
Abstract
The arthropod-transmitted chikungunya virus (CHIKV) causes a flu-like disease that is characterized by incapacitating arthralgia. The re-emergence of CHIKV and the continual risk of new epidemics have reignited research in CHIKV pathogenesis. Virus-specific antibodies have been shown to control virus clearance, but antibodies present at sub-neutralizing concentrations can also augment virus infection that exacerbates disease severity. To explore this occurrence, CHIKV infection was investigated in the presence of CHIKV-specific antibodies in both primary human cells and a murine macrophage cell line, RAW264.7. Enhanced attachment of CHIKV to the primary human monocytes and B cells was observed while increased viral replication was detected in RAW264.7 cells. Blocking of specific Fc receptors (FcγRs) led to the abrogation of these observations. Furthermore, experimental infection in adult mice showed that animals had higher viral RNA loads and endured more severe joint inflammation in the presence of sub-neutralizing concentrations of CHIKV-specific antibodies. In addition, CHIKV infection in 11 days old mice under enhancing condition resulted in higher muscles viral RNA load detected and death. These observations provide the first evidence of antibody-mediated enhancement in CHIKV infection and pathogenesis and could also be relevant for other important arboviruses such as Zika virus.
Similar content being viewed by others


Introduction
Chikungunya virus (CHIKV) is a member of the Alphavirus genus of the Togaviridae family1,2. It is responsible for chikungunya fever (CHIKF), a disease characterized by the presence of incapacitating arthralgia3. CHIKV is transmitted by arthropod vectors, such as the Aedes aegypti and Aedes albopictus mosquitoes, with the latter being implicated in the transmission of CHIKV during the 2005–2006 Indian Ocean outbreak and in Europe4. For the past decade, re-emergence of CHIKV has led to numerous outbreaks in different parts of the world: Asia5,6,7,8,9,10,11,12, Europe4,13,14 and islands in the Indian Ocean15,16. Outbreaks of CHIKV infections have also been reported in the Caribbean islands17,18 and CHIKV has since successfully invaded North, Central and South America19.
Enhancement of arbovirus infections via antibodies was first demonstrated in 196420. This is a paradoxical phenomenon of antibodies forming complexes by binding to viruses, which then interact with cell surface receptors and promote entry into susceptible host cells, subsequently increasing virus replication21,22. This was observed for rabies virus23, influenza virus24, dengue virus (DENV)25,26, Ross River virus (RRV)27, human immunodeficiency virus (HIV)28 and Marburg virus29. Among alphaviruses, although virus enhancement was documented only in RRV infections27,30,31,32, most of these studies were conducted using in vitro murine cell line-based systems27,31,32. The development of a suitable infection system with primary human cells and an in vivo model allows the study of antibody enhancement in clinically important viruses, such as the recently emerged Zika virus (ZIKV), which infection is enhanced with cross-reactive anti-DENV antibodies33.
Here, we demonstrate antibody-mediated enhancement of CHIKV attachment and infection in primary human monocytes and B cells and a relevant murine cell line in the presence of sub-neutralizing levels of anti-CHIKV antibodies obtained from CHIKV-infected patients or animals. This enhancement was further demonstrated to mediate through the Fc receptors (FcγRs), with FcγRII being the key mediator. Importantly, two complementary animal models demonstrated enhanced CHIKV infections in the presence of sub-neutralizing levels of anti-CHIKV antibodies, with severe disease outcome and increase lethality. This study brings also caution to the importance of such undesired effects in anti-CHIKV vaccine designs.
Results
CHIKV-specific polyclonal antibodies mediate CHIKV infection enhancement in primary human cells
To investigate if sub-neutralizing concentrations of CHIKV-specific antibodies enhance CHIKV infection, diluted CHIKV-specific patients’ plasma obtained from a CHIKV cohort8,34,35 were mixed with CHIKV before being used to infect human primary monocytes and B cells. At low antibody concentration, antibody-mediated enhancement was shown to occur at antibody concentrations of 3.6 ± 2.9 μg/ml (Table 1). The presence of CHIKV antigen was detected by flow cytometry, where detection was increased by ~5 fold in monocytes (Fig. 1a) and by ~20 fold in B cells (Fig. 1b). However, active virus replication was not observed (Fig. 1c,d) in both cell types. Next, a Zs-Green tagged CHIKV variant was used for the infection of human whole blood. With this virus, a successful infection would lead to the production of the Zs-Green protein. Levels of infection can therefore be known through the detection of Zs-Green positive cells. It was observed that infection in the presence of patients’ plasma (total IgG concentrations of 1.8 ± 1.45 µg/ml) led to an increase in the numbers of Zs-Green positive monocytes. However, this was not observed in the B cells and plasmacytoid dendritic cells (pDCs) (Fig. S1a). Once again, the viral RNA load did not concur with enhanced infection (Fig. S1b).

Enhancement of CHIKV infection in purified primary human cells. Primary human (a,c) monocytes and (b,d) B cells (2 × 106 cells per infection) were infected with CHIKV (moi 10) in the presence (enhanced) or absence (non-enhanced) of diluted CHIKV-specific patient plasma containing total IgG at a concentration of 3.6 ± 2.9 μg/ml. (a,b) Detection of CHIKV antigen and (c,d) viral RNA load were determined at 0 and 6 hpi. Level of CHIKV antigen detected is expressed as fold enhancement relative to the non-enhanced infection controls. Data shown are mean ± SD from 3 independent experiments by parametric unpaired t test (**P = 0.0015 for 0 hpi monocytes; *P = 0.0458 for 0 hpi B cells and **P = 0.0053 for 6 hpi B cells). Expression of FcγRs in (e) monocytes and (f) B cells. Primary human (g) monocytes and (h) B cells (2 × 106 cells per infection) were treated with 10 μg of FcγRs blocking agent prior to CHIKV infection (moi 10) under enhancing conditions (as above). Percentage of infection is expressed as level of CHIKV antigen detected relative to the non-treated infection controls at 0 hpi. Data are presented as mean ± SD from 4 independent experiments by Mann-Whitney U test (*P = 0.0286 for monocytes and *P = 0.0143 for B cells).
Enhancement in CHIKV infection is mediated by FcγRII
Fcγ receptors (FcγRs)-dependent pathway is the most common mechanism in antibody-mediated enhancement of infections22. Monocytes express FcγRI and FcγRII equally (Fig. 1e), while B cells express only FcγRII (Fig. 1f). To investigate if FcγRs play a role in antibody-mediated enhancement, cells were pre-incubated with 10 μg of FcγR blocking agent prior to CHIKV infection under enhancement. Blocking of FcγRs significantly reduced the level of CHIKV antigen detected in monocytes (Fig. 1g) and B cells (Fig. 1h).
To elucidate which class of FcγRs enhances CHIKV infection, four different FcγR-expressing stable human B lymphocyte cell line, ST486, that have been modified to stably express a single class of FcγR driven by the same promoter36 were used (Fig. 2a). Parental ST486 cells naturally deficient in FcγRs expression were used as controls and were not susceptible to CHIKV infection (Fig. 2b). A significant increase in CHIKV antigen was detected in ST486 cells over-expressing FcγRI or FcγRII when infection was performed in the presence of low levels of CHIKV-specific antibodies (Fig. 2c,d). ST486 cells over-expressing FcγRIII neither supported nor enhanced CHIKV infection (Fig. 2e).

Enhancement of CHIKV infection is mediated via Fcγ receptors. (a) Expression of FcγRI, FcγRII and FcγRIII was determined in FcγR-expressing ST486 cell lines by flow cytometry (n = 3). CHIKV (moi 10) was used to infect (b) non-FcγR-expressing, (c) FcγRI-expressing, (d) FcγRII-expressing and (e) FcγRIII-expressing ST486 cell lines (2 × 106 cells per infection) in the presence (enhanced) or absence (non-enhanced) of diluted patient plasma containing total IgG at a concentration of 3.6 ± 2.9 μg/ml (n = 9). Level of CHIKV antigen was determined at 0 and 6 hpi. Level of CHIKV antigen detected is expressed as fold enhancement relative to the non-enhanced infection controls. Data are presented as mean ± SD by Mann-Whitney U test (***P < 0.0001 for level of CHIKV infection observed at both 0 and 6 hpi in FcγRI and FcγRII expressing cells).
Antibody-mediated enhancement of CHIKV infection in human macrophages
Macrophages have been hypothesized to act as a possible cellular vehicle and reservoir for virus dissemination and persistence37,38,39. Using human monocytes-derived macrophages (MDMs), which express high levels of FcγRI and II (Fig. 3a), antibody-mediated enhancement of CHIKV infection was investigated with diluted patients’ plasma (total IgG concentrations of 1.8 ± 1.45 µg/ml. In this experiment, infection was once again performed with the Zs-Green tagged CHIKV variant. It was observed that elevated levels of ZS-green positive cells were being detected at 24 hpi (Fig. 3b). Likewise, it was also increased in primary monocytes isolated from the same donor (Fig. S2a). Despite the increased level of Zs-green positive cells, viral RNA load remained unchanged (Fig. 3c and S2b). Similarly, the enhancement observed was mediated via FcγRII, as blocking with FcγRII-specific antibodies completely abrogated infection (Fig. S3).

CHIKV infection enhancement in macrophages from humans and mice. (a) Expression of FcγRs in human monocytes-derived macrophages (MDMs) was detected with flow cytometry. Human MDMs (2 × 106 cells per infection) were infected with Zs-Green tagged CHIKV (moi 10) either in the presence (enhanced) or absence (non-enhanced) of diluted CHIKV patient plasma containing total IgG at a concentration of 1.8 ± 1.45 μg/ml. (b) Level of infection and (c) viral RNA load were determined at 24 and 48 hpi. Level of infection was determined by the amount of Zs-Green positive cells and is expressed as fold enhancement relative to the non-enhanced infection controls. Data shown are mean ± SD from 4 independent experiments by Mann-Whitney U test (*P = 0.0317 for level of infection at 24 hpi). (d) FcγRs expression in RAW264.7 cells was determined by flow cytometry. CHIKV (moi 10) was used to infect RAW264.7 cells (2 × 106 cells per infection) in the presence (enhanced) or absence (non-enhanced) of diluted CHIKV-specific mice sera at concentration of ~2 µg/ml. (e) Detection of CHIKV antigen and (f) viral RNA load were determined in these infected cells at 0, 6 and 12 hpi. Level of CHIKV antigen detected is expressed as fold enhancement relative to non-enhanced infection controls. Data shown are mean ± SD from 3 independent experiments by parametric unpaired t test (*P = 0.0341 and 0.0350 for 0 and 6 hpi respectively for level of infection in RAW264.7 cells; ***P = 0.0006 for 6 hpi and *P = 0.0269 for 12 hpi for viral RNA load in infected RAW264.7 cells).
Antibody-mediated enhancement of CHIKV infection in mouse macrophages
Before studying the impact of CHIKV antibody-mediated enhancement in mice, in vitro CHIKV infections were first performed in the RAW264.7 mouse macrophage cell line. RAW264.7 cells have been used in several studies to investigate the effects of antibody-mediated enhancement of infection in RRV31,32, a closely related alphavirus. RAW264.7 cells have high levels of FcγRII/III (Fig. 3d). As a result, an increased in CHIKV infection was observed at 6 hpi in the presence of sub-neutralizing levels of CHIKV-specific mouse sera containing ~2 μg/ml of total IgG antibodies (Table 1) (Fig. 3e). The increased in CHIKV infection was further complemented with higher viral RNA load (Fig. 3f).
Given that enhanced CHIKV infection in RAW264.7 cells led to both increased levels of CHIKV antigen detected and viral RNA load, the consequences of this enhancement was further studied via gene expression analyses on the Type-I Interferon (IFN) and pro-inflammatory pathways (Fig. 4a). Expectedly, high levels of several pro-inflammatory and anti-viral genes such as IL-6, iNOS, IFNα, IFNβ, IRF3, IRF9, Viperin and ISG15 were detected starting at 6 hpi when CHIKV infection was enhanced (Fig. 4b).

Levels of Type I IFN and pro-inflammatory genes upon CHIKV infection under enhancing conditions. RAW264.7 cells (2 × 106 cells per infection) were infected with CHIKV (moi 10) under both enhancing and non-enhancing conditions and infected cells were harvested at 0, 6 and 12 hpi for gene expression study on selected pro-inflammatory and Type I IFN related genes by qRT-PCR. (a) Signature of gene expression across all stipulated time points displayed as a two-way clustered heat plot. Data are normalized to mock-infected controls. (b) Elevated expression of key mediators during enhanced CHIKV infection at 12 hpi. Data are normalized relative to mock-infected controls. Data are presented as mean ± SD from 3 independent experiments by parametric unpaired t test (*P = 0.0299 for IL-6; *P = 0.0117 for iNOS; *P = 0.0184 for IFNα; *P = 0.0498 for IRF3; *P = 0.0259 for IRF9; *P < 0.0001 for Viperin; *P = 0.0466 for ISG15).
Antibody-mediated enhancement leads to increased infection and exacerbation of disease severity in CHIKV-infected adult mice
To further establish and evaluate the contribution of antibody-mediated enhancement in CHIKV infection, in vivo infections were performed in two different and well-characterized CHIKV mouse models that mimic disease in humans40,41,42. Using the joint footpad model41,42,43,44,45,46,47,48, 3 weeks old wild-type (WT) C57BL/6 mice were inoculated with 106 plaque forming unit (PFU) of CHIKV in the right footpad, followed by intra-peritoneal administration of diluted CHIKV-specific mice sera containing total IgG at a concentration of ~2 μg/ml. Disease progression was monitored daily for viremia and degree of joint inflammation at the footpad. Mice infected under enhancing conditions had significantly higher viral RNA load during the acute phase of the disease (Fig. 5a) and endured a more pronounced inflammation at the infected joint footpad (Fig. 5b). Tissue immune-phenotyping (Fig. S4) further revealed higher levels of infiltration of neutrophils and possibly CD4+ T cells into the joint footpad at 6 days post-infection (dpi) during enhanced CHIKV infection (Fig. 5c). Moreover, a much lower number of CD4+ T cells and monocytes were present in the popliteal lymph node (pLN) draining the right footpad (Fig. 5d), indicating the possible egress of these cells from the pLN into the circulation or infected tissues during enhanced infection. Gene expression studies also revealed higher levels of IFNγ as well as IL-10 in the inflamed footpad during enhanced infection. This was coupled with a lower expression of β-defensin 14 (DEF14) (Fig. 5e).

Exacerbation of joint inflammation in adult mice. (a,b) Three-weeks old, C57BL/6 WT female mice (n = 5 animals per group) were infected via footpad inoculation with 106 PFU of CHIKV. Immediately, these mice were administered intraperitoneally with ~2 µg/ml (total IgG concentration) mice sera from CHIKV-infected mice (enhanced) or PBS (non-enhanced). Daily assessment of (a) viremia and (b) disease score were performed. All Data are presented as mean ± SD by Mann-Whitney U test (*P = 0.0278, *P = 0.0159, *P = 0.0159 and *P = 0.0297 for 2, 3, 4 and 5 dpi viremia respectively; **P = 0.0040, **P = 0.0079, **P = 0.0060, **P = 0.0079, **P = 0.0079, *P = 0.0104, *P = 0.0172, **P = 0.0040, **P = 0.0060, *P = 0.0106 and *P = 0.0106 for 3, 4, 5, 6, 7, 8, 9, 10, 11, 13 and 14 dpi disease score respectively). Immune-phenotyping was performed for (c) footpad and (d) popliteal lymph node on 6 dpi. Results are displayed as number of neutrophils, CD4+ T cells and monocytes per organ from 4 to 10 animals per group. (e) Total RNA was extracted from the joint footpads of infected mice (n = 5 to 10 animals per group) and qRT-PCR was performed to detect for the expression of crucial immune genes. Gene expression data are expressed as fold expression relative to the mock-infected mice. Mice sacrificed for immune-phenotyping and gene expression studies were infected as described for (a,b). All Data are presented as mean ± SD by Mann-Whitney U test (*P = 0.0186, *P = 0.0326 and *P = 0.0185 for neutrophils, monocytes and CD4+ T cells infiltration into respective organs; *P = 0.0276, *P = 0.0376 and *P = 0.0376 for IFNγ, DEF14 and IL-10 expression respectively).
In a second model, intradermal CHIKV infection in IFNαR+/− mice leads to the development of a mild infection, with CHIKV targeting the muscles, joint and skin fibroblasts40. Utilizing this model, 3 weeks old IFNαR+/− mice were infected with 106 PFU CHIKV via the joint footpad to assess the outcome of antibody-mediated enhancement. While a similar degree of joint footpad inflammation (Fig. 6a) was observed in IFNαR+/− mice as compared to WT mice, higher viremia was detected in the IFNαR+/− animals (Fig. 6b). However, when CHIKV infection was performed in the presence of diluted CHIKV-specific mice sera at a concentration of ~2 μg/ml total IgG, increased viremia (Fig. 6a) and more pronounced joint footpad inflammation (Fig. 6b) was observed in the IFNαR+/− mice. These observations reinforce the significance of sub-neutralizing concentrations of CHIKV-specific antibodies in enhancing CHIKV infection and aggravating disease pathology.

Aggravated disease outcome in CHIKV-infected IFNαR+/− mice. Three-weeks old IFNαR+/− female mice (n = 7 to 11 animals per group) were infected via footpad inoculation with 106 PFU of CHIKV. Immediately, these mice were administered intraperitoneally with ~2 µg/ml (total IgG concentration) mice sera from CHIKV-infected mice (enhanced) or PBS (non-enhanced). Daily assessment of (a) viremia and (b) disease score were performed. Data are presented as mean ± SD by Mann-Whitney U test (**P = 0.0027, **P = 0.0011, ***P = 0.0009, ***P = 0.0005, **P = 0.0017 and *P = 0.0159 for 2, 3, 4, 5, 6 and 7 dpi viremia respectively; +P = 0.0125, +++P < 0.0001, ++P = 0.0015 and +P = 0.0277 for 4, 5, 6 and 7 dpi viremia respectively; *P = 0.0109 and *P = 0.0306 for 10 and 11 dpi disease score respectively; +++P < 0.0001, +++P = 0.0006, +P = 0.0109, +P = 0.0277, +P = 0.0031, +P = 0.0095 and +P = 0.0031 for 1, 6, 7, 8, 9, 10 and 11 dpi disease score respectively. (*) indicates significance comparing between CHIKV-infected IFNαR+/− (blue) and WT animals (black); (+) indicates significance comparing between IFNαR+/− mice infected under enhanced (red) and non-enhanced (blue) conditions.
CHIKV-specific polyvalent human antibodies aggravate CHIKV infection in young mice, leading to death
It has been demonstrated that prophylactic administration of human purified polyvalent immunoglobulins G (IgG) containing anti-CHIKV antibodies (CHIKVIGs) protected susceptible IFNα/βR−/− adult animals and wild-type neonates during CHIKV infection49. To assess if these antibodies could exacerbate CHIKV disease severity at sub-neutralizing concentrations, different concentrations of CHIKVIGs were administered intra-peritoneally to CHIKV-infected 11 days old C57BL/6 WT mice at the ventral thorax40 (Fig. 7). Mortality was observed in mice receiving CHIKVIGs at concentrations of 1–100 µg/kg (Fig. 7a), whereas mice receiving either 1000 µg/kg or 0.1 µg/kg of CHIKVIGs survived the infection. Unexpectedly, approximately 30% of mice receiving a control non-CHIKV-specific polyvalent IgG also died from infection (Fig. 7a). Nevertheless, viral RNA load in serum, liver and muscles was determined in CHIKV-infected mice receiving 10 µg/kg of CHIKVIGs. It was observed that enhanced CHIKV infection led to a more significant viral burden in the muscles at 3 dpi (Fig. 7b), but not at 6 dpi (Fig. 7c). Taken together, these findings highlight that enhanced CHIKV infection is detrimental in young animals.

Enhanced infection-associated lethality in young mice. (a) 11-day old C57BL/6 (female, n = 10 animals per group) were inoculated intradermally with 106 PFU of CHIKV-21. Immediately after, these mice were given intraperitoneally either human naive polyvalent antibodies or purified CHIKVIGs at different concentrations. Survival of these animals was monitored over a period of 3 weeks. (b,c) 11 days old C57BL/6 (female, n = 3 animals per group) were infected as described in (a) but were intraperitoneally given purified human CHIKVIGs at 10 µg/kg. Control animals were given 10 µg/kg of human naive polyvalent antibodies. Viral RNA load in the serum, liver and muscle were determined at both (b) 3 and (c) 6 dpi. All data are presented as mean ± SD. *p < 0.05 by parametric unpaired t test.
Discussion
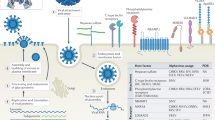
This study provides the first documentation of antibody-mediated enhancement in CHIKV infection in vitro and in relevant in vivo mouse models. Despite the increased detection of CHIKV antigen in primary human monocytes, B cells and MDMs when infection was performed in the presence of sub-neutralizing levels of CHIKV-specific antibodies, viral replication was not increased. Antibody-mediated enhancement in viral infections has been reported to mostly involve FcγRs22. The involvement of FcγRs in enhancing CHIKV infection may lead to two outcomes: the extrinsic and intrinsic pathways (Fig. 8). In the extrinsic pathway, virus attachment and possibly entry is facilitated without much significance to viral replication and manipulation of host immune responses22,50. Results obtained with infection in the whole blood, primary human monocytes, B cells and MDMs supported that the extrinsic pathway is at play. The increase detection of Zs-Green signal in MDMs during the later time points is suggestive of viral entry and initiation of viral replication, as the Zs-Green is cloned under a duplicated sub-genomic promoter42,51. However, no further increase in both Zs-Green signal and viral RNA load may suggest that enhanced CHIKV infection do not necessary augments viral replication. This quick shutdown of viral replication may actually indicate a putative mechanism employed by CHIKV for chronic persistence. While it is known that CHIKV could persist chronically using macrophages as possible cellular reservoirs38,39, its exact mechanism is not known. As such, the restricted replication observed in monocytes and MDMs could be a form of immune subversion by the virus to achieve chronic persistency52.

Impact of antibody-mediated enhancement in CHIKV infection. Virus-antibody complexes are formed when antibodies are present in sub-neutralizing concentrations and are taken up by cells expressing FcγRs via either extrinsic or intrinsic pathways. Extrinsic pathways result in increased virus attachment and possibly entry with no visible benefits to virus replication. Whereas in intrinsic pathways, it can lead to enhanced anti-inflammatory or enhanced pro-inflammatory infection. Enhanced anti-inflammatory infection downplays the inflammatory response associated with normal infection and instead upregulates the anti-inflammatory response leading to heightened infection and viral replication. In enhanced pro-inflammatory infection, it leads to an increased in active infection, without negative suppression of pro-inflammatory immune response.
On the other hand, the intrinsic pathway typically results in increased virus entry and replication, often accompanied by the alterations of host immune responses31,32,53,54,55,56 (Fig. 8). Intriguingly, enhanced CHIKV infection in murine RAW264.7 macrophages was even more prominent when the virus started replicating (6 hpi). Subsequently, this led to the high expression of several pro-inflammatory and anti-viral genes. This observation is enthralling as similar studies in DENV and RRV reported the down-regulation of these pro-inflammatory responses31,32,53,54,55,56. While the data in this report differs, it opens up another compelling perspective in which enhanced CHIKV infection in susceptible cells can lead to augmented virus entry and replication, resulting in an active pro-inflammatory response from the infected cells (Fig. 8). While the consequences of the elevated immune response remains to be investigate, it is plausible that CHIKV manipulates this immune outcome to its own advantage to achieve chronicity, as observed for other viral infections57.
Whether antibodies are enhancing or neutralizing depends on numerous factors such as possible future infection with closely-related viruses (e.g., other alphaviruses), the virus strain, virus titer and concentrations, epitopes specificities, isotypes and FcγRs-binding affinities of the antibodies22,58,59,60. Patient plasma and mice serum samples used in this study at a low dilution factor have been previously shown to be strongly neutralizing44,46,61,62 and recognized epitopes located mainly on the CHIKV E2 glycoprotein44,46. Furthermore, these CHIKV-specific antibodies were of IgG3 isotype62, and hence capable of moving across the placenta63. It remains to be seen if infants born to mothers infected with CHIKV during their pregnancy may suffer from a more severe disease due to the low levels of maternal-acquired CHIKV-specific antibodies. It was reported that newborn mice infected with DENV in the presence of maternally acquired anti-DENV antibodies had a more severe disease outcome64.
Using the joint footpad mouse model of CHIKV infection41,42,43,44,45,46,47,48, disease severity was greatly enhanced in both WT and IFNαR+/− mice upon passive administration of low levels of CHIKV-specific antibodies. The exact mechanisms of this phenomenon remains to be fully elucidated. However as presented in this study, the disruption of host immune response could be a crucial driving force behind the augmented disease severity. This brings caution to the phenomenon that sub-neutralizing concentrations of virus-specific antibodies can enhance severity of infection and advocates the need for careful vaccine design and extensive pre-clinical trials. In fact, it was reported that CHIKV severity was increased in vaccinated mice that presented sub-optimal immune responses65. Unfortunately, it was also observed that a proportion of mice receiving the naïve human polyvalent antibodies succumbed to CHIKV infection. This was definitely an unexpected observation and could be due to the presence of unknown artifacts present in the control antibodies that resulted in this outcome. These naïve polyvalent antibodies had previously been shown not to exhibit any immuno-reactivity against CHIKV49.
To conclude, the relevance of antibody-mediated enhancement in aggravating viral disease severity should not be underestimated. The recent 2016 outbreaks of ZIKV infections across the globe and the potential of anti-DENV antibodies cross-reacting with ZIKV33 highlights the relevance of this anomaly. Therefore, there is a need to expand research efforts in the understanding and prevention of antibody-mediated enhancement in viral infections.
Methods
Ethics approval
This study was reviewed and approved by the institutional review board at the National Healthcare Group with Domain Specific Review Board no. DSRE E/08/414. Written informed consent was obtained from all CHIKF patients prior to the collection of samples. Human blood samples were obtained from healthy donors with written informed consent in accordance with the guidelines from the Health Sciences Authority of Singapore (study approval number: NUS IRB number 10–250). Animal studies protocols were approved: (1) by the Institutional Animal Care and Use Committee (IACUC) of the Agency for Science, Technology and Research (A*STAR) (IACUC number: 151018); and (2) by the Institut Pasteur Animal Ethics Committee according to project #2014-0019 using level 3 isolators.
Patient plasma samples
Plasma samples from patients, who were admitted with acute CHIKF to the Communicable Disease Centre at Tan Tock Seng Hospital (CDC/TTSH), during the outbreak from 1 August to 23 September 20088,9,35 were used in this study. These plasma have previously been characterized44,61,62,66. In this study, plasma samples collected 2–3 months post-illness onsets were used. All patient plasma were aliquoted and stored at −80 °C. Plasma samples were also collected from 8 healthy volunteers as controls.
Primary cells (B cells and monocytes) isolation
Peripheral blood mononuclear cells (PBMCs) were extracted from 50 ml of donors’ blood as described previously37. Extracted PBMCs were subjected to either B cells or monocytes isolation using B cell Isolation Kit II and Monocytes Isolation Kit II (Miltenyi Biotec) respectively following manufacturer’s instructions. Purity was > 95% as verified by flow cytometry.
Cell culture
African green monkey kidney epithelial cells (Vero-E6) and mouse macrophage cell line RAW264.7 cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco) supplemented with 10% Fetal Bovine Serum (FBS) (Gibco). Human B lymphocytic ST486 and Fcγ Receptors (FcγRs) over-expressing ST486 cell lines were cultured in Roswell Park Memorial Institute medium (RPMI 1640) supplemented with 10% FBS. Aedes albopictus monolayer (C6/36) cells were cultured in Leibovitz’s medium (L-15) (Gibco) supplemented with 10% FBS. Extracted primary human cells were maintained in Iscove’s Modified Dulbecco’s Medium (IMDM) (Hyclone) supplemented with 10% Human serum (HS) (I-DNA Biotechnology). All cultures were incubated at 37 °C with 5% CO2 supplied with the exception of C6/36 which was incubated at 28 °C with no CO2 supplied. All media and reagents were tested negative for endotoxins.
MDMs differentiation
Isolated monocytes were differentiated into MDMs as described67,68 with some modifications. Briefly, isolated monocytes were allowed to differentiate over a period of 5 days in IMDM (Hyclone) supplemented with 10% HS (I-DNA Biotechnology). A change of fresh medium was performed on the third day, in which the non-adhering cells (presumably non-differentiated monocytes or dead cells) would have been removed, leaving behind the adhering MDMs.
Virus stocks
CHIKV-SGP11 was isolated from an outbreak in Singapore in 2008 and prepared in Vero-E6 cells as described37. This was used for all in vitro experiments involving isolated primary human monocytes, B cells and the RAW264.7 cells. For the whole blood and MDMs (together with their corresponding precursor monocytes), a full-length CHIKV infectious clone based on the CHIKV LR2006-OPY169 expressing the Zs-Green protein was used. CHIKV-SGP11 was further propagated in C6/36 cells and purified by ultra-centrifugation44 before being used in infection studies in 3 weeks old mice. In infection studies in 11 days old mice, CHIKV-21 isolate obtained from a patient during the 2005–2006 outbreak of CHIKV infection in La Réunion was used49.
In vitro FcγR expression assay
Cells were first stained with 10 μl of Human FcγR blocking reagent (Miltenyi Biotec), before staining with the following anti-FcγR antibodies and respective fluorophore isotype controls following manufacturer’s recommendations: FITC-conjugated mouse anti-human CD64 (eBioscience), APC-conjugated mouse anti-human CD32 (eBioscience), PE-conjugated mouse anti-human CD16 (eBioscience), mouse IgG1κ isotype control FITC (eBioscience), mouse IgG1κ isotype control APC (eBioscience) and mouse IgG1κ isotype control PE (eBioscience). For RAW264.7 cells, staining was performed with FITC-conjugated rat anti-mouse FcγRI and anti-mouse FcγRII antibodies (R&D). Data were acquired with BD FACS Calibur (BD Biosciences) using BD CellQuest Pro software (BD Biosciences). Results were analyzed with FlowJo (version 10) software (Tree Star).
In vitro CHIKV infection assay
CHIKV (moi 10) was first incubated with diluted heat-inactivated (56 °C for 30 min) CHIKV-specific patient plasma or animal sera in serum-free IMDM (Hyclone) or DMEM (Gibco) respectively, for 2 h on a shaking heat-block (37 °C; 350 rpm) before being used to infect the respective primary cells or cells lines (2 × 106 cells per infection) for 1.5 h in a 37 °C incubator, with atmosphere of 5% (v/v) CO2. Virus overlay was removed and cells were washed once with appropriate serum-free medium before they were re-suspended in appropriate complete medium. Cells were further incubated at 37 °C, with atmosphere of 5% (v/v) CO2,, before being harvested at indicated time points. During harvesting, 140 μl of infected cell suspension was aliquoted for viral RNA extraction. For gene expression studies, an aliquot of cell suspension was spun down and the resultant cell pellet was dried before being stored in −80 °C for downstream total RNA extraction. Remaining cells were collected by centrifugation and fixed with FACS lysing buffer (BD Biosciences) and stored for downstream staining procedures. Mock and non-enhanced control infections were performed by incubating cells with serum-free DMEM (Gibco) and viruses respectively. For ex vivo infection in human whole blood, Zs-Green tagged CHIKV variant (2 × 107 PFU) was first incubated with diluted heat-inactivated CHIKV-specific patient plasma in serum free IMDM (Hyclone) for 2 h on a shaking heat-block (37 °C; 350 rpm) before being added to 1 ml of fresh citrate whole blood. 1 ml of whole blood typically contains between 1.5–2 × 106 leukocytes. Infection was incubated in 37 °C, with atmosphere of 5% (v/v) CO2 until being harvested at the indicated time points. During harvesting, the mixture was spun down and 140 μl of plasma was obtained for viral RNA extraction. Red blood cells were subseqeuntly lyzed and cell pellet washed and subsequently fixed with FACs lysing buffer (BD Biosciences and stored for downstream staining procedures. Mock and non-enhanced control infections were performed by incubating cells with serum-free IMDM (Hyclone) and viruses respectively. Results are expressed as fold enhancement relative to non-enhanced infections. Fold enhancement is calculated according to the equation: Fold enhancement = (Level of CHIKV antigen detected from enhanced infection group / level of CHIKV antigen detected from non-enhanced infection group).
In vitro FcγR blocking assay
FcγR blocking agent (Miltenyi Biotec) was added to the cells for 30 min prior to CHIKV infection under enhancing conditions as described above. Immediately after the virus overlay was removed, cells were washed and harvested for downstream procedures as mentioned above. For FcγRII blocking, mouse anti-human CD32 antibodies (Stemcell) were used instead. Results are expressed as % infectivity relative to non-treated-enhanced infections. Reduction in infectivity was calculated according to the equation: % infectivity = 100 × Level of CHIKV antigen detected from treated-enhanced infection group / Level of CHIKV antigen detected from non-treated-enhanced infection group).
Viral RNA load assay
Viral RNA was extracted using QIAamp Viral RNA Mini Kit (QIAGEN), following manufacturer’s instructions. Viral RNA load was subsequently measured using real time quantitative reverse transcription PCR (qRT-PCR) utilizing QuantiTect® Probe RT-PCR kit (QIAGEN) modified from a previously described method to detect negative-strand nsP1 RNA70,71.
Flow cytometry
Fixed cells were permeabilized with FACS permeabilizing solution 2 (BD Biosciences). For detection of CHIKV antigens in primary human cells (B cells, monocytes), staining was done as described previously using a commercially available anti-alphavirus mAb (Santa Cruz; SC-58088)37,47. For whole blood and human MDMs, CHIKV infection was directly quantified by the detection of Zs-Green signal under the FITC channel. For detection of CHIKV antigen in RAW264.7 cells, staining was performed with rabbit anti-CHIKV nsP2 antibodies followed by secondary staining with Alexa Fluor 488-conjugated Goat anti-rabbit IgG (H + L) (Invitrogen). For primary human cells, an additional staining step was performed. Surface markers Pacific-Blue-conjugated mouse anti-human CD45, Qdot-605-conjugated mouse anti-human CD19, PerCP Cy5.5 mouse anti-human CD14 and PE-Cy7-conjugated mouse anti-human BDCA2 (all antibodies were from eBiosciences) were stained following manufacturer’s protocol. Human monocytes, B cells and pDCs are defined by the specific surface expression of CD14, CD19 and BDCA2 respectively. Data was acquired using either BD FACS Calibur or BD FACS Canto II (BD Biosciences). Softwares used include BD CellQuest Pro software (for FACSCalibur) and BD FACSDiva software (for FACSCanto II) (BD Biosciences). Gating strategy is shown in Fig. S5. A total of 30,000–50,000 cells were acquired and results were analyzed with FlowJo (version 10) (Tree Star).
Gene expression
Total RNA from cells was extracted using RNeasy® Mini Kit (QIAGEN) according to manufacturer’s instructions, without any DNAse treatment. Eluted RNA was quantified and diluted to 10 ng/µl before being stored in −80 °C. Gene expression profiling was performed via RT-PCR using QuantiFASTTM SYBR® Green RT-PCR Kit (QIAGEN) as described47. Results are expressed as relative fold change in expression level compared to mock infections, as previously described47. Gene expression data from infected RAW264.7 cells are displayed by two-way hierarchical clustering generated with Multi Experiment Viewer (version 4.9) (Microarray Software Suite TM4)72. Sequences of primers used are displayed in Table S1.
Animal studies
CHIKV infection in 3 weeks old WT and IFNαR+/− C57BL/6 mice was performed. Briefly, 106 PFU of CHIKV isolate (SGP11), diluted in 30 µl of PBS, were inoculated subcutaneously in the ventral side of the right hind footpad, towards the ankle42,43,45,46,47,48. Following this, PBS diluted (1:1000) CHIKV-specific mice sera were passively administered intraperitoneally into the infected animals. Control mice were inoculated with PBS alone. Viremia and the degree of footpad inflammation were monitored as described previously42,43,45,46,47,48. Three weeks old animals were bred and kept under specific pathogen-free conditions in the Biological Resource Centre, A*STAR, Biopolis, Singapore. For in vivo model with human polyvalent immunoglobulins, 11 days old female C57BL/6 mice were inoculated intra-dermally in the ventral thorax with 106 PFU CHIKV isolates (CHIKV-21) diluted in 30 µl of sterile PBS. Human polyvalent immunoglobulins (0.1 µg/kg to 1000 µg/kg) were given intraperitoneally following virus inoculation. Control animals were given 10 µg/kg of human naive polyvalent antibodies. Survival of all experimental animals was monitored daily and were handled in accordance with the guidelines described above.
Extraction of animal tissues
Extractions the joint footpad were performed as previously described42,43,45,48. Excised joint footpads were shredded and digested in digestion medium containing dispase (2 U/ml; Invitrogen), collagenase IV (20 mg/ml; Sigma-Aldrich), and DNase I mix (50 mg/ml; Roche Applied Science) in complete RPMI medium for 3 h at 37 °C. Cell debris and skin tissues were removed by passing through 40 μm cell strainer (BD Falcon) followed by RBC lysis (R&D system). Cells were further purified by spinning down in 35% v/v Percoll (Sigma) solution in RPMI prior to staining. The popliteal lymph node (pLN), located at the area to the back of the mice knee joint, was delicately retrieved and briefly digested in 1 ml of digestion medium containing dispase (2 U/ml), collagenase IV (20 mg/ml), and DNase I mix (50 mg/ml) in complete RPMI medium for 30 min at 37 °C. Disintegration of pLN was encouraged by gentle pipetting before passing contents through a 70 μm nylon mesh cloth (Sefar). RBCs were further lyzed with RBC lysis buffer (R&D system) prior to further staining procedures.
For total RNA extraction from excised joint footpad, specimens were first shredded in TRIzol (Invitrogen) with Micro SmashTM (Digital Biology), a micro-homogenizing system, following these conditions: 2 rounds of homogenization at 5000 rpm for 45 s followed by 30 s on ice. Subsequently, the sample was spun to remove the unwanted cell debris. Chloroform was then added to the TRIzol fraction causing a phase separation into the aqueous and organic phase. The aqueous phase, which contains the RNA, was carefully removed into a clean eppendorf tube. It was then further processed with the RNeasy® Mini Kit (QIAGEN) according to manufacturer’s instructions, before being used for gene expression studies as described above. No DNAse treatment was performed during the total RNA extraction.
Leukocytes profiling
Footpad and pLN cells were resuspended in blocking buffer (1% v/v rat and mouse serum) for 20 min. Live cells were stained with a Live/Dead Fixable Aqua Dead Cell Stain Kit (Invitrogen) for 30 min before staining with the following cell-specific markers following manufacturer’s protocol: APC-Cy7-conjugated rat anti- mouse CD45 (BD Biosciences), PE-Cy7–conjugated rat anti-mouse CD3 (Biolegend), Pacific Blue-conjugated rat anti-mouse CD4 (Biolegend), PE-conjugated rat anti-mouse CD11b (BD Biosciences) and APC-conjugated rat anti-mouse Ly6G (Biolegend) for 20 min at room temperature. Cells were subsequently washed and fixed with 100 µl neat IC fixation buffer (eBioscience) for 5 min. Cells were then washed and resuspended for flow cytometry data acquisition. Data were acquired BD FACSCanto II (BD Biosciences) with FACSDiva software. Analyses were performed using FlowJo (Version 10) (Tree Star).
Quantification of Total IgG antibodies
The total amount of IgG present in both heat-inactivated CHIKV-infected patient’s plasma and CHIKV-infected mice sera were quantified with IgG Human ELISA Kit and IgG Mouse ELISA Kit (Abcam) respectively following manufacturer’s protocol.
Statistical analysis
Comparisons between different groups were performed using either non-parametric Mann-Whitney rank sum test or parametric unpaired t-test (two tail analyses), when data follow a normal distribution. Analyzes were performed with GraphPad PRISM (version 6) (GraphPad Software). P values of < 0.05 are considered to be statistically significant.
References
Ross, R. W. The Newala epidemic. III. The virus: isolation, pathogenic properties and relationship to the epidemic. The Journal of hygiene 54(2), 177–191 (1956).
Strauss, J. H. & Strauss, E. G. The alphaviruses: gene expression, replication, and evolution. Microbiol Rev 58(3), 491–562 (1994).
Lanciotti, R. S. et al. Chikungunya virus in US travelers returning from India, 2006. Emerg Infect Dis 13(5), 764–767 (2007).
Angelini, R. et al. Chikungunya in north-eastern Italy: a summing up of the outbreak. Euro Surveill 12(11), E071122.071122 (2007).
Chua, K. B. Epidemiology of chikungunya in Malaysia: 2006-2009. Med J Malaysia 65(4), 277–282 (2010).
Kumarasamy, V. et al. Re-emergence of Chikungunya virus in Malaysia. Med J Malaysia 61(2), 221–225 (2006).
Laras, K. et al. Tracking the re-emergence of epidemic chikungunya virus in Indonesia. Trans R Soc Trop Med Hyg 99(2), 128–141 (2005).
Leo, Y. S. et al. Chikungunya outbreak, Singapore, 2008. Emerg Infect Dis 15(5), 836–837 (2009).
Ng, L.-C. et al. Entomologic and virologic investigation of Chikungunya, Singapore. Emerg Infect Dis 15(8), 1243–1249 (2009).
Ravi, V. Re-emergence of chikungunya virus in India. Indian J Med Microbiol 24(2), 83–84 (2006).
Seneviratne, S. L. & Perera, J. Fever epidemic moves into Sri Lanka. Br Med J (Clin Res Ed) 333(7580), 1220–1221 (2006).
Watanaveeradej, V. et al. Transplacental Chikungunya virus antibody kinetics, Thailand. Emerg Infect Dis 12(11), 1770–1772 (2006).
Beltrame, A. et al. Imported Chikungunya Infection, Italy. Emerg Infect Dis 13(8), 1264–1266 (2007).
Rezza, G. et al. Infection with chikungunya virus in Italy: an outbreak in a temperate region. Lancet 370(9602), 1840–1846 (2007).
Her, Z. et al. Chikungunya: a bending reality. Microbes Infect 11(14), 1165–1176 (2009).
Renault, P. et al. A major epidemic of chikungunya virus infection on Reunion Island, France, 2005-2006. Am J Trop Med Hyg 77(4), 727–731 (2007).
Enserink, M. Crippling Virus Set to Conquer Western Hemisphere. Science 344(6185), 678–679 (2014).
Leparc-Goffart, I. et al. Chikungunya in the Americas. Lancet 383(9916), 514 (2014).
Weaver, S. C. & Forrester, N. L. Chikungunya: Evolutionary history and recent epidemic spread. Antiviral Res 120, 32–39 (2015).
Hawkes, R. A. Enhancement of the infectivity of arboviruses by specific antisera produced in domestic fowls. Aust J Exp Biol Med Sci 42, 465–482 (1964).
Pierson, T. C. Modeling antibody-enhanced dengue virus infection and disease in mice: protection or pathogenesis? Cell Host Microbe 7(2), 85–86 (2010).
Takada, A. & Kawaoka, Y. Antibody-dependent enhancement of viral infection: molecular mechanisms and in vivo implications. Rev Med Virol 13(6), 387–398 (2003).
Porterfield, J. S. Antibody-mediated enhancement of rabies virus. Nature 290(5807), 542 (1981).
Ochiai, H. et al. Infection enhancement of influenza A NWS virus in primary murine macrophages by anti-hemagglutinin monoclonal antibody. J Med Virol 36(3), 217–221 (1992).
Morens, D. M. & Halstead, S. B. Measurement of antibody-dependent infection enhancement of four dengue virus serotypes by monoclonal and polyclonal antibodies. J Gen Virol 71(12), 2909–2914 (1990).
Halstead, S. B. & O’Rourke, E. J. Antibody-enhanced dengue virus infection in primate leukocytes. Nature 265(5596), 739–741 (1977).
Linn, M. L. et al. Antibody-dependent enhancement and persistence in macrophages of an arbovirus associated with arthritis. J Gen Virol 77(3), 407–411 (1996).
Robinson, W. E. et al. Antibody-dependent enhancement of human immunodeficiency virus type 1 infection. Lancet 331(8589), 790–794 (1988).
Nakayama, E. et al. Antibody-dependent enhancement of Marburg virus infection. J Infect Dis 204(Suppl 3), S978–985 (2011).
Halstead S. B. Dengue Antibody-Dependent Enhancement: Knowns and Unknowns. Microbiol Spec 2(6), (2014).
Lidbury, B. A. & Mahalingam, S. Specific ablation of antiviral gene expression in macrophages by antibody-dependent enhancement of Ross River virus infection. J Virol 74(18), 8376–8381 (2000).
Mahalingam, S. & Lidbury, B. A. Suppression of lipopolysaccharide-induced antiviral transcription factor (STAT-1 and NF-kappa B) complexes by antibody-dependent enhancement of macrophage infection by Ross River virus. Proc Natl Acad Sci USA 99(21), 13819–13824 (2002).
Dejnirattisai, W. et al. Dengue virus sero-cross-reactivity drives antibody-dependent enhancement of infection with zika virus. Nature immunology 17(9), 1102–1108 (2016).
Ng, K. W. et al. Clinical features and epidemiology of chikungunya infection in Singapore. Singapore Med J 50(8), 785–790 (2009).
Win, M. K. et al. Chikungunya fever in Singapore: acute clinical and laboratory features, and factors associated with persistent arthralgia. J Clin Virol 49(2), 111–114 (2010).
Jaume, M. et al. Anti-severe acute respiratory syndrome coronavirus spike antibodies trigger infection of human immune cells via a pH- and cysteine protease-independent FcγR pathway. J Virol 85(20), 10582–10597 (2011).
Her, Z. et al. Active infection of human blood monocytes by Chikungunya virus triggers an innate immune response. J Immunol 184(10), 5903–5913 (2010).
Hoarau, J.-J. et al. Persistent chronic inflammation and infection by Chikungunya arthritogenic alphavirus in spite of a robust host immune response. J Immunol 184(10), 5914–5927 (2010).
Labadie, K. et al. Chikungunya disease in nonhuman primates involves long-term viral persistence in macrophages. J Clin Invest 120(3), 894–906 (2010).
Couderc, T. et al. A mouse model for Chikungunya: young age and inefficient type-I interferon signaling are risk factors for severe disease. PLoS Pathog 4(2), e29 (2008).
Gardner, J. et al. Chikungunya virus arthritis in adult wild-type mice. J Virol 84(16), 8021–8032 (2010).
Teo, T.-H. et al. A Pathogenic Role for CD4+T Cells during Chikungunya Virus Infection in Mice. J Immunol 190(1), 259–269 (2013).
Her, Z. et al. Loss of TLR3 aggravates CHIKV replication and pathology due to an altered virus-specific neutralizing antibody response. EMBO Mol Med 7(1), 24–41 (2015).
Kam, Y.-W. et al. Early neutralizing IgG response to Chikungunya virus in infected patients targets a dominant linear epitope on the E2 glycoprotein. EMBO Mol Med 4(4), 330–343 (2012b).
Lee W. W. L. et al. Expanding regulatory T cells alleviates chikungunya virus-induced pathology in mice. J Virol; https://doi.org/10.1128/JVI.00998-15, (2015).
Lum, F.-M. et al. An essential role of antibodies in the control of chikungunya virus infection. J Immunol 190(12), 6295–6302 (2013).
Teng, T.-S. et al. Viperin restricts chikungunya virus replication and pathology. J Clin Invest 122(12), 4447–4460 (2012).
Teo T.-H. et al. Caribbean and La Réunion Chikungunya virus isolates differ in their capacity to induce pro-inflammatory Th1 and NK cell responses and acute joint pathology. J Virol; https://doi.org/10.1128/JVI.00909-15, (2015).
Couderc, T. et al. Prophylaxis and Therapy for Chikungunya Virus Infection. J Infect Dis 200(4), 516–523 (2009).
Halstead, S. B. et al. Intrinsic antibody-dependent enhancement of microbial infection in macrophages: disease regulation by immune complexes. The Lancet Infectious diseases 10(10), 712–722 (2010).
Tsetsarkin, K. et al. Infectious clones of Chikungunya virus (La Reunion isolate) for vector competence studies. Vector borne and zoonotic diseases 6(4), 325–337 (2006).
Sallie, R. Replicative homeostasis: a mechanism of viral persistence. Med Hypotheses 63(3), 515–523 (2004).
Chareonsirisuthigul, T. et al. Dengue virus (DENV) antibody-dependent enhancement of infection upregulates the production of anti-inflammatory cytokines, but suppresses anti-DENV free radical and pro-inflammatory cytokine production, in THP-1 cells. J Gen Virol 88(2), 365–375 (2007).
Rolph, M. S. et al. Downregulation of interferon-β in antibody-dependent enhancement of dengue viral infections of human macrophages is dependent on interleukin-6. J Infect Dis 204(3), 489–491 (2011).
Suhrbier, A. & La Linn, M. Suppression of antiviral responses by antibody-dependent enhancement of macrophage infection. Trends Immunol 24(4), 165–168 (2003).
Ubol, S. et al. Mechanisms of immune evasion induced by a complex of dengue virus and preexisting enhancing antibodies. J Infect Dis 201(6), 923–935 (2010).
Zuniga, E. I. et al. Innate and Adaptive Immune Regulation During Chronic Viral Infections. Annu Rev Virol 2(1), 573–597 (2015).
Boonnak, K. et al. Role of dendritic cells in antibody-dependent enhancement of dengue virus infection. J Virol 82(8), 3939–3951 (2008).
Hohdatsu, T. et al. The role of IgG subclass of mouse monoclonal antibodies in antibody-dependent enhancement of feline infectious peritonitis virus infection of feline macrophages. Arch Virol 139(3-4), 273–285 (1994).
Midgley, C. M. et al. Structural analysis of a dengue cross-reactive antibody complexed with envelope domain III reveals the molecular basis of cross-reactivity. J Immunol 188(10), 4971–4979 (2012).
Kam, Y.-W. et al. Longitudinal analysis of the human antibody response to chikungunya virus infection: implications for sero-diagnosis assays and vaccine development. J Virol 86(23), 13005–13015 (2012c).
Kam, Y.-W. et al. Early appearance of neutralizing immunoglobulin G3 antibodies is associated with chikungunya virus clearance and long-term clinical protection. J Infect Dis 205(7), 1147–1154 (2012a).
Palmeira, P. et al. IgG placental transfer in healthy and pathological pregnancies. Clin Dev Immuno 2012, 985646 (2012).
Ng, J. K. et al. First experimental in vivo model of enhanced dengue disease severity through maternally acquired heterotypic dengue antibodies. PLoS Pathog 10(4), e1004031 (2014).
Hallengärd, D. et al. Prime-boost immunization strategies against Chikungunya virus. J Virol 88(22), 13333–13343 (2014b).
Kam, Y. W. et al. Sero-prevalence and cross-reactivity of chikungunya virus specific anti-E2EP3 antibodies in arbovirus-infected patients. PLoS Negl Trop Dis 9(1), e3445 (2015).
Liu, H. et al. Transcriptional diversity during monocyte to macrophage differentiation. Immunol Lett 117(1), 70–80 (2008).
Triggiani, M. et al. Differentiation of monocytes into macrophages induces the upregulation of histamine H1 receptor. J Allergy Clin Immunol 119(2), 472–481 (2007).
Pohjala, L. et al. Inhibitors of alphavirus entry and replication identified with a stable Chikungunya replicon cell line and virus-based assays. PLoS One 6(12), e28923 (2011).
Pastorino, B. et al. Development of a TaqMan RT-PCR assay without RNA extraction step for the detection and quantification of African Chikungunya viruses. J Virol Methods 124(1-2), 65–71 (2005).
Plaskon, N. E. et al. Accurate strand-specific quantification of viral RNA. PLoS One 4(10), e7468 (2009).
Saeed, A. I. et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34(2), 374–378 (2003).
Acknowledgements
We acknowledge the study participants and healthy volunteers for providing the plasma and blood samples. We would also like to express our gratitude to the research staffs and clinical staffs from CDC/TTSH for patient enrolment, study coordination, and collection of patient samples. We would also want to thank Martial Jaume (Hong Kong University Pasteur Research Center) for providing the transformed ST486 cell lines. We would also like to thank Andres Merits for providing the Zs-Green tagged CHIKV infectious clone and the rabbit anti-CHIKV nsP2 polyclonal antibodies. In addition, we express our gratitude to the SIgN Flow Cytometry core and to Kenneth Low and Adeline Lin for their technical assistance. We thank P. Bergeat, Director of the Laboratoire Français du Fractionnement et des Biotechnologies, for his support. This project was funded in part by the EU project FP7-ICRES (grant no. 261202), and LabEx IBEID, Institut Pasteur and Inserm. Lastly, we are grateful to Kai-Er Eng for editing of the manuscript. The sponsors were not involved in the design of the study, gathering of the data, analysis or drafting of the manuscript.
Author information
Authors and Affiliations
Contributions
F.M.L., T.C., B.S.C., R.Y.O., Z.H. and Y.W.K. performed the experiments. F.M.L., T.C., A.C., Y.S.L., Y.W.K., L.R., M.L. and L.F.P.N. analyzed the data. A.C. and Y.S.L. provided the patient samples. F.M.L., T.C., Y.W.K., L.R., M.L. and L.F.P.N. conceptualized the study and wrote the manuscript. All authors read and approved of the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lum, FM., Couderc, T., Chia, BS. et al. Antibody-mediated enhancement aggravates chikungunya virus infection and disease severity. Sci Rep 8, 1860 (2018). https://doi.org/10.1038/s41598-018-20305-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-20305-4
This article is cited by
-
New Vaccines on the Immediate Horizon for Travelers: Chikungunya and Dengue Vaccines
Current Infectious Disease Reports (2023)
-
Investigations on the effects of anti-Leishmania major serum on the progression of Leishmania infantum infection in vivo and in vitro – implications of heterologous exposure to Leishmania spp
Parasitology Research (2021)
-
Antibody-based therapeutic interventions: possible strategy to counter chikungunya viral infection
Applied Microbiology and Biotechnology (2020)
-
Effects of Chikungunya virus immunity on Mayaro virus disease and epidemic potential
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
