
Abstract
Vibrio cholerae inhabits aquatic environments worldwide and has over 200 recognized serogroups classified by O-polysaccharide specificity. Here, we report that V. cholerae selects either of two genetic traits during their evolution. Sequencing of the specific gene locus MS6_A0927 revealed that 339 of 341 strains of V. cholerae and closely related Vibrio species originating from 34 countries over a century carried either metY (M) (~1,269 bp) or luxR-hchA (LH) (~1,600 bp) genes, and consequently those vibrios were separated into two clusters, M (45.4%) and LH (54.6%). Only two strains contained both M and LH in the same locus. Moreover, extensive polymorphisms in those genes were detected in M and LH with 79 and 46 sequence variations, respectively. V. cholerae O1 strains isolated from cholera outbreaks worldwide, and some non-O1 strains evolving from O1 via exchange of genes encoding cell surface polysaccharides possessed LH alleles. Analysis of polymorphisms in the gene locus implicated a high degree of genetic diversity and identical subpopulations among the V. cholerae species.
Similar content being viewed by others

Introduction
Vibrio cholerae is a gram-negative bacterial pathogen responsible for cholera, and several million cholera cases including 21,000–143,000 deaths occur worldwide each year1. Serological grouping of V. cholerae has identified up to 206 O-serogroups2. Epidemic/pandemic cholera is typically ascribed to serogroup O1; however, in 1992, a novel serogroup O139 V. cholerae caused outbreaks in Asian countries3. V. cholerae carries several virulence-related genes to provoke pathogenic processes in the infected hosts. The key virulence factors of serogroups O1 and O139 include cholera toxin (CT), which is responsible for profuse watery diarrhea, and a pilus colonization factor known as toxin-coregulated pilus (TCP). Although most non-O1/non-O139 or environmental isolates of V. cholerae do not produce CT and lack the cholera toxin genes, some strains possess heat-stable enterotoxin (Stn)4, hemolysin (HlyA)5,6, repeat in toxin (RTX)7, Cholix toxin (ChxA)8,9, hemagglutinin protease (HAP)10, type 6 secretion system (T6SS)11, or type III secretion system (TTSS)12. However, the pathogenic mechanisms of these isolates remain to be elucidated.
High throughput sequencing facilitates the rapid and accurate identification of virulence factors of pathogenic bacteria, and can be used to identify the pathways of infectious disease transmission13,14,15. Although genomic technologies are rapidly evolving, their widespread implementation in clinical microbiology laboratories and for monitoring public health is limited owing to the need for effective semi-automated pipelines, standardized quality control and data interpretation, bioinformatics expertise, and infrastructure16. Relatedness and differences among V. cholerae isolates have been investigated by several molecular fingerprinting methods for a prolonged duration17. Pulsed field gel electrophoresis (PFGE) has been used frequently for typing of the O1 and O139 serogroups of V. cholerae18,19. Although PFGE is highly reproducible and its discriminatory power is sufficiently high, it is laborious, and is limited with regard to intra- and inter- laboratory comparison compared to sequence-based methods17,20. Multilocus sequence typing (MLST) overcomes the poor portability of traditional and older molecular typing approaches20. It is a technique whereby several internal control genes (loci) are sequenced, and relatedness among isolates is displayed as a dendrogram constructed using the matrix of pairwise differences between the allelic profiles. This approach provides high discriminatory power and is informative for the study of V. cholerae21,22. Single locus sequence typing (SLST) also has been widely used to determine the relationships in other organisms23,24,25.
We found that V. cholerae O1 genomes possess either metY or hchA/luxR on the specific gene locus MS6_A0927 on a conserved syntenic region of the chromosome II26. The locus was evidence that a unique O1 strain MS6 isolated from a diarrheal patient was distinguished from pandemic O1 strains and was most closely related to US Gulf Coast strains. MS6 and US Gulf Coast strains carried the metY gene in the locus, whereas the seventh cholera pandemic O1 strains carried luxR-hchA genes. In this paper, we report the prevalence, distribution, and sequence diversity of alternative genes located in MS6_A0927 among a large population of diverse V. cholerae and discuss their future evolutionary aspects.
Results and Discussion
V. cholerae separates into two clusters based on the locus MS6_A0927
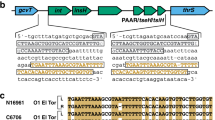
We investigated the distribution of the metY (M) gene and luxR-hchA (LH) genes among vibrios. First, BLAST searches were performed using M (1,269 bp) from V. cholerae strain MS6 and LH from V. cholerae strain O395 (1,600 bp) as query sequences against 186 genomes including 178 V. cholerae, 6 V. mimicus, 1 V. metecus, and 1 V. parilis obtained from the NCBI database (www.ncbi.nlm.nih.gov). All strains carried either M (n = 57) or LH (n = 128), except for strain 87395 that was revealed to carry both M and LH (Table S1). We then designed a multiplex PCR system for detection of M, LH, toxR, VC2346, tcpA, and ctxAB, and determined the sequences of M and LH in the locus MS6_A0927 of 153 strains of non-O1 V. cholerae and 2 strains of V. mimicus (Table 1). Eleven genotypes were obtained by the multiplex PCR assay, including toxR/M (53.5%), toxR/LH (30.3%), ctxAB/tcpA/toxR/VC2346/LH (3.2%), ctxAB/tcpA/toxR/M (2.6%), ctxAB/tcpA/toxR/LH (2.6%), toxR/VC2346/M (2.6%), tcpA/toxR/M (1.9%), tcpA/toxR/LH (1.3%), tcpA/toxR/VC2346/M (0.6%), tcpA/toxR/VC2346/LH (0.6%), and M (0.6%). Two major virulence genes, ctxAB and tcpA, were detected in 13 strains from 9 different serogroups (O8, O26, O37, O44, O48, O49, O75, O141, and O191). Moreover, the M gene was detected in 94 V. cholerae and 2 V. mimicus (62%, 96/155), whereas 59 V. cholerae (38%, 59/155) carried LH. PCR using the primers MS6_A0926F and MS6_A0928R prior to sequencing of M or LH amplified target regions with expected molecular sizes, 1.96 kb or 2.13 kb, in all test strains except for V. cholerae O35 N2_17 (Fig. 1). The DNA sequences of the PCR products were determined and the existence of M and LH was confirmed. Strain N2_17 carried both M and LH in the locus, although the multiplex PCR failed to detect its M gene. L and H were always detected together.

Strategy for determination of the M and LH sequences in the locus MS6_A0927. (A) Schematic diagram of M (upper) and LH (lower) regions between MS6_A0926 and MS6_A0928 and positions of sequencing primers. (B) PCR products obtained using the universal primers MS6_A0926 and MS6_A0928. Lane M, 1 kb DNA ladder; lane 1, Vibrio cholerae O1 El Tor MS6; lane 2, V. cholerae O1 El Tor N16961; lane 3, V. cholerae O35 N2_17. The expected sizes of PCR products for MS6 (M) and N16961 (LH) were 1.96 and 2.13 kb, respectively. Arrowhead indicates the amplicon that contained both sequences M and LH. All serotyped strains of non-O1 V. cholerae (n = 153) and V. mimicus (n = 2) from our bacterial stock were examined by this strategy and the results are presented in Table 1.
Overall, 339 of 341 strains of vibrios, mostly V. cholerae, carried either M or LH in the locus MS6_A0927, and consequently those vibrios were separated into two clades: M (45.4%) and LH (54.6%) (Fig. 2). The remaining two strains of V. cholerae carried both M and LH in the same locus; thus, all V. cholerae strains carried either M or LH, or both. The M/LH sequence profiling further classified the 341 strains of vibrios into 127 subclades.

Vibrio cholerae organisms were classified into two clusters, M and LH. Dendrograms were constructed based on the genes M or LH from 341 Vibrio strains by the neighbor-joining method using MEGA v.6.0. Scale bars indicate nucleotide substitutions per site. Color coding is based on the presence of genes for cholera toxin (ctxAB) and toxin-coregulated pilus (tcpA): red, ctxAB+, tcpA+; blue, ctxAB−, tcpA+; green, ctxAB+, tcpA−; black, ctxAB−, tcpA−. The M and LH genes exhibited 79 and 46 sequence variations, respectively. The predominant subclades of LH and M were LH1, LH2, M4, and M6 in the order of description.
Evolutionary selection of M or LH in V. cholerae
We detected LH only in V. cholerae and not in other vibrios. In contrast, M was present in strains of V. cholerae, V. mimicus, V. parilis, V. metecus, V. splendidus, V. cyclitrophicus, V. tasmaniensis, V. tubiashii, and V. jasicida, but not in V. fluvialis, V. furnissii, V. parahaemolyticus, V. vulnificus, and V. anguillarum in publicly available database. The nucleotide sequence of M showed higher similarity within a single species than between different species (Fig. S1). Exceptionally, M from V. cholerae CP1037(10) showed higher similarity to that from in V. mimicus than to those of most V. cholerae. V. mimicus, V. parilis, and V. metecus carry M on the MS6_A0927 in chromosome II, similarly to V. cholerae, whereas V. splendidus, V. tasmaniensis, and V. cyclitrophicus carry M near the homologue of the glutamine-fructose-6-phosphate transaminase gene (MS6_0339), which is located on chromosome I. The latter three species and V. tubiashii and V. jasicida harbored homologous of MS6_A0926 and MS6_A0928 at various distances (0.2 kb to 10 kb) from the two loci. V. fluvialis and V. furnissii, which are more closely related to V. cholerae and V. mimicus27, carried homologues of the aldehyde dehydrogenase (MS6_1585) and sigma 54-dependent transcriptional regulator (MS6_1586) genes between the two loci, although they did not carry M and LH. The alternative selection of M/LH at MS6_A0927 would have occurred in ancestral populations of V. cholerae.
The distributions of M and LH in the strains of V. cholerae were generally associated with genome-based phylogeny (Fig. 3). The V. cholerae O1 lineage carried LH, except for the four strains in phylogenetic groups C and D. All 112 strains of groups A and B except for CP1046 exhibited subclade LH2, and the four strains of the group E showed LH1. The difference in these two subclades, LH1 and LH2, was ascribed to the absence of thymine in the 8-bp poly-T region of the H gene, which caused a frameshift to generate a modified protein that was shorter by 83 amino acids. The four strains in groups C and D showed subclades M2 and M1, respectively. Subclade M1 was represented by a Thai strain, MS6. This strain was very similar to the Russian strain P-1878528. The US gulf coast strains 2740–80 and 3569-08 were designated M2, which differed from M1 by one nucleotide. The sequences of the neighboring genes kbl (MS6_A0926) and lysR (MS6_A0928) were mostly identical among the strains of groups AB and E, but they were different from those in groups C and D, corresponding to the subclustering results for the targeted gene sequence of MS6_A0927 (Table 2). LH was likely replaced with M in the strains of groups C and D after they diverged from a common ancestor of classical and El Tor-biotype organisms.

Phylogenetic relationships among Vibrio cholerae and other Vibrio spp. and distribution of genes M and LH. A maximum likelihood tree showed phylogenetic relationships among 178 strains of V. cholerae, 6 V. mimicus, 1 V. metecus, and 1 V. parilis. Color coding is based on the presence of M: red; LH: blue; and M and LH: green. Bootstrap supports (%) are indicated at branching points. Branch lengths are proportional to sequence differences. Pathogenic O1 strains were classified into five phylogenic groups, A to E. Asterisk indicates a possible recombination event for the M gene through horizontal gene transfer.
We found a similar gene arrangement in the loci between MS6_A0926 and MS6_A0928 containing M and LH in the two strains N2_17 and 87395 (Fig. S2). Strain 87395 was phylogenetically closely related with HE-09, which exhibited M5 in common. In addition, the DNA sequence of LH was clearly different from those in V. cholerae, except for the four strains N56, N79, N80, and N83 of subclade LH4.
Based on these observations, V. cholerae commonly carried a single copy of the M and/or LH genes in the specific locus. It is likely that V. cholerae originally carried the M gene; however, horizontal gene transfer led to the emergence of V. cholerae carrying LH in ancient times. In some subclades of V. cholerae, such as those in groups C and D and CP1037(10), gene replacement with M occurred in succession. As observed in strains 87395 and N2.17, an incomplete choice between the two alternatives of M and LH could occur, resulting in incidental possession of both M and LH in these strains. The alternative choice of M or LH might be of benefit to the survival of V. cholerae in different environmental conditions. Very recently, Das et al. (29) reported that the product of H in V. cholerae O1 classical biotype strain O395 was endowed with molecular chaperone, aminopeptidase, and robust methylglyoxalase activities. The functional roles of these genes in V. cholerae are now under investigation.
Genetic diversity and population structure of V. cholerae revealed by M/LH sequence profiling
The above-described data show that V. cholerae have maintained M or LH in MS6_A0927 on their genomes and indicate that the subclades corresponding well to clusters generated from a genome-wide phylogenetic analysis. Therefore, we considered the advantages and limitations of using our targeted gene sequencing for V. cholerae investigations. The M/LH sequence profiling exhibited the highest discrimination index (D = 0.63) as compared with those of the nine SLST analyses that targeted the two gene loci kbl and lysR and seven housekeeping genes adk, gyrB, mdh, metE, pntA, purM, and pyrC (Table 2). The kbl and lsyR genes encode 2-amino-3-ketobutyrate coenzyme A ligase and the LysR family of transcriptional regulators, respectively. M/LH sequence profiling differentiated between the four groups, i.e., A/B, C, D, and E, whereas the 9 other SLSTs failed to differentiate these groups. The conventional MLST analysis based on the seven loci was able to differentiate between groups A and B, but not between C and D. The ability to distinguish C from D was critical to trace the pathogen MS6 back to its likely origin in China or vice versa. Recently, sporadic cholera outbreaks caused by US Gulf-like V. cholerae O1 (non-7th pandemic clone) occurred in Zhejiang province, China29. MLST analysis separated 13 Zhejiang strains into 3 sequence types—ST75, ST169, and ST170—and of them, 10 strains with ST75 sequence type were identical to US Gulf coast strains, while M/LH sequence profiling showed that all the 13 strains belonged to group D (subclade M1), differentiating them from US Gulf coast strains (M2). The results of M/LH sequence profiling corresponded well with those of the clusters deduced by their whole genome sequence-based phylogenetic analysis, and consequently, it demonstrated higher epidemiological relevance than MLST did.
Vibrios with M or LH were isolated from clinical and environmental sources. Subclades LH1, LH2, M4, and M6 were dominantly found in test strains (Fig. 2). Among them, LH2 mostly comprised the seventh cholera pandemic El Tor and toxigenic O139 strains, whereas the classical type of V. cholerae, the major player in the sixth cholera pandemic, belonged to subclade LH1. Interestingly, there have been no reports on the toxigenic and non-toxigenic O1 strains in group C and D that replaced LH with M causing epidemics and severe diarrhea (Fig. 3)28,29,30,31,32. Furthermore, all 20 clinical strains of non-O1/non-O139 V. cholerae obtained in the Haiti cholera epidemic belonged to two subclades M4 and M6 (Table S1), which corresponded to clusters HC-1 and −2 as indicated by the comparative genomic analysis of Hassan et al. (29). The three environmental strains from Haiti from 2010 were in subclade M7. In addition, eight strains of serogroup O75 CP1110-CP1117 from an oyster-borne cholera outbreak in Florida33 belonged to subclade M3.
In this study, 130 strains including V. cholerae O1 and O139 were separated into 13 subclades. Subclade LH2 contained current pandemic/epidemic clone and normally possessed the genes ctxAB/tcpA/toxR/VC2346/LH, which were targeted by our multiplex PCR. Most of the non-toxigenic V. cholerae of O1 serogroup, such as strains 12129(1) and TM11079-80, belonged to different subclades, and phylogenetic lineages from the toxigenic O1 strains and their O1 antigen phenotype probably arose from horizontal gene exchange in the evolution of V. cholerae13. The alterations in the cell surface antigens of V. cholerae can lead to new epidemics/pandemics, especially in populations without adequate immunity against the serogroup. V. cholerae O139 Bengal evolved from a V. cholerae O1 El Tor strain by exchange of genes encoding cell surface polysaccharides34,35, and cholera caused by V. cholerae O139 Bengal has rapidly spread in southeast Asian countries following its initial isolation in Madras, India, in 1992. A high incidence of cholera has been observed in both adults and children in the areas where cholera is endemic36,37.
In conclusion, our targeted gene sequencing of MS6_A0927 revealed divergent genetic traits among V. cholerae species.
Methods
Bacterial strains and genomes
This study analyzed 341 genomes consisting of i) O1/O139 (n = 128), non-O1/non-O139 (n = 48), and unknown serogroups (n = 2) of V. cholerae strains; V. mimicus (n = 6); V. metecus (n = 1); and V. parilis (n = 1) deposited in GenBank; and ii) serotyped strains of non-O1 V. cholerae (n = 153) and V. mimicus (n = 2). Detailed information on the genomes/strains used in this study is presented in Table S1 and Table 1.
Multiplex PCR method
In total, 186 genomes of Vibrio species (Table S1) were referenced for the in-house design of the multiplex PCR method and targeted gene sequencing. Primers for the six target genes toxR, ctxAB, luxR-hchA (LH), metY (M), VC234638,39, and tcpA were designed to target each consensus region. The multiplex PCR assay for detection of those genes was performed in a 25-µl reaction mixture containing 0.2 mM dNTPs, 1 × Ex Taq buffer, 2 mM MgCl2, each primer at 1 µM, 0.75 U of Ex Taq DNA polymerase (Takara Bio, Otsu, Japan), and 100 ng of genomic DNA extracted using the NucleoSpin Tissue kit (Macherey-Nagel, Düren, Germany). Primer sequences are shown in Table S2. Thermal cycling conditions were as follows: 94 °C for 30 s and 30 cycles of 94 °C for 30 s, 59 °C for 1 min, and 72 °C for 1 min. Amplicons were separated by 2% agarose gel electrophoresis and bands were visualized under ultraviolet transillumination after staining of the gel with ethidium bromide (Fig. S3).
Targeted gene sequencing
PCR amplification of the locus MS6_A0927 was performed in a 50-µl reaction mixture containing 1 × Ex Taq buffer, 2 mM MgCl2, 0.2 mM dNTPs, each external primer at 1 µM (MS6_A0926F and MS6_A0928R) (Fig. 1), 1.25 U of Ex Taq DNA polymerase, and 100 ng of purified genomic DNA. Thermal cycling conditions were as follows: 95 °C for 4 min and 30 cycles of 95 °C for 30 s, 62 °C or 55 °C for 30 s, and 72 °C for 1.5 min. PCR products were purified with the NucleoSpin Gel and PCR Clean-up kit (Macherey-Nagel) or the QIAquick Gel Extraction kit (Qiagen, Valencia, CA, USA).
Samples positive for either M or LH were sequenced within these regions (Fig. 1). The PCR amplification for sequencing was performed in a 10-µl reaction mixture containing 1.5 µl of BigDye Terminator v.3.1 Ready Reaction Mix, 1.25 µl of 5 × BigDye Sequencing Buffer, each internal primer at 0.32 µM (lh_uni1–4 and m_uni1–4), and 40 ng of purified PCR product. The cycling condition was as follows: 96 °C for 1 min and 30 cycles of 96 °C for 10 s, 50 °C for 5 s, and 60 °C for 4 min. The reaction product was sequenced on the ABI 3130xl Genetic Analyzer platform (Applied Biosystems, Foster City, CA, USA). Sequence data determined in this study were submitted to DDBJ (DNA Data Bank of Japan, National Institute of Genetics, Mishima, Shizuoka, Japan) and published with the accession no. LC202659-LC202813.
For M/LH sequence profiling, DNA sequences of the targeted locus were aligned to one of two references: the 1,296-bp region of the MS6 strain (MS6_A0927) or the 1,600-bp region of the classical O1 strain O395 (from VC395_A0912 to VC395_A0913). The subclades were numbered by sequential assignment to each nucleotide sequence variation.
The sequence variations based on M and LH were compared with those from the nine SLST analyses. The sequence data of kbl (MS6_A0926) and lysR (MS6_A0928) and of the seven gene regions targeted by a multilocus sequence typing (MLST) procedure21 were extracted from the 186 genomes. Regions of sequences of the primer sets in each locus of the MLST were used. DNA sequences of each target gene were aligned to each reference sequence from MS6 (kbl and lysR) or N16961 (adk, gyrB, mdh, metE, pntA, purM, and pyrC). Arabic numbers were sequentially assigned to each sequence variation in each target region as described in Table 2. In addition, the number of alleles for MLST were determined based on the profile definitions in the Vibrio cholerae MLST databases (https://pubmlst.org/vcholerae/). The discrimination index was determined by calculation of the Simpson index of diversity, D40.
Phylogenetic tree
Dendrograms were constructed by the neighbor-joining method using MEGA v.6.041. Relationships among strains were assessed by genome-wide phylogenetic analysis. Coding sequences present as a single copy in genomes were analyzed using the Pan-genomes Analysis Pipeline v.1.0226,42.
References
Ali, M., Nelson, A. R., Lopez, A. L. & Sack, D. A. Updated global burden of cholera in endemic countries. PLoS Negl Trop Dis 9, e0003832, https://doi.org/10.1371/journal.pntd.0003832 (2015).
Yamai, S., Okitsu, T., Shimada, T. & Katsube, Y. Distribution of serogroups of Vibrio cholerae non-O1 non-O139 with specific reference to their ability to produce cholera toxin, and addition of novel serogroups. Kansenshogaku Zasshi 71, 1037–1045 (1997).
Ramamurthy, T., Yamasaki, S., Takeda, Y. & Nair, G. B. Vibrio cholerae O139 Bengal: odyssey of a fortuitous variant. Microbes Infect 5, 329–344 (2003).
Sarkar, B. et al. Preferential association of the heat-stable enterotoxin gene (stn) with environmental strains of Vibrio cholerae belonging to the O14 serogroup. Epidemiol Infect 129, 245–251 (2002).
Figueroa-Arredondo, P. et al. Cell vacuolation caused by Vibrio cholerae hemolysin. Infect Immun 69, 1613–1624, https://doi.org/10.1128/IAI.69.3.1613-1624.2001 (2001).
Saka, H. A. et al. Vibrio cholerae cytolysin is essential for high enterotoxicity and apoptosis induction produced by a cholera toxin gene-negative V. cholerae non-O1, non-O139 strain. Microb Pathog 44, 118–128, https://doi.org/10.1016/j.micpath.2007.08.013 (2008).
Lin, W. et al. Identification of a vibrio cholerae RTX toxin gene cluster that is tightly linked to the cholera toxin prophage. Proc Natl Acad Sci USA 96, 1071–1076 (1999).
Jorgensen, R. et al. Cholix toxin, a novel ADP-ribosylating factor from Vibrio cholerae. J Biol Chem 283, 10671–10678, https://doi.org/10.1074/jbc.M710008200 (2008).
Awasthi, S. P. et al. Novel cholix toxin variants, ADP-ribosylating toxins in Vibrio cholerae non-O1/non-O139 strains, and their pathogenicity. Infect Immun 81, 531–541, https://doi.org/10.1128/iai.00982-12 (2013).
Wu, Z., Nybom, P. & Magnusson, K. E. Distinct effects of Vibrio cholerae haemagglutinin/protease on the structure and localization of the tight junction-associated proteins occludin and ZO-1. Cell Microbiol 2, 11–17 (2000).
Pukatzki, S. et al. Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc Natl Acad Sci USA 103, 1528–1533, https://doi.org/10.1073/pnas.0510322103 (2006).
Shin, O. S. et al. Type III secretion is essential for the rapidly fatal diarrheal disease caused by non-O1, non-O139 Vibrio cholerae. mBio 2, e00106–e00111, doi:10.1128/ (2011).
Chun, J. et al. Comparative genomics reveals mechanism for short-term and long-term clonal transitions in pandemic Vibrio cholerae. Proc Natl Acad Sci USA 106, 15442–15447, https://doi.org/10.1073/pnas.0907787106 (2009).
Mutreja, A. et al. Evidence for several waves of global transmission in the seventh cholera pandemic. Nature 477, 462–465, https://doi.org/10.1038/nature10392 (2011).
Ceccarelli, D., Spagnoletti, M., Cappuccinelli, P., Burrus, V. & Colombo, M. M. Origin of Vibrio cholerae in Haiti. Lancet Infect Dis 11, 262, https://doi.org/10.1016/S1473-3099(11)70078-0 (2011).
Kwong, J. C., McCallum, N., Sintchenko, V. & Howden, B. P. Whole genome sequencing in clinical and public health microbiology. Pathology 47, 199–210, https://doi.org/10.1097/PAT.0000000000000235 (2015).
Rahaman, M. H., Islam, T., Colwell, R. R. & Alam, M. Molecular tools in understanding the evolution of Vibrio cholerae. Front Microbiol 6, 1040, https://doi.org/10.3389/fmicb.2015.01040 (2015).
Mahalingam, S. et al. Molecular Epidemiologic Analysis of Vibrio cholerae 01 Isolates by Pulsed-Field Gel Electrophoresis. J Clin Microbiol 32, 2975–2979 (1994).
Cooper, K. L. et al. Development and validation of a PulseNet standardized pulsed-field gel electrophoresis protocol for subtyping of Vibrio cholerae. Foodborne Pathog Dis 3, 51–58 (2006).
Sabat, A. J. et al. Overview of molecular typing methods for outbreak detection and epidemiological surveillance. Euro Surveill 18, 20380 (2013).
Octavia, S. et al. Population structure and evolution of non-O1/non-O139 Vibrio cholerae by multilocus sequence typing. PLoS One 8, e65342, https://doi.org/10.1371/journal.pone.0065342 (2013).
Salim, A., Lan, R. & Reeves, P. R. Vibrio cholerae pathogenic clones. Emerg Infect Dis 11, 1758–1760 (2005).
Steer, A. C., Law, I., Matatolu, L., Beall, B. W. & Carapetis, J. R. Global emm type distribution of group A streptococci: systematic review and implications for vaccine development. Lancet Infect Dis 9, 611–616, https://doi.org/10.1016/s1473-3099(09)70178-1 (2009).
Frenay, H. M. et al. Molecular typing of methicillin-resistant Staphylococcus aureus on the basis of protein A gene polymorphism. Eur J Clin Microbiol Infect Dis 15, 60–64 (1996).
Scholz, C. F., Jensen, A., Lomholt, H. B., Bruggemann, H. & Kilian, M. A novel high-resolution single locus sequence typing scheme for mixed populations of Propionibacterium acnes in vivo. PLoS One 9, e104199, https://doi.org/10.1371/journal.pone.0104199 (2014).
Okada, K. et al. Comparative genomic characterization of a Thailand-Myanmar isolate, MS6, of Vibrio cholerae O1 El Tor, which is phylogenetically related to a “US Gulf Coast” clone. PLoS One 9, e98120, https://doi.org/10.1371/journal.pone.0098120 (2014).
Sawabe, T. et al. Updating the Vibrio clades defined by multilocus sequence phylogeny: proposal of eight new clades, and the description of Vibrio tritonius sp. nov. Front Microbiol 4, 414, https://doi.org/10.3389/fmicb.2013.00414 (2013).
Kuleshov, K. V. et al. Draft Genome Sequences of Vibrio cholerae O1 ElTor Strains 2011EL-301 and P-18785, Isolated in Russia. Genome Announc 1, https://doi.org/10.1128/genomeA.00659-13 (2013).
Luo, Y. et al. US Gulf-like toxigenic O1 Vibrio cholerae causing sporadic cholera outbreaks in China. J Infect 72, 564–572, https://doi.org/10.1016/j.jinf.2016.02.005 (2016).
Wachsmuth, I. K., Bopp, C. A., Fields, P. I. & Carrillo, C. Difference between toxigenic Vibrio cholerae O1 from South America and US gulf coast. Lancet 337, 1097–1098 (1991).
DePaola, A. et al. Isolation of Latin American epidemic strain of Vibrio cholerae O1 from US Gulf Coast. Lancet 339, 624 (1992).
Okada, K., Roobthaisong, A., Swaddiwudhipong, W., Hamada, S. & Chantaroj, S. Vibrio cholerae O1 isolate with novel genetic background, Thailand-Myanmar. Emerg Infect Dis 19, 1015–1017, https://doi.org/10.3201/eid1906.120345 (2013).
Haley, B. J. et al. Genome Sequences of Clinical Vibrio cholerae Isolates from an Oyster-Borne Cholera Outbreak in Florida. Genome Announc 1, https://doi.org/10.1128/genomeA.00966-13 (2013).
Bik, E. M., Bunschoten, A. E., Gouw, R. D. & Mooi, F. R. Genesis of the novel epidemic Vibrio cholerae O139 strain: evidence for horizontal transfer of genes involved in polysaccharide synthesis. EMBO J 14, 209–216 (1995).
Comstock, L. E. et al. The capsule and O antigen in Vibrio cholerae O139 Bengal are associated with a genetic region not present in Vibrio cholerae O1. Infect Immun 63, 317–323 (1995).
Ramamurthy, T. et al. Emergence of novel strain of Vibrio cholerae with epidemic potential in southern and eastern India. Lancet 341, 703–704 (1993).
Faruque, S. M. et al. Emergence of a new clone of toxigenic Vibrio cholerae O1 biotype El Tor displacing V. cholerae O139 Bengal in Bangladesh. J Clin Microbiol 35, 624–630 (1997).
Grim, C. J. et al. Occurrence of the Vibrio cholerae seventh pandemic VSP-I island and a new variant. OMICS 14, 1–7, https://doi.org/10.1089/omi.2009.0087 (2010).
Talkington, D. et al. Characterization of toxigenic Vibrio cholerae from Haiti, 2010-2011. Emerg Infect Dis 17, 2122–2129, https://doi.org/10.3201/eid1711.110805 (2011).
Hunter, P. R. & Gaston, M. A. Numerical index of the discriminatory ability of typing systems: an application of Simpson’s index of diversity. J Clin Microbiol 26, 2465–2466 (1988).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30, 2725–2729, https://doi.org/10.1093/molbev/mst197 (2013).
Zhao, Y. et al. PGAP: pan-genomes analysis pipeline. Bioinformatics 28, 416–418, https://doi.org/10.1093/bioinformatics/btr655 (2012).
Acknowledgements
This research was supported by the Japan Initiative for Global Research Network on Infectious Diseases from Ministry of Education, Culture, Sports, Science & Technology (MEXT) in Japan, and Japan Agency for Medical Research and Development (AMED); Grant-in-Aid for Scientific Research (C) (No.17K08826) from the MEXT; and the Department of Medical Sciences, the Ministry of Public Health, Thailand. The funders had no role in study design, data collection or analysis, decision to publish, or preparation of the manuscript. We are grateful to Dr. Eiji Arakawa, NIID, Japan, for providing the strains of non-O1 V. cholerae and to Dr. Munir Alam, icddr, b, Bangladesh, for valuable suggestions and discussions. We also thank to Dr. Mathukorn Na-Ubol, RCC-ERI, for her technical assistance.
Author information
Authors and Affiliations
Contributions
K.O. designed the study. K.O., W.W., S.C., W.N., A.R., W.K., F.M., and T.T. performed the experiments. All the authors analyzed the data. S.C., T.T., I.N., M.O., and S.K. contributed reagents, materials, and/or analysis tools. K.O. and S.H. wrote the manuscript, All the authors contributed to the research and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Okada, K., Wongboot, W., Chantaroj, S. et al. Vibrio cholerae embraces two major evolutionary traits as revealed by targeted gene sequencing. Sci Rep 8, 1631 (2018). https://doi.org/10.1038/s41598-018-19995-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-19995-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
