africa. BMC Microbiol. 15, 1–17 (2015).
Boykin, L. M., Bell, C. D., Evans, G., Small, I. & De Barro, P. J. Is agriculture driving the diversification of the Bemisia tabaci species complex (Hemiptera: Sternorrhyncha: Aleyrodidae)?: dating, diversification and biogeographic evidence revealed. BMC Evol. Biol. 13, 228 (2013).
Tahiri, A., Halkett, F., Granier, M., Gueguen, G. & Peterschmitt, M. Evidence of gene flow between sympatric populations of the Middle East-Asia Minor 1 and Mediterranean putative species of Bemisia tabaci. Ecol. Evol. 3, 2619–2633 (2013).
Somoza, R. & Zaffarana, C. B. Mid-Cretaceous polar standstill of South America, motion of the Atlantic hotspots and the birth of the Andean cordillera. Earth Planet. Sci. Lett. 271, 267–277 (2008).
Veevers, J. J. Break of Australia and Antractica estimated as midcretaceous (95+/−5ma) from magnetic and seismic data at the continental margin. Earth Planet. Sci. Lett. 77, 91–99 (1986).
Torsvik, T. H. et al. Late Cretaceous India-Madagascar fit and timing of break-up related magmatism. Terra Nov. 12, 220–224 (2000).
Santos-Garcia, D., Vargas-Chavez, C., Moya, A., Latorre, A. & Silva, F. J. Genome evolution in the primary endosymbiont of whiteflies sheds light on their divergence. Genome Biol. Evol. 7, 873–888 (2015).
Campbell, B. C., Steffen-Campbell, J. D. & Gill, R. J. Evolutionary origin of whiteflies (Hemiptera: Sternorrhyncha: Aleyrodidae) inferred from 18S rDNA sequences. Insect Mol. Biol. 3, 73–88 (1994).
Drohojowska, J. & Szwedo, J. A new whitefly from lower Cretaceous Lebanese amber (Hemiptera: Sternorrhyncha: Aleyrodidae). Insect Syst. Evol. 42, 179–196 (2011).
Pletsch, T. et al. Cretaceous separation of Africa and South America: the view from the West African margin. J. South Am. Earth Sci. 14, 147–174 (2001).
Lee, W., Park, J., Lee, G.-S., Lee, S. & Akimoto, S. Taxonomic status of the Bemisia tabaci complex (Hemiptera: Aleyrodidae) and reassessment of the number of its constituent species. PLoS One 8, e63817 (2013).
Legg, J. P. Emergence, spread and strategies for controlling the pandemic of cassava mosaic virus disease in East and Central Africa. Crop Prot. 18, 627–637 (1999).
Maruthi, M. N., Colvin, J. & Seal, S. Mating compatibility, life-history traits, and RAPD-PCR variation in Bemisia tabaci associated with the cassava mosaic disease pandemic in East Africa. Entomol. Exp. Appl. 99, 13–23 (2001).
De Barro, P. J., Liu, S.-S., Boykin, L. M. & Dinsdale, A. B. Bemisia tabaci: a statement of species status. Annu. Rev. Entomol. 56, 1–19 (2011).
Ueda, S., Kitamura, T., Kijima, K., Honda, K. I. & Kanmiya, K. Distribution and molecular characterization of distinct Asian populations of Bemisia tabaci (Hemiptera: Aleyrodidae) in Japan. J. Appl. Entomol. 133, 355–366 (2009).
Lefeuvre, P. et al. Evolutionary time-scale of the begomoviruses: evidence from integrated sequences in the Nicotiana genome. PLoS One 6, e19193 (2011).
Delatte, H. et al. A new silverleaf-inducing biotype Ms of Bemisia tabaci (Hemiptera: Aleyrodidae) indigenous to the islands of the south-west Indian Ocean. Bull. Entomol. Res. 95, 29–35 (2005).
Karut, K., Kaydan, M. B., Tok, B., Döker, İ. & Kazak, C. A new record for Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae) species complex of Turkey. J. Appl. Entomol. 139, 158–160 (2015).
Tay, W. et al. The trouble with MEAM2: Implications of pseudogenes on species delimitation in the globally invasive Bemisia tabaci (Hemiptera: Aleyrodidae) cryptic species complex. Genome Biol. Evol. 9, 2732–2738 (2017).
Maruthi, M. N. et al. Reproductive incompatibility and cytochrome oxidase I gene sequence variability amongst host-adapted and geographically separate Bemisia tabaci populations (Hemiptera: Aleyrodidae). Syst. Entomol. 29, 560–568 (2004).
Saito, T. Geologic significance of coiling direction in the planktonic foraminifera Pulleniatina. Geology 4, 305 (1976).
Keigwin, L. D. Pliocene closing of the Isthmus of Panama, based on biostratigraphic evidence from nearby Pacific ocean and Caribbean sea cores. Geology 6, 630–634 (1978).
Sanmartin, I., Enghoff, H. & Ronquist, F. Patterns of animal dispersal, vicariance and diversification in the Holarctic. Biol. J. Linn. Soc. 73, 345–390 (2001).
Hsieh, C. H., Ko, C. C., Chung, C. H. & Wang, H. Y. Multilocus approach to clarify species status and the divergence history of the Bemisia tabaci (Hemiptera: Aleyrodidae) species complex. Mol. Phylogenet. Evol. 76, 172–180 (2014).
Thao, M. & Baumann, P. Evolutionary relationships of primary prokaryotic endosymbionts of whiteflies and their hosts. Appl. Environ. Microbiol. 70, 3401–3406 (2004).
Wang, H.-L. et al. The characteristics and expression profiles of the mitochondrial genome for the Mediterranean species of the Bemisia tabaci complex. BMC Genomics 14, 401 (2013).
Tay, W. T., Elfekih, S., Court, L., Gordon, K. H. & De Barro, P. J. Complete mitochondrial DNA genome of Bemisia tabaci cryptic pest species complex Asia I (Hemiptera: Aleyrodidae). Mitochondrial. DNA 27, 972–973 (2016).
Wang, H.-L. et al. The complete mitochondrial genome of Bemisia afer (Hemiptera: Aleyrodidae). Mitochondrial. DNA 27, 98–99 (2016).
Stearns, B. P. & Stearns, C. S. Watching from the edge of extinction. (Yale University Press, 1999).
Stearns, B. P. & Viers, S. C. S. Still watching from the edge of extinction. Bioscience 60, 141–146 (2010).
Drohojowska, J. & Szwedo, J. Early Cretaceous Aleyrodidae (Hemiptera: Sternorrhyncha) from the Lebanese amber. Cretac. Res. 52, 368–389 (2015).
Walsh, P. S., Metzger, D. A. & Higuchi, R. Chelex-100 as a meidum for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques 10, 506–513 (1991).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Beitz, E. T(E)Xshade: shading and labeling of multiple sequence alignments using (LTEX)-T-A 2(epsilon). Bioinformatics 16, 135–139 (2000).
Edgar, R. C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 113 (2004).
Posada, D. & Crandall, K. A. MODELTEST: testing the model of DNA substitution. Bioinformatics 14, 817–818 (1998).
Ronquist, F. & Huelsenbeck, J. P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 (2003).
Drummond, A. J. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214 (2007).
Acknowledgements
This work was funded by grants from the Bill & Melinda Gates Foundation: Enabling Research Tools for Cassava Virologists and Breeders (OPP1080766), Disease Diagnostics for Sustainable Cassava Productivity in Africa Phase II (OPP1052391) and the African Cassava Whitefly Project (OPP1058938). We would like to thank farmers in Uganda for allowing us to collect whitefly from their fields. Computational resources provided by the Pawsey Supercomputing Centre with funding from the Australian Government and the Government of Western Australia are well appreciated. We are particularly grateful to Jim C. Weale and Colin Hanbury who guided H.M. in developing his scientific writing skills.
Author information
Authors and Affiliations
Contributions
J.C., S.S., C.A.O., T.A., F.T., J.N. and P.S. designed the study. H.M. and R.K. collected material, DNA extraction and amplification for sequencing. S.S., M.P. and H.M. designed and tested the new primer set. H.M., J.C., S.S., H.W. and P.S. analysed the data. H.M., S.S., P.S. and J.C. wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mugerwa, H., Seal, S., Wang, HL. et al. African ancestry of New World, Bemisia tabaci-whitefly species. Sci Rep 8, 2734 (2018). https://doi.org/10.1038/s41598-018-20956-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-20956-3
This article is cited by
-
Composition of the whiteflies microbiome in populations with and without insecticide applications in Yucatan Mexico
Biologia (2024)
-
Is polyphagy of a specific cryptic Bemisia tabaci species driving the high whitefly populations on cassava in eastern Africa?
Journal of Pest Science (2024)
-
Genotyping of Bemisia tabaci (Hemiptera: Aleyrodidae) affirmed a new record of Asia II 7, China 3 and dominance of Asia I cryptic species in Bihar, India
International Journal of Tropical Insect Science (2023)
-
Comparative evolutionary analyses of eight whitefly Bemisia tabaci sensu lato genomes: cryptic species, agricultural pests and plant-virus vectors
BMC Genomics (2023)
-
Transcriptional analysis of Bemisia tabaci MEAM1 cryptic species under the selection pressure of neonicotinoids imidacloprid, acetamiprid and thiamethoxam
BMC Genomics (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
