
Abstract
Type I interferon (IFN) pathways are significant in SLE pathogenesis. Less is known about the utility of measuring markers of IFN activity in patients, or whether patient subsets with different profiles exist. We explored the longitudinal associations of IFN-induced chemokines with disease activity in a cohort of SLE patients. We calculated a validated composite score (IFN-CK) of three type I IFN-inducible chemokines (CCL2/CXCL10/CCL19) measured in 109 SLE patients (median 7 occasions over 3.2 years). Longitudinal associations of IFN-CK score with disease activity (SLEDAI-2K) and other variables were assessed using general estimating equation (GEE) methods. IFN-CK was detectable in all patients. SLEDAI-2K was significantly associated with IFN-CK, damage score and prednisolone dose. SLEDAI-2K remained significantly associated with IFN-CK over time after adjustment of covariates. Patients with high time-adjusted mean IFN-CK had lower complement and higher time-adjusted disease activity. Concordance between IFN-CK and SLEDAI-2K varied widely among patients, with some individuals having none, others weak, and a subset very high concordance. In summary in our cohort of SLE patients, serum IFN-CK varied over time with disease activity, but with wide variation in concordance. Differing relationships between IFN pathway activation and disease activity may be valuable in assigning patients to emerging IFN-pathway targeting treatments.
Similar content being viewed by others


Introduction
Systemic lupus erythematosus (SLE; lupus) is a chronic autoimmune disease characterised by immunologically-mediated inflammatory activity across multiple organ systems and the potential for irreversible end organ damage1. Striking clinical diversity is a hallmark of SLE, with variation in clinical manifestations, disease activity, and long term outcomes1,2. Similarly, a wide range of genetic, transcriptional, immunological and cellular events have been associated with SLE, but findings do not uniformly apply across patients. It is likely that this intrinsic biological diversity among patients has contributed to disappointing results in studies of promising targeted therapies, when applied to a pooled disease population3,4.
One of the best characterised biological pathways in SLE is the type I interferon (IFN) pathway, with finding of an IFN “gene signature”, a pattern of increased transcriptional activity of selected IFN-inducible genes, reported in a majority of SLE patients and replicated across multiple cohorts5. Given these associations, type I IFN is an attractive therapeutic target in SLE and multiple treatment strategies are under investigation6. A recent Phase II trial of anifrolumab, an anti-IFN receptor monoclonal antibody, found that responses were predicted by baseline IFN signature, suggesting a role for biological stratification in selecting patients for targeted therapies7. The association of changes in such signatures with disease activity is less clear8. In contrast, serum concentrations of certain chemokines induced by type I IFN that associate tightly with IFN-inducible gene expression9,10 have been reported to correlate with disease activity in previous studies10,11. A previous cross sectional study by the authors investigating the association of IFN-CK with disease parameters in SLE revealed findings including an association between IFN-CK score and disease activity12. Such previous studies have not included paired longitudinal serum biomarker levels with clinical data. It is not clear, therefore, whether variation in IFN-induced serum proteins vary with disease activity across time, or behave similarly in all patients.
The aim of the current study was to determine whether a composite index of type I IFN induced chemokines was associated with SLE disease activity over time, and particularly whether the strength of such relationships varied among patients, by using data from a large longitudinal set of paired clinical visits and serum samples. Our findings indicate a longitudinal association between type I IFN-induced chemokines and SLE disease activity, and identify the existence of divergence among patients according to the level of concordance between these measures. These findings suggest that measurement of IFN-induced chemokines over time may have utility in evaluating associations between disease activity and IFN pathway activation in SLE.
Results
Patient characteristics
Data from 944 visits in 109 patients were used in this analysis. Table 1 summarises the characteristics of the study population. In brief, 83% of the study group were female with a mean age at enrolment of 41.7 years (13.2). Half the subjects were of Asian ethnicity, with most others of European descendent. Median length of study follow-up was 3.2 years, and patients had a median of 7 clinic visits and matched serum samples during the study period. The time-adjusted mean SLEDAI-2K (AMS) of the study group was 4.4, with a TAM-PGA of 0.5. Patients used an average (TAM) prednisolone dose of 5.0 mg/day. Over 61% had organ damage (median SDI = 1) and 75% experienced flares during the study period. Assessed using SLEDAI-2K domains, serological activity was the most common manifestation of active disease (88%) followed by cutaneous (64%) and renal (42%) activity.
Longitudinal associations of disease activity
Univariable GEE analyses showed several factors to be statistically significantly associated with SLEDAI-2K at each visit, as shown in Table 2. Increasing age was associated with a small but significant reduction in SLEDAI-2K. Both PGA and SDI score were positively associated with an increase in SLEDAI-2K, as was prednisolone dose; an increase of dose by 10 mg/d was associated with an increase of disease activity by 1.1 SLEDAI-2K units. We also observed a positive association between IFN-CK score and SLEDAI-2k. An increase of one unit in IFN-CK score was significantly associated with an increase in SLEDAI-2K of 0.7 (RC = 0.73, (95% CI: 0.12, 1.43) p = 0.02). Gender and ethnicity were not statistically significantly associated with disease activity. After adjustment using multivariable GEE analysis, prednisolone dose, PGA, SDI, and age remained statistically significantly associated with SLEDAI-2K (Table 3). After adjustment, IFN-CK score also remained significantly associated with SLEDAI-2K, wherein one unit increase in IFN-CK was associated with a SLEDAI-2K increase of 0.5 ((95% CI: 0.04, 0.98), p-value = 0.03). This method confirms the longitudinal association of IFN-CK with disease activity in SLE.
Comparison of low and high IFN-CK groups
We next compared clinical characteristics between patients with high and low time-adjusted mean IFN-CK scores. Low IFN-CK was deemed ≤0.3 and high IFN-CK > 0.3 based on the median value of all time-adjusted mean IFN-CK scores. Serum complement levels were significantly lower in the high IFN-CK group (Table 4). As expected given the results above, there was also numerically greater disease activity over time in the high IFN-CK group, with a median AMS in the high IFN-CK group of 5.2 compared with 3.6 in the low IFN-CK group (p = 0.06). Frequency of dsDNA positivity was also numerically higher in the high IFN-CK group (83% vs. 68%, p = 0.07).
Concordance between IFN-CK and SLEDAI in individual patients
When plotting individual patient time series, it was noticeable that some patients had concordance over time between IFN-CK and SLEDAI-2K scores, while others did not. In order to categorise patients according to IFN-CK:SLEDAI-2K concordance, a correlation coefficient for each patient was calculated. Of the 109 patients, 7 patients had identical SLEDAI-2K scores at all visits, therefore correlation coefficients were not calculated. Of the remaining 102 patients, 39 (38%) had a correlation coefficient (r) less than or equal to zero (r ≤ 0). Of the 63 patients with r > 0, 15 patients had r ≥ 0.7, demonstrating strong concordance between IFN-CK and SLEDAI-2K (Fig. 1). Interestingly, Bland-Altman graphs, used to examine the extent of agreement between two variables by plotting the differences between the pairs of measurements against the mean of each pair, indicated greater concordance for lower values of SLEDAI-2K and IFN-CK score, even among patients where these was a strong correlation over time between these variables (r ≥ 0.7) (Fig. 2).

IFN-CK SLEDAI-2K concordance in patients with r ≥ 0.7. Concordance between type I interferon inducible chemokine score (IFN-CK) and disease activity (SLEDAI-2k) was assessed in individual patients. A strong concordance between these variables over time (r ≥ 0.7) was seen in a subset of 15 patients.

Bland-Altman’s limits-of-agreement for (a) overall study population; (b) patient group with correlation coefficient r ≥ 0.7 for IFN-CK and SLEDAI-2K. Bland-Altman graphs were generated to examine the extent of agreement between type I interferon inducible chemokine score (IFN-CK) and disease activity (SLEDAI-2k) in the overall study population (graph 2a) and in the patient group with correlation coefficient r ≥ 0.7 for IFN-CK and SLEDAI-2k (graph 2b). Greater concordance was seen for lower values of SLEDAI-2k and IFN score.
We further investigated whether patients in this sub-group differed from the groups with r ≤ 0 and those with r > 0 but < 0.7). No differences were observed in standard indicators of disease activity and severity such as AMS, PGA, or number of ACR criteria. However, patients in the high correlation group (r ≥ 0.7) had significantly fewer visits and lower time adjusted mean and cumulative prednisolone doses (Table 5).
Discussion
Serum concentrations of IFN-induced chemokines are a surrogate marker of activation of the type I IFN system, which has been strongly implicated in the pathogenesis of SLE. IFN-induced gene transcriptional signatures measured in peripheral blood have been most often used to interrogate the IFN system in human SLE, but the most thorough longitudinal studies to date have not demonstrated strong associations of IFN gene signatures with disease activity over time8. Using a validated composite score derived from serum concentrations of IFN-induced chemokines, in a prospectively-followed multi-ethnic lupus cohort, we have demonstrated a significant longitudinal association between this marker of type I IFN activity and SLE disease activity. Moreover, we demonstrate that the strength of the relationship between disease activity and IFN-CK varies markedly between patients, with some patients showing high, and others no, concordance between these variables.
While several studies have suggested an association between the type I IFN system and SLE disease activity, very few have had the longitudinal design necessary to investigate fluctuations with disease activity in patients over time. In a large longitudinal study of paediatric SLE, no strong associations of IFN transcriptional signatures with disease activity over time were found8. We have here demonstrated, using the GEE method, that an increase in type I IFN activity as measured by IFN-CK score is associated with an increase in SLEDAI, and this association remains significant on multivariable analysis adjusting for other variables associated with disease activity. This adds support to prior observations unadjusted for such confounders which suggested that patients with high IFN-induced chemokines at baseline were more likely to flare over the subsequent year, and that IFN-CK score may rise with disease flares11. The current study advances on our previous work associating IFN-CK with increased disease activity, by analysing paired longitudinal biomarker and clinical data, better reflecting the association between fluctuations among patients in IFN-CK score and SLEDAI, while accounting for correlations within patients.
Our categorical analysis also suggested patients in the high IFN-CK group had higher markers of disease activity over time, including higher AMS, reduced complement levels and higher anti-dsDNA. Serological markers of disease activity such as complement (C3 and C4) have been found to inversely correlate with type I IFN activity in SLE in previous studies of IFN-induced gene transcripts13,14. The biological basis of this association may relate to the induction of the type I IFN system by immune complexes, neutrophil extracellular traps or other endogenous stimuli in SLE15. Type I IFN transcriptional signatures have also been strongly linked to renal and to a lesser extent CNS and haematological manifestations in SLE16. In our study IFN-CK scores had the strongest association with overall SLEDAI-2k rather than individual organ manifestations, although cutaneous and haematological manifestations were the strongest contributors to this association (data not shown). This discrepancy in findings between studies may be due to the IFN gene signature and IFN induced serum chemokines representing two slightly different measures of the type I IFN system17. Gene transcription peripheral blood signatures appear to be a more static measure, suitable for predicting disease phenotype even though not associated with disease activity over time8,18,19,20. Consistent with this, standard doses of corticosteroids which reduce disease activity do not suppress the IFN signature21.
In addition to confirming a longitudinal association between IFN-CK score and SLEDAI in an overall cohort, our study also identified the important finding that patients vary widely in their concordance between these variables, with the rise and fall of these variables mirroring one another closely in some patients but not at all in others. Interestingly, concordance appeared to be greater in patients with lower IFN-CK scores and less active disease. One possible explanation is the effect of glucocorticoids on the expression of CCL2, CCL19, and CXCL10 in response to type I IFN. The induction of these chemokines by Type I IFN is mediated by promoters that are highly sensitive to glucocorticoids, such as nuclear factor kappa B22,23. In a previous study investigating the serum IFN-CK score used here, patients with inactive disease had significantly lower IFN-CK scores when on high doses of prednisolone (≥10 mg daily) compared with low doses or no prednisolone24. In our study, doses of prednisolone were significantly lower in the high concordance group. Increased doses of prednisolone could therefore have impacted on the ability to discern correlations with IFN-CK in higher disease activity groups.
Importantly, markers that relate IFN system activity to disease activity may be of utility in assigning patients to treatment with emerging anti-IFN-pathway drugs. In the Phase II trial of anti-IFN receptor monoclonal antibody anifrolumab, IFN gene signature was used to biologically stratify patients, with a high gene signature predicting response to therapy25. This raises interest in the role of a more downstream marker of type I IFN activity, such as IFN-CK, in such trials. Given that IFN-CK (in contrast to IFN gene signature) appears to correlate not only with IFN activity but also fluctuations in disease activity, such a marker conceivably has utility in stratifying patients and monitoring response to treatment.
A unique feature of our cohort is its ethnic composition, with close to half of all patients being of Asian background. While type I IFN activity has been confirmed in Asian SLE patients using IFN induced gene transcripts9,26, previous studies of IFN-induced chemokines have been limited mainly to patients of Caucasian, African American and Hispanic ethnicity. We recently reported a cross-sectional study of serum IFN-CK in SLE in which the association of Asian ethnicity with higher disease activity was independent of IFN-CK12. Given this it is interesting to note that Asian patients represented only 27% of those with high concordance between IFN-CK and SLEDAI, despite comprising close to 50% of the overall cohort. This lends further support to the hypothesis that type I IFN may be a less predominant biological driver of disease activity, or that IFN-CK less well reflects IFN pathway activation, in SLE in Asians compared to other ethnicities. This is also the longest-duration study to date investigating associations between type I IFN and disease activity in SLE, and was performed using a serum archive tightly linked to prospectively acquired disease activity data. Limitations of the current study include being limited to 109 patients; a larger cohort may have allowed demonstration of additional significant associations of IFN-induced chemokines.
In conclusion, we have confirmed a longitudinal association between serum concentration of type I IFN-induced chemokines and disease activity in SLE. Importantly, Type I IFN induced chemokines were detectable in 100% of patients, in contrast to many other cytokines that are only detectable in a subset of cases27,28. Our data also suggest the existence of subgroups of patients with widely variable concordance between IFN-CK score and SLEDAI, indicating that the relationship between IFN-CK and disease activity is present only in some patients. These data lend further support to the potential of type IFN-induced serum pro-inflammatory proteins, as opposed to IFN-induced gene signatures, to identify the subgroup of patients with both active disease and activation of the Type I IFN system. These findings may be relevant to the stratification of patients undergoing therapy with treatments targeting the Type I IFN system.
Methods
Study design and participants
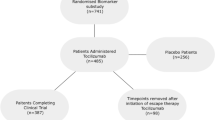
Data were prospectively acquired between June 2007 and January 2012 from patients who attended the SLE Clinic at Monash Medical Centre, a tertiary referral public hospital in Melbourne, Australia, who fulfilled the American College of Rheumatology (ACR) criteria for the classification of SLE29, were over 18 years of age, had complete data available and provided written informed consent27,30. Patients were included in the current study if they had complete clinical data and a matched serum sample available for at least three separate clinic visits. Ethics approval for this study was obtained from, and the study carried out in accordance with, the Monash Health Human Research Ethics Committee.
Patient information
Patients were seen at 3–6 monthly intervals, or more frequently according to clinical need. At each clinic visit disease activity was documented using the 2000 modification of the SLE disease activity index (SLEDAI-2K)31. A measure of disease activity over time was generated using the adjusted mean SLEDAI-2K (AMS)31. Disease-related damage was assessed at baseline and annually using the Systemic Lupus International Collaborating Clinics (SLICC) Damage Index (SDI)32. Birth date, gender, year of disease onset and ethnicity were recorded at baseline. Autoantibody positivity was documented at baseline and included ANA titre, anti-double stranded DNA (anti-dsDNA) positivity and antibodies to a range of extractable nuclear antigens (ENA) including ribonucleoprotein (RNP), Sm, Ro, and La.
Measurement of serum concentrations of IFN induced chemokines (IFN-CK)
Patient serum samples were obtained and stored at −800C until use as described12,27,28. Activation of type I IFN pathways was assessed by measurement of three type I IFN inducible chemokines (CCL2, CXCL10 and CCL19) as described by Bauer11. Concentrations of serum CCL2, CXCL10 and CCL19 were determined in each sample using sandwich ELISA, as previously described12,27. Briefly, 96-well plates (Immunoplates, Nunc, Roakilde, Denmark) were coated with primary antibody (anti-human CCL2, CXCL10 or CCL19; R&D Systems, Minneapolis, MN, USA) and incubated overnight before being blocked by 1% bovine serum albumin. After washing, recombinant human protein standards and serum samples were added in duplicate and incubated overnight. Binding was detected using a biotinylated goat anti-human antibody (R&D Systems) and streptavidin conjugated to horseradish peroxidase (Silenus, Melbourne, Australia). Colour was developed with 3,3′5,5′-tetramethylbensidine (Sigma, Sydney, Australia) and read at 450 nm. In order to integrate the results obtained for the three type I IFN induced chemokines, a composite IFN-CK score was derived for each sample, in the manner validated by Bauer et al.11: concentrations above the 95th centile for each chemokine were assigned a value of one, with the remaining concentrations scaled to this percentile. Scaled values for each chemokine were then added to produce a final IFN-CK score ranging from 0 to 3.
Statistical Analysis
Statistical analyses were performed using Stata version 14 (StataCorp, College Station, Texas, USA). Continuous variables were described either as mean (standard deviation (SD)) or median (interquartile range [IQR], range) according to data distribution; categorical variables were described as frequency (%). Time adjusted means were calculated for several continuous variables to account for varying time intervals between visits. High time adjusted mean IFN-CK was defined as a score above the median value of 0.3. Several factors including demographics and disease characteristics were compared between low (<0.3) and high time adjusted means IFN-CK (≥0.3) and p-values were derived using t-tests, Wilcoxon rank sum tests and Pearson’s chi-squared tests to compare means, medians and percentages respectively.
The generalised estimating equation (GEE) method was used to examine longitudinal associations of SLEDAI-2K with several variables measured repeatedly (e.g. complement, PGA, IFN-CK). This is in contrast to previous cross-sectional studies where associations between IFN-CK and disease activity were investigated using linear and logistic regression models12. The GEE approach specifies how the outcome of a subject changes with covariates from one measurement to the next, while allowing for the correlation between repeated measurements on the same subject over time. Since SLEDAI-2K is a continuous variable, we specified Gaussian distribution for the family along with an identity link, and exchangeable correlation matrix in the model. Robust standard errors were derived adjusting for patient clustering. The QIC (quasilikelihood under the independence model criterion) method was used to assess the best working correlation structures and best subsets of covariates for GEE analyses. Univariable GEE models were performed for each independent variable and a p-value threshold of 0.1 was applied for variable selection for the multivariable model. Potential collinearity between independent variables was also assessed before including these in the multivariable model. Results were reported as regression coefficients (RC, exponentiated beta coefficients) with corresponding 95% confidence intervals (95% CI). A p-value < 0.05 was considered statistically significant.
In addition, we determined the concordance between IFN-CK and SLEDAI-2K in individual study participants using Pearson’s correlation coefficients (r). Based on the r values, patients were grouped in to three categories: r < 0 (no correlation); 0 < r < 0.7 (intermediate correlation), and r > = 0.7 (strong correlation). We used Bland-Altman plots to determine the degree of concordance between SLEDAI-2K and IFN-CK throughout the range of possible values for these variables, in patients where the correlation between these variables was strong (r > 0.7). Patient characteristics were compared among these three categories: means were compared using ANOVA, medians were compared using the Kruskal-Wallis test and proportions were compared using Pearson’s chi-Squared test.
References
Tsokos, G. C. Systemic lupus erythematosus. in N Engl J Med 365, 2110–2121 (2011).
Smith, P. P. & Gordon, C. Systemic lupus erythematosus: Clinical presentations. in Autoimmun Rev 10, 43–45 (2009).
Franklyn, K., Hoi, A., Nikpour, M. & Morand, E. F. The need to define treatment goals for systemic lupus erythematosus. in Nature Reviews Rheumatology 10, 567–571 (2014).
Jourde-Chiche, N., Chiche, L. & Chaussabel, D. Introducing a New Dimension to Molecular Disease Classifications. Trends in Molecular Medicine 22, 451–453 (2016).
Crow, M. K. Type I interferon in the pathogenesis of lupus. J Immunol 192, 5459–5468 (2014).
Crow, M. K., Olferiev, M. & Kirou, K. A. Targeting of type I interferon in systemic autoimmune diseases. in Transl Res 165, 296–305 (2015).
Furie, R. et al. Anifrolumab, an Anti-Interferon-Alpha Receptor Monoclonal Antibody, in Moderate to Severe SLE. Arthritis and rheumatism 69, 376–386 (2017).
Banchereau, R. et al. Personalized Immunomonitoring Uncovers Molecular Networks that Stratify Lupus Patients. Cell 165, 1548–1550 (2016).
Fu, Q. et al. Association of elevated transcript levels of interferon-inducible chemokines with disease activity and organ damage in systemic lupus erythematosus patients. in Arthritis Research & Therapy 10, R112 (2008).
Bauer, J. W. et al. Elevated serum levels of interferon-regulated chemokines are biomarkers for active human systemic lupus erythematosus. in PLoS Med. 3, e491 (2006).
Bauer, J. W. et al. Interferon-regulated chemokines as biomarkers of systemic lupus erythematosus disease activity: A validation study. in Arthritis Rheum 60, 3098–3107 (2009).
Connelly, K. L., Kandane-Rathnayake, R., Hoi, A., Nikpour, M. & Morand, E. F. Association of MIF, but not type I interferon-induced chemokines, with increased disease activity in Asian patients with systemic lupus erythematosus. Sci Rep 6, 29909 (2016).
Petri, M. et al. Longitudinal expression of type I interferon responsive genes in systemic lupus erythematosus. in Lupus 18, 980–989 (2009).
Landolt-Marticorena, C. et al. Lack of association between the interferon-{alpha} signature and longitudinal changes in disease activity in systemic lupus erythematosus. in Ann. Rheum. Dis. 68, 1440–1446 (2009).
Elkon, K. Type I Interferon and Systemic Lupus Erythematosus. in Journal of Interferon & Cytokine Research (2011).
Bennett, L. et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. in J. Exp. Med. 197, 711–723 (2003).
Crow, M. K. & Kirou, K. A. Interferon-induced versus chemokine transcripts as lupus biomarkers. Arthritis Research & Therapy 10, 126 (2008).
Petri, M. et al. The systemic lupus erythematosus interferon signature is associated with current activity and is also predictive of hematologic and mucocutaneous disease activity at the next visit. Arthritis Rheum S2, S464 (2005).
Petri, M. et al. Longitudinal expression of type I interferon responsive genes in systemic lupus erythematosus. Lupus 18, 980–989 (2009).
Landolt-Marticorena, C. et al. Lack of association between the interferon-{alpha} signature and longitudinal changes in disease activity in systemic lupus erythematosus. Ann. Rheum. Dis. 68, 1440–1446 (2009).
Guiducci, C. et al. TLR recognition of self nucleic acids hampers glucocorticoid activity in lupus. Nature 465, 937–941 (2010).
Pietila, T. E. et al. Multiple NF-kappaB and IFN regulatory factor family transcription factors regulate CCL19 gene expression in human monocyte-derived dendritic cells. J Immunol 178, 253–261 (2007).
Buttmann, M., Berberich-Siebelt, F., Serfling, E. & Rieckmann, P. Interferon-beta is a potent inducer of interferon regulatory factor-1/2-dependent IP-10/CXCL10 expression in primary human endothelial cells. J Vasc Res 44, 51–60 (2007).
Bauer, J. W. et al. Interferon-regulated chemokines as biomarkers of systemic lupus erythematosus disease activity: A validation study. Arthritis Rheum 60, 3098–3107 (2009).
Furie, R. et al. A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. in Arthritis Rheum 63, 3918–3930 (2011).
Yuan, Y. J., Luo, X. B. & Shen, N. Current advances in lupus genetic and genomic studies in Asia. in Lupus 19, 1374–1383 (2010).
Vincent, F. B., Northcott, M., Hoi, A., Mackay, F. & Morand, E. F. Clinical associations of serum interleukin-17 in systemic lupus erythematosus. in Arthritis Research & Therapy 15, R97–R97 (2013).
Rudloff, I. et al. Brief Report: Interleukin-38 Exerts Antiinflammatory Functions and Is Associated With Disease Activity in Systemic Lupus Erythematosus. in Arthritis Rheumatol 67, 3219–3225 (2015).
Hochberg, M. C. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. in Arthritis Rheum 40, 1725 (1997).
Vincent, F. B., Northcott, M., Hoi, A., Mackay, F. & Morand, E. F. Association of serum B cell activating factor from the tumour necrosis factor family (BAFF) and a proliferation-inducing ligand (APRIL) with central nervous system and renal disease in systemic lupus erythematosus. in Lupus 22, 873–884 (2013).
Gladman, D., Ibañez, D. & Urowitz, M. Systemic lupus erythematosus disease activity index 2000. in J. Rheumatol. 29, 288–291 (2002).
Gladman, D. D. et al. The Systemic Lupus International Collaborating Clinics/American College of Rheumatology (SLICC/ACR) Damage Index for Systemic Lupus Erythematosus International Comparison. in J. Rheumatol. 27, 373–376 (2000).
Acknowledgements
The authors gratefully acknowledge the contributions of patients of the Monash Lupus clinic, and of Monash Lupus clinicians including Ms Sue Morton RN, Professor Richard Kitching, and Dr Joanne Ghali. Financial support: EFM and MN have received research grants from the National Health and Medical Research Council of Australia. EFM has received research support from Astra-Zeneca, Pfizer, Bristol-Myers Squibb, Janssen, UCB, CSL, and GSK. MN has received research support from Actelion, Bayer, CSL Biotherapies, GlaxoSmithKline and Pfizer. Industry affiliations: EFM has received honoraria from or had paid advisory roles with Astra-Zeneca, Pfizer, Bristol-Myers Squibb, Janssen, UCB, Baxalta, and Mitsubishi Tanabe.
Author information
Authors and Affiliations
Contributions
K.C., R.K. and E.M. wrote the main manuscript text. R.K. and M.H. performed statistical analysis and prepared the tables and figures. A.H. and M.N. reviewed the manuscript. All authors approved the manuscript prior to submission.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Connelly, K.L., Kandane-Rathnayake, R., Huq, M. et al. Longitudinal association of type 1 interferon-induced chemokines with disease activity in systemic lupus erythematosus. Sci Rep 8, 3268 (2018). https://doi.org/10.1038/s41598-018-20203-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-20203-9
This article is cited by
-
Whole blood hydroxychloroquine: Does genetic polymorphism of cytochrome P450 enzymes have a role?
Clinical and Experimental Medicine (2023)
-
Potential relevance of type I interferon-related biomarkers for the management of polygenic autoimmune rheumatic diseases with childhood onset
Clinical Rheumatology (2023)
-
Investigation of type I interferon responses in ANCA-associated vasculitis
Scientific Reports (2021)
-
The early local and systemic Type I interferon responses to ultraviolet B light exposure are cGAS dependent
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
