
Abstract
Microglia are known to engage in physical interactions with neurons. However, our understanding of the detailed mechanistic regulation of microglia-neuron interactions is incomplete. Here, using high resolution two photon imaging, we investigated the regulation of NMDA receptor-induced microglia-neuron physical interactions. We found that the GluN2A inhibitor NVPAAM007, but not the GluN2B inhibitor ifenprodil, blocked the occurrence of these interactions. Consistent with the well-known developmental regulation of the GluN2A subunit, these interactions are absent in neonatal tissues. Furthermore, consistent with a preferential synaptic localization of GluN2A subunits, there is a differential sensitivity of their occurrence between denser (stratum radiatum) and less dense (stratum pyramidale) synaptic sub-regions of the CA1. Finally, consistent with differentially expressed GluN2A subunits in the CA1 and DG areas of the hippocampus, these interactions could not be elicited in the DG despite robust microglial chemotactic capabilities. Together, these results enhance our understanding of the mechanistic regulation of NMDA receptor-dependent microglia-neuronal physical interactions phenomena by the GluN2A subunit that may be relevant in the mammalian brain during heightened glutamatergic neurotransmission such as epilepsy and ischemic stroke.
Similar content being viewed by others

Introduction
Microglia are resident immune cells in the central nervous system (CNS) and their functions have become appreciated in normal physiology from development through adulthood. Microglial involvement in CNS physiology ranges from the regulation of neural circuit development in the immature brain1,2,3 to neurogenesis and learning in the adult brain4,5,6,7. Furthermore, microglial activities have been implicated during CNS dysfunction in diseases as well as in normal aging8,9,10. These observations suggest that microglia and neurons engage in a delicate balance of intercellular communication that needs to be carefully regulated1,11,12,13. Indeed, microglia are the most dynamic resident cells of the CNS and make transient physical contact with neuronal boutons, dendrites and somata14,15,16,17,18. Moreover, excitatory and inhibitory neurotransmission have been shown to reciprocally modulate this microglial surveillance19. However, our understanding of the repertoire of microglia-neuron physical interactions and its underlying mechanisms are not exhaustive.
Microglia have also now been suggested to regulate neurotransmission. For example, genetically perturbing microglial fractalkine receptors during development alters neurotransmission20,21 that could have long lasting effects into adulthood22. Moreover, selectively activating microglia enhances excitatory neurotransmission23. In this light, microglia are integrated into neuronal synapse function leading to a further revision of the synapse model into the “quad-partite” model24,25. Recently, we and others17,18,26 were able to uncover physical microglia-neuron interaction axes that occur during intense glutamatergic activation of neuronal NMDA receptors (NMDAR). We found that NMDAR activation triggers purine release that elicits microglial process extension (MPEs) and microglial process convergence (MPCs) through P2Y12 receptors17,18. In the current study, we continued these investigations to provide further insights into the mechanisms showing the requirement for the GluN2A NMDAR subunit in these microglia-neuron physical interactions.
Results
Regulation of NMDAR-Induced Microglia-Neuron Physical Interactions by the GluN2A Subunit
We and others recently reported that glutamate activation of neuronal NMDARs elicits MPEs through microglial P2Y12 receptors18,26. To provide further insights into its underlying mechanisms, we investigated the role of specific subunits of the NMDAR on MPEs using a pharmacological approach. NMDARs are composed of several subunits including obligatory GluN1 and GluN2 subunits—the GluN2A and GluN2B. With a wealth of interest in the role of the GluN2A and GluN2B subunits in NMDAR function in the CNS27,28, we investigated the potential subtype-dependent regulation of microglia-neuron interactions by NMDAR activation. To this end, we used Ifenprodil (“ifen”, 3 µM), a well-recognized GluN2B antagonist, and NVPAAM007 (“NVP”; 0.4 µM), a GluN2A antagonist29.
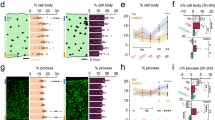
We first tested for GluN2A and GluN2B components of the NMDA-induced currents in pyramidal neurons using these drugs. Consistent with our previous results29, we show that NVP blocks a majority (~60%) of the NMDA-induced current amplitude while ifen further blocked about 10% of the NMDA-induced current (Fig. 1a-c). Pre-incubation with either drug did not alter basal microglial motile dynamics (data not shown). As expected, the perfusion of NMDA (30 µM, 15 min) induced robust microglial process extensions (MPEs) to hippocampal CA1 area (Fig. 2a,b). Interestingly, co-application of NMDA with NVP but not ifen abolished NMDAR-induced MPEs (Fig. 2a,b; see also Supplementary Video 1). This indicates significant roles for the GluN2A subunit of the NMDAR in MPEs.

GluN2A/B Subunit Contributions to NMDAR-induced Currents. (a) Sample tracings of NMDA(100 µM)-induced currents under basal conditions (top), following 10 min of NVPAAM007 (0.4 µM, middle) and combined NVPAAM007 and Ifenprodil (3 µM, bottom). (b) Graph of NMDA-induced current amplitude with applications of GluN2 antagonist. (c) Graph showing the average percent of NVP- and ifenprodil-sensitive components of the NMDA-induced current after sequential application of NVP and ifenprodil. All data are presented as mean ± S.E.M. n = 3 slices each.

Regulation of NMDAR-induced Microglia-Neuron Physical Interactions by the GluN2A Subunit. (a) Representative z-stack two-photon images of GFP-expressing microglia in hippocampal CA1 of acute brain slices before (left) and after 15 min of NMDA (30 µM) treatment (center) in the presence of GluN2A antagonist (top) or GluN2B antagonist (bottom). Rightmost images are merged images of the before (red) and after (green) images. Extending microglial processes can be visualized in the stratum pyramidale layer (dashed lines) in green. (b) Quantitative summary of corresponding data to (a); insert: high magnification image collected from a double trangenic mouse brain slices following NMDA-treatment. The images show microglial processes (GFP-labelled) making bulbous tipped contact (white arrows) with a neuron (YFP-labelled) following NMDA treatment. (c–f) Representative image of the field of view with boxed region that is expanded to show timelapse images of converging microglial processes (green) that terminate on a neuronal dendrite (white arrow) that occurs after NMDAR activation. (g,h) A schematic of sites of microglial process convergence events during a 30 minute imaging period from three representative experiments (g) and quantified summary (h) showing significantly reduced events with NVP but not Ifen during NMDA treatment. All data are presented as mean ± S.E.M. n = 4–6 slices each. ***P < 0.001.
We recently reported the existence of another form of microglia-neuron physical interaction which we termed microglial process convergence (MPCs)30, a phenomenon that was observed in epileptic conditions in an NMDAR-dependent manner17. Though distinct from MPEs, we speculated that MPCs are also regulated by the GluN2A subunit. Consistent with this hypothesis, compared to ifen, NVP significantly reduced the occurrence of MPCs following a 10 minute glutamate (1 mM) treatment (Fig. 2c–h). Together, these results indicate that the GluN2A subunit regulates NMDAR-induced microglia-neuron physical interactions including both MPEs and MPCs.
Developmental Regulation of NMDAR-Induced Microglia-Neuron Physical Interactions
GluN2A and GluN2B subunits exhibit a differential developmental regulation such that GluN2B predominate during early postnatal development which eventually gives way to the later predomination of GluN2A subunits during latter postnatal development into adulthood31,32,33. Considering that the GluN2A but not the GluN2B subunit is required for MPEs, we investigated the possibility of a developmental regulation of MPEs. Interestingly, we found that NMDA (30 µM) failed to induce MPEs in P7 tissues although we could observe the phenomena in tissues from P30 mice (Fig. 3a,b; see also Supplementary Video 2) and even as early as P12 (data not shown). Similarly, glutamate (1 mM), the physiological agonist of NMDARs, failed to elicit MPEs in P7 tissues though it induced robust MPEs in P30 tissues (Fig. 3c,d; see also Supplementary Video 3). Likewise, MPCs did not occur in response to glutamate treatment in slices from P7 mice while a robust occurrence was observed in P30 and P60 brain slices (Fig. 3e,f). Together, these results indicate that there is a developmental regulation of NMDAR-induced microglia-neuron physical interactions that is consistent with the well-documented developmental switch from predominant GluN2B expression during early postnatal development to later predominant GluN2A expression. For simplicity and given the similarities in the GluN2A and developmental regulation of both MPEs and MPCs, we focused on MPEs for the rest of our studies.

Developmental Regulation of NMDAR-induced Microglia-Neuron Physical Interactions. (a–d) Representative z-stack two-photon images of GFP-expressing microglia in hippocampal CA1 of acute brain slices before (left) and after 15 min of NMDA (a, 30 µM) or glutamate (c, 1 mM) treatment (center) in a P7 slice (top) or P30 slice (bottom). Rightmost images are merged images of the before (red) and after (green) images. Extending microglial processes can be visualized in the stratum pyramidale layer (dashed lines) in green. Quantitative summary of corresponding data to (a) and (c) are provided in (b) and (d), respectively. (e,f), Schematic showing microglial process convergence event sites (e) and summary (f). Results indicate the failure of glutamate to increased microglial process convergence events in P7 but not P30 or P60 slices. All data are presented as mean ± S.E.M. n = 4–6 slices each. ***P < 0.001.
Differential Sensitivity to NMDAR-Induced Microglial Process Extension in the Stratum Radiatum and Stratum Pyramidale
Previously, MPEs were shown to require brief, repeated NMDAR activation in the stratum radiatum (SR) of the CA126. However, in our prolonged perfusion of NMDA/glutamate paradigm, we found that MPEs only occurred after at least 5 minutes of global NMDA/glutamate application in the stratum pyramidale (SP) of the CA118. To address this seeming discrepancy, we performed experiments in which NMDA (30 µM) was applied for 4 minutes followed by a washout of the drug. Interestingly, under these conditions, while we could elicit MPEs in the SR, MPEs were not detectable in the SP (Fig. 4; see also Supplementary Video 4). In general, MPEs were not obvious in the stratum oriens in either the 4 minute or 15 minute NMDA application paradigm (Fig. 4). These results thus suggest that there is a differential sensitivity to NMDAR-induced MPEs in the CA1 region. Since GluN2A subunits are predominantly localized to synaptic regions27,34 such as the SR over extrasynaptic regions such as the SP and SO of the CA1, these results are consistent with a role for the GluN2A subunit in the differential sensitivity of MPEs in the SR compared to the SP.

Differential Sensitivity in Somatic and Dendritic Responses during NMDAR-Induced Microglial Process Extension. (a) Representative two-photon z-stack merged color-coded images from 15 (top) and 4 (bottom) min NMDA (30 µM) treatment. Color code indicate microglial morphologies before (red) and after (green) NMDA treatment. (b) Higher magnification two-photon z-stack images of the stratum oriens (SO) stratum pyramidale (SP) and stratum radiatum (SR) regions of the corresponding 15 (left images) and 4 (right images) min NMDA treatment images from (a). (c,d) Quantitative summary of microglial process extension in the different regions during a 15 min (c) or a 4 min (d) NMDA-treatment protocol. All data are presented as mean ± S.E.M. n = 3–4 slices each.
Differential Regulation of Microglial Process Extension between the DG and CA1
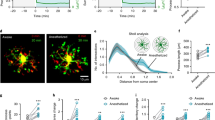
The GluN2A/GluN2B subunit ratio is higher in CA1 neurons compared to dentate gyrus (DG) neurons. Moreover, the GluN2A/B ratio remains unchanged in the DG while it is increased in the CA1 between neonatal development and adulthood35. On this basis, we speculated that DG neurons, given the maintenance of the immature GluN2A/GluN2B expression ratio even in adults, would fail to elicit MPEs. Indeed, upon the application of NMDA (30 µM), we detected a significant difference between the response of microglia in the DG and the CA1. Microglial processes exhibited a strong extension in the CA1 but lacked a response in the DG (extension index: 1.89 ± 0.08 in the CA1 and 1.1 ± 0.13 in the DG; Fig. 5a,b; See also Supplementary Video 5). Similar microglial responses were obtained with glutamate application (extension index: 2.83 ± 0.30 in CA1 and 0.99 ± 0.05 in the DG; Fig. 5c,d; see also Supplementary Video 6).

Regional Regulation of Microglial Process Extension. (a) Representative z-stack two-photon images of GFP-expressing microglia of acute brain slices before (left) and after 15 min of NMDA (30 µM) treatment (center) from the CA1 (top) and DG (bottom) regions of the hippocampus. Rightmost images are merged images of the before (red) and after (green) images. Extending microglial processes can be visualized in the neuronal cell body layer (dashed lines) in green. (b) Quantitative summary of corresponding data to (a); n = 4 slices each. (c) Representative z-stack two-photon images of GFP-expressing microglia of acute brain slices before (left) and after 15 min of glutamate (1 mM) treatment (center) from the CA1 (top) and dentate gyrus (DG, bottom) regions of the hippocampus. Rightmost images are merged images of the before (red) and after (green) images. Extending microglial processes can be visualized in the neuronal cell body layer (dashed lines) in green. (d) Quantitative summary of corresponding data to (c) All data are presented as mean ± S.E.M. n = 4–6 slices.
The differences in the ability to elicit MPEs in the DG compared to the CA1 could be explained by one of the following possibilities. On the one hand, it is possible that neuronal NMDAR activation in the DG differs from that in the CA1. On the other hand, it is possible that microglial chemotactic responses to released ATP may differ between these two hippocampal regions. To rule out the possibility that DG microglia may lack chemotactic responses, a laser injury was induced in the CA1 (n = 5 slices) and DG (n = 4 slices) regions, and the extent and rate of microglial process extension to the injury were measured36. We found that microglia in both hippocampal regions are capable of responding robustly to laser-induced purinergic signals (Fig. 6; see also Supplementary Video 7). These results indicate that DG microglia are not deficient in their ability to respond to ATP. Together, our results indicate that the weaker MPE induction in the DG might result from differences in neuronal physiology rather than a microglial chemotactic function.

Microglial Chemotactic Responses are not Deficient in the Dentate Gyrus. (a,b) Microglial processes respond robustly to a laser-induced tissue injury (asterisks) by chemotaxis in both the CA1 (top panel) and the DG (bottom panel). n = 4 slices each.
Discussion
Microglial process extension (MPEs) and microglial process convergence (MPCs) in response to neuronal NMDAR activation was recently discovered in the murine brain17,18,26. In the current study, we show that GluN2A subunits of the NMDAR are critical for these microglia-neuron physical interaction phenomena. In this light, we show by proof of principle consistent with GluN2A roles that these interactions are (i) developmentally regulated, (ii) synaptically sensitive and (iii) differentially expressed in the CA1 and DG. These observations enhance our understanding of the mechanisms guiding physical microglia-neuron interactions that may be relevant during diseased contexts such as epilepsy and ischemic stroke.
Previous work had implicated NMDAR activation in the coupling of ATP release via pannexin channels during pathology37 and suggested this as a novel signaling modality for NMDARs38. Our findings here provide another possible mechanism that suggests the coupling of NMDAR activation to ATP release via the GluN2A subunit to elicit microglial process extension/convergence by P2Y12R activation. The precise avenue for ATP release remains to be determined, as previous work ruled out pannexin and connexin channels17,18,26. At the concentrations of glutamate/NMDA used, GluN2B subunits were also activated in slices. However, the lack of microglial morphological responses during GluN2B inhibition with its potent inhibitor (ifenprodil) suggest that the GluN2B subunits are not (or less) coupled to the ATP release pathway.
Neuroprotective function of NMDAR-induced Microglia-Neuron Physical Interactions
We investigated the contribution of either the GluN2A or the GluN2B subunit to MPEs and found that the GluN2A receptor is predominantly responsible for NMDAR-induced MPEs/MPCs (Fig. 2). Emerging concepts (though without a full consensus) suggest that NMDARs with the GluN2A subunit are predominantly localized to the synapse, while those with the GluN2B subunit are predominantly extrasynaptic i.e. localized outside the synapse27. Similarly, studies suggest that synaptically localized GluN2A subunits mediate neuronal survival by inhibiting apoptotic mechanism mediated by synaptic calcium influx through these NMDARs. Conversely, extrasynaptic NMDARs with the GluN2B subunit are suggested to mediate neuronal demise through excessive calcium influx and pro-apoptotic mechanisms39. If this hypothetical framework is assumed for our studies and because we found a requirement for the GluN2A subunit of the NMDAR in MPEs, it is tempting to speculate that MPEs serve a neuroprotective function. This is consistent with our previous studies using P2Y12 and CX3CR1 knockout mice in the context of acute seizures where these receptors positively regulate these interactions17,18,40. There, we found that a genetic inhibition of MPEs and MPCs in these knockouts within an epileptic context correlated with a worsened behavioral phenotype during seizures. Since MPEs/MPCs require GluN2A activation, it is tempting to speculate that MPEs may serve neuroprotective functions.
Developmental Regulation of NMDAR-Induced Microglia-Neuron Physical Interactions
We show that MPEs and MPCs are developmentally regulated since they are absent in immature neonatal tissues (Fig. 3). MPEs and MPCs continue to be detected from the second week of hippocampal development (data not shown) into adulthood26. Several microglia-neuron communication mechanisms are developmentally regulated. For example, microglial regulation of neurotransmission through CX3CR1 is present during early postnatal development but disappears in young mice in both the hippocampus21 and barrel cortex20. Moreover, microglial colonization of CNS tissues is also developmentally regulated through fractalkine signaling20,21.
In our study, the developmental regulation of MPEs and MPCs seems to be dependent on initiating neuronal mechanisms that require neuronal maturation. Microglia in neonatal tissues can respond robustly to purines through the engagement of their P2Y12 receptors41,42 whose mobilization is critical for MPEs18,26. This developmental regulation of MPEs/MPCs is consistent with the well-documented developmental shift from predominant GluN2B expression during early postnatal development to predominant GluN2A expression beginning in the second week of postnatal development31,32,33. Although we have not excluded other possibilities to be responsible for the differences observed with regards to MPEs/MPCs between neonatal and mature tissues, our data with regards to GluN2A NMDAR subunit roles is consistent with the observation of a developmental sensitivity to microglia-neuron physical interactions.
Regional Regulation of NMDAR-induced Microglial Process Extension
One of the remarkable findings of this study is the differential regulation of MPEs in different brain regions. We found two levels of differential regulation: a localized differential regulation in the CA1 and a regionalized differential regulation between the CA1 and DG. Concerning the first level of regulation, we document a greater sensitivity to glutamate for MPEs in the stratum radiatum of the CA1 than the stratum pyramidale. This greater sensitivity for NMDAR-induced MPEs is consistent with GluN2A roles given that the subunit is predominantly localized to synaptic sites located in the stratum radiatum27,39. It is also possible that the ATP release mechanism may require a lower threshold for activation in this region than in the stratum pyramidale.
The second level of differential regulation of MPEs is between different regions of the hippocampus. Previously, we observed that MPEs occur in both the CA1 region of the hippocampus and layer II/III of the cortex18,26. However, we found that the phenomena are not present in all regions of the hippocampus as we could not elicit MPEs in the DG by either glutamate or NMDA application. We found that microglia resident in the DG were capable of functional chemotaxis in response to a laser-induced injury (Fig. 6b). Indeed, NMDA elicits differential release of neurotransmitters between the DG and CA1 e.g. for norepinephrine43 suggesting that DG neurons function differently than CA1 neurons to NMDAR activation. Consistently, DG neurons maintain an immature GluN2A/GluN2B expresssion ratio that does not change during development35 and DG express lower levels of GluN2A mRNA than in the CA1 of the adult rat44.
In summary, we have extended previous findings on microglia-neuron physical interactions in response to NMDAR activation and found its regulation predominantly through the GluN2A subunit of the NMDAR. Our study was performed in an entirely ex vivo slice system and raises questions as to the relevance of these findings in vivo, which will be addressed in future studies. Our previous17,18 and current results suggest that these interactions may serve a neuroprotective function, and the current findings provide novel insights into the mechanisms that may enhance our understanding of the dynamic interractions between microglia and neurons. Particularly, this neuroprotective interaction could be harnessed especially in conditions of excessive glutamatergic neuronal signalling such as seizures, epilepsy, and ischemic stroke.
Methods
Animals
Both male and female mice were used in accordance with guidelines of the Institutional Animal Care and Use Committee (IACUC) at Rutgers University and Mayo Clinic as approved by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International. Heterozygous reporter mice expressing GFP under the control of the fractalkine receptor promoter (CX3CR1-GFP+/−)45 and YFP under the control of the Thy1 promoter46,47 were obtained from The Jackson Laboratory.
Slice Preparation
Freshly isolated cortical or hippocampal slices were prepared from mice at various ages as stated in the results. Briefly, mice were anesthetized in an isofluorane (5%) chamber and swiftly decapitated. Brains from decapitated mice were carefully removed and placed in ice-cold, oxygenated (95%O2 and 5%CO2) artificial cerebrospinal fluid (ACSF) with the following composition (in mM): 124 NaCl, 25 NaHCO3, 2.5 KCl, 1 KH2PO4, 2 CaCl2, 2 MgSO4, 10 glucose, and sucrose added to make 300–320 mOsmol. Coronal slices (300 µm) were prepared and transferred to a recovery chamber for 30 or more minutes with oxygenated ACSF with the same composition as above at room temperature before imaging or electrophysiological studies.
Whole cell patch Recording
Whole-cell patch recordings were performed in pyramidal neurons in live brain slices. The slices were placed in a stage and perfused with oxygenated (95% O2 and 5% CO2) artificial cerebrospinal fluid (ACSF) with the same composition as above. The recording pipette (3–5 MΩ) were filled with solution containing (mM): 115 Cs-MeSO3, 5 NaCl, 10 HEPES, 1 MgCl2, 0.2 EGTA, and 10 Na2Phosphocreatine (pH 7.2; 280–300 mOsmol). Data were amplified and filtered at 2 kHz by a patch-clamp amplifier (Multiclamp 700B), digitalized (DIGIDATA 1440 A), stored, and analyzed by pCLAMP (Molecular Devices, Union City, CA). NMDA (100 µM) was puff applied (10 psi, 10 ms, ~15 µm from patched cell) to induce EPSC. Access resistance of 15–30 MΩ was monitored throughout the experiment and data was discarded when changes more than 15% were observed. Antagonists were applied through the perfusion system.
Two-photon Imaging
Experiments were conducted at room temperature with slices maintained in oxygenated ACSF with the same composition as above in a perfusion chamber at a flow rate of ~2 ml/min. Microglia were typically imaged using a two-photon microscope (Scientifica) with a Ti:sapphire laser (Mai Tai; Spectra Physics) tuned to 900 nm. Fluorescence was detected using two photomultiplier tubes in whole-field detection mode and a 565 nm dichroic mirror with 525/50 nm (green channel). The laser power was maintained at ~25 mW or below. We imaged microglia between 50 and 100 µm from the slice surface.
Drugs
Glutamate, Ifenprodil and NMDA were purchased from Sigma. NVPAAM007 was a gift from Dr. Min Zhuo (University of Toronto). Stock solutions of all drugs were diluted to the working concentrations in ACSF and applied to the slices through a bath.
Extension Index Analysis
Image analysis was done using ImageJ. Max projection images were collated to form time-lapse movies. Where necessary, movies were registered using the StackReg plugin to eliminate any x-y drift. For extension index, ROIs in the cell body layer of the CA1 or dentate gyrus (DG) were cropped from movies to be analyzed. These regions were then binarized and an automated threshold was set on the ROI. The area of the suprathresholded regions of the projection stack was then measured through time and normalized to the area of the first frame of the movie to a starting normalized index value of 1.0. The index through time of each time-lapse movie was then determined.
Process Convergence Analysis
Quantification of microglial process convergence events was done manually as previously reported17. Events were identified when microglial processes spontaneously converged toward a focal point. To avoid arbitrary selection of these events, analysis was done by counting all the observed events irrespective of size so as not to bias our analysis/quantification. The frequency of occurrence of these events was determined in 330 × 330 × 45 µm field of view from 30-min long imaging sessions.
Statistical Analysis
For all experimental analyses, 3–9 slices from different mice were analyzed and averaged to determine significance. Data are presented as mean ± SEM at the final recorded time point after drug application. Student’s T-test and one-way ANOVA with Bonferonni corrections where appropriate were used to establish significance.
References
Ueno, M. & Yamashita, T. Bidirectional tuning of microglia in the developing brain: from neurogenesis to neural circuit formation. Curr Opin Neurobiol 27, 8–15, https://doi.org/10.1016/j.conb.2014.02.004 (2014).
Squarzoni, P. et al. Microglia modulate wiring of the embryonic forebrain. Cell reports 8, 1271–1279, https://doi.org/10.1016/j.celrep.2014.07.042 (2014).
Wake, H., Moorhouse, A. J., Miyamoto, A. & Nabekura, J. Microglia: actively surveying and shaping neuronal circuit structure and function. Trends Neurosci 36, 209–217, https://doi.org/10.1016/j.tins.2012.11.007 (2013).
Rogers, J. T. et al. CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J Neurosci 31, 16241–16250, https://doi.org/10.1523/JNEUROSCI.3667-11.2011 (2011).
Parkhurst, C. N. et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609, https://doi.org/10.1016/j.cell.2013.11.030 (2013).
Sierra, A. et al. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 7, 483–495, https://doi.org/10.1016/j.stem.2010.08.014 (2010).
Miyamoto, A. et al. Microglia contact induces synapse formation in developing somatosensory cortex. Nat Commun 7, 12540, https://doi.org/10.1038/ncomms12540 (2016).
Beggs, S. & Salter, M. W. The known knowns of microglia-neuronal signalling in neuropathic pain. Neurosci Lett 557(Pt A), 37–42, https://doi.org/10.1016/j.neulet.2013.08.037 (2013).
Mosher, K. I. & Wyss-Coray, T. Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem Pharmacol 88, 594–604, https://doi.org/10.1016/j.bcp.2014.01.008 (2014).
Norden, D. M. & Godbout, J. P. Review: microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol Appl Neurobiol 39, 19–34, https://doi.org/10.1111/j.1365-2990.2012.01306.x (2013).
Eyo, U. B. & Wu, L. J. Bidirectional Microglia-Neuron Communication in the Healthy Brain. Neural Plast 2013, 456857, https://doi.org/10.1155/2013/456857 (2013).
Paolicelli, R. C. & Gross, C. T. Microglia in development: linking brain wiring to brain environment. Neuron Glia Biol 7, 77–83, https://doi.org/10.1017/S1740925X12000105 (2011).
Paolicelli, R. C., Bisht, K. & Tremblay, M. E. Fractalkine regulation of microglial physiology and consequences on the brain and behavior. Front Cell Neurosci 8, 129, https://doi.org/10.3389/fncel.2014.00129 (2014).
Li, Y., Du, X. F., Liu, C. S., Wen, Z. L. & Du, J. L. Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Dev Cell 23, 1189–1202, https://doi.org/10.1016/j.devcel.2012.10.027 (2012).
Tremblay, M. E., Lowery, R. L. & Majewska, A. K. Microglial interactions with synapses are modulated by visual experience. PLoS Biol 8, e1000527, https://doi.org/10.1371/journal.pbio.1000527 (2010).
Wake, H., Moorhouse, A. J., Jinno, S., Kohsaka, S. & Nabekura, J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci 29, 3974–3980, https://doi.org/10.1523/JNEUROSCI.4363-08.2009 (2009).
Eyo, U. B. et al. Regulation of Physical Microglia-Neuron Interactions by Fractalkine Signaling after Status Epilepticus. eNeuro 3, https://doi.org/10.1523/ENEURO.0209-16.2016 (2017).
Eyo, U. B. et al. Neuronal Hyperactivity Recruits Microglial Processes via Neuronal NMDA Receptors and Microglial P2Y12 Receptors after Status Epilepticus. J Neurosci 34, 10528–10540, https://doi.org/10.1523/JNEUROSCI.0416-14.2014 (2014).
Fontainhas, A. M. et al. Microglial morphology and dynamic behavior is regulated by ionotropic glutamatergic and GABAergic neurotransmission. PLoS One 6, e15973, https://doi.org/10.1371/journal.pone.0015973 (2011).
Hoshiko, M., Arnoux, I., Avignone, E., Yamamoto, N. & Audinat, E. Deficiency of the microglial receptor CX3CR1 impairs postnatal functional development of thalamocortical synapses in the barrel cortex. J Neurosci 32, 15106–15111, https://doi.org/10.1523/JNEUROSCI.1167-12.2012 (2012).
Paolicelli, R. C. et al. Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458, https://doi.org/10.1126/science.1202529 (2011).
Zhan, Y. et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci 17, 400–406, https://doi.org/10.1038/nn.3641 (2014).
Pascual, O., Ben Achour, S., Rostaing, P., Triller, A. & Bessis, A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc Natl Acad Sci USA 109, E197–205, https://doi.org/10.1073/pnas.1111098109 (2012).
Schafer, D. P., Lehrman, E. K. & Stevens, B. The “quad-partite” synapse: microglia-synapse interactions in the developing and mature CNS. Glia 61, 24–36, https://doi.org/10.1002/glia.22389 (2013).
Ji, K., Miyauchi, J. & Tsirka, S. E. Microglia: an active player in the regulation of synaptic activity. Neural plasticity 2013, 627325, https://doi.org/10.1155/2013/627325 (2013).
Dissing-Olesen, L. et al. Activation of neuronal NMDA receptors triggers transient ATP-mediated microglial process outgrowth. J Neurosci 34, 10511–10527, https://doi.org/10.1523/JNEUROSCI.0405-14.2014 (2014).
Paoletti, P., Bellone, C. & Zhou, Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 14, 383–400, https://doi.org/10.1038/nrn3504 (2013).
Wu, L. J. & Zhuo, M. Targeting the NMDA receptor subunit NR2B for the treatment of neuropathic pain. Neurotherapeutics 6, 693–702, https://doi.org/10.1016/j.nurt.2009.07.008 (2009).
Wu, L. J., Xu, H., Ren, M., Cao, X. & Zhuo, M. Pharmacological isolation of postsynaptic currents mediated by NR2A- and NR2B-containing NMDA receptors in the anterior cingulate cortex. Mol Pain 3, 11, https://doi.org/10.1186/1744-8069-3-11 (2007).
Eyo, U. B. et al. Modulation of microglial process convergence toward neuronal dendrites by extracellular calcium. J Neurosci 35, 2417–2422, https://doi.org/10.1523/JNEUROSCI.3279-14.2015 (2015).
Williams, K., Russell, S. L., Shen, Y. M. & Molinoff, P. B. Developmental switch in the expression of NMDA receptors occurs in vivo and in vitro. Neuron 10, 267–278 (1993).
Sheng, M., Cummings, J., Roldan, L. A., Jan, Y. N. & Jan, L. Y. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature 368, 144–147, https://doi.org/10.1038/368144a0 (1994).
Quinlan, E. M., Olstein, D. H. & Bear, M. F. Bidirectional, experience-dependent regulation of N-methyl-D-aspartate receptor subunit composition in the rat visual cortex during postnatal development. Proc Natl Acad Sci USA 96, 12876–12880 (1999).
Sanz-Clemente, A., Nicoll, R. A. & Roche, K. W. Diversity in NMDA receptor composition: many regulators, many consequences. Neuroscientist 19, 62–75, https://doi.org/10.1177/1073858411435129 (2013).
Le Bail, M. et al. Identity of the NMDA receptor coagonist is synapse specific and developmentally regulated in the hippocampus. Proc Natl Acad Sci USA 112, E204–213, https://doi.org/10.1073/pnas.1416668112 (2015).
Davalos, D. et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 8, 752–758, https://doi.org/10.1038/nn1472 (2005).
Thompson, R. J. et al. Activation of pannexin-1 hemichannels augments aberrant bursting in the hippocampus. Science 322, 1555–1559, https://doi.org/10.1126/science.1165209 (2008).
Weilinger, N. L. et al. Metabotropic NMDA receptor signaling couples Src family kinases to pannexin-1 during excitotoxicity. Nat Neurosci 19, 432–442, https://doi.org/10.1038/nn.4236 (2016).
Zhang, X. M. & Luo, J. H. GluN2A versus GluN2B: twins, but quite different. Neurosci Bull 29, 761–772, https://doi.org/10.1007/s12264-013-1336-9 (2013).
Eyo, U. B., Murugan, M. & Wu, L. J. Microglia-Neuron Communication in Epilepsy. Glia 65, 5–18, https://doi.org/10.1002/glia.23006 (2017).
Kurpius, D., Nolley, E. P. & Dailey, M. E. Purines induce directed migration and rapid homing of microglia to injured pyramidal neurons in developing hippocampus. Glia 55, 873–884, https://doi.org/10.1002/glia.20509 (2007).
Haynes, S. E. et al. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci 9, 1512–1519, https://doi.org/10.1038/nn1805 (2006).
Andres, M. E., Bustos, G. & Gysling, K. Regulation of [3H]norepinephrine release by N-methyl-D-aspartate receptors in minislices from the dentate gyrus and the CA1-CA3 area of the rat hippocampus. Biochem Pharmacol 46, 1983–1987 (1993).
Liu, Z., Zhao, W., Xu, T., Pei, D. & Peng, Y. Alterations of NMDA receptor subunits NR1, NR2A and NR2B mRNA expression and their relationship to apoptosis following transient forebrain ischemia. Brain Res 1361, 133–139, https://doi.org/10.1016/j.brainres.2010.09.035 (2010).
Jung, S. et al. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol 20, 4106–4114 (2000).
Chen, Q. et al. Imaging neural activity using Thy1-GCaMP transgenic mice. Neuron 76, 297–308, https://doi.org/10.1016/j.neuron.2012.07.011 (2012).
Feng, G. et al. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28, 41–51 (2000).
Acknowledgements
This work is supported by National Institute of Health (R01NS088627, R21DE025689, R01NS089578, K22NS104392), the New Jersey Commission on Spinal Cord Research (CSCR15ERG015), and New Jersey Commission on Brain Injury Research (CBIR14FEL001). We thank Min Zhuo (University of Toronto) for providing us NVPAAM007.
Author information
Authors and Affiliations
Contributions
U.B.E. and L.J.W. designed the research; U.B.E., A.B., J.L., S.S., J.Z. and M.M. performed the research; U.B.E., A.B., J.L., S.S., R.W., V.L.D., J.Z. and M.M. performed analysis for the research; H.Z. and Y.T. contributed analytic tools; U.B.E. and L.J.W. wrote the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Eyo, U.B., Bispo, A., Liu, J. et al. The GluN2A Subunit Regulates Neuronal NMDA receptor-Induced Microglia-Neuron Physical Interactions. Sci Rep 8, 828 (2018). https://doi.org/10.1038/s41598-018-19205-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-19205-4
This article is cited by
-
APOE4 genotype and aging impair injury-induced microglial behavior in brain slices, including toward Aβ, through P2RY12
Molecular Neurodegeneration (2024)
-
Microglial process convergence onto injured axonal swellings, a human postmortem brain tissue study
Scientific Reports (2024)
-
Modulating Expression of Endogenous Interleukin 1 Beta in the Acute Phase of the Pilocarpine Model of Epilepsy May Change Animal Survival
Cellular and Molecular Neurobiology (2023)
-
The semantics of microglia activation: neuroinflammation, homeostasis, and stress
Journal of Neuroinflammation (2021)
-
Hippocampal neurons isolated from rats subjected to the valproic acid model mimic in vivo synaptic pattern: evidence of neuronal priming during early development in autism spectrum disorders
Molecular Autism (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
