
Abstract
Understating the microbial communities and ecological processes that influence their structure in permafrost soils is crucial for predicting the consequences of climate change. In this study we investigated the bacterial and archaeal communities along depth profiles of four soil cores collected across Alaska. The bacterial and archaeal diversity (amplicon sequencing) overall decreased along the soil depth but the depth-wise pattern of their abundances (qPCR) varied by sites. The community structure of bacteria and archaea displayed site-specific pattern, with a greater role of soil geochemical characteristics rather than soil depth. In particular, we found significant positive correlations between methane trapped in cores and relative abundance of methanogenic archaeal genera, indicating a strong association between microbial activity and methane production in subsurface soils. We observed that bacterial phylogenetic community assembly tended to be more clustered in surface soils than in deeper soils. Analyses of phylogenetic community turnover among depth profiles across cores indicated that the relative influence of deterministic and stochastic processes was mainly determined by soil properties rather than depth. Overall, our findings emphasize that the vertical distributions of bacterial and archaeal communities in permafrost soils are to a large extent determined by the variation in site-specific soil properties.
Similar content being viewed by others


Introduction
Approximately 25% of Earth’s terrestrial surface is underlain by permafrost1, which encompasses ∼50% of the total global soil carbon (~1672 Pg;2,3). However, permafrost soils are warming rapidly due to global climate change4, potentially resulting in extensive thaw and increased thickness of the seasonally thawed soil active layer. The thawing of the permafrost soils will expose frozen carbon pool to microbial decomposition5,6, which could further enhance the emission of carbon dioxide (CO2) and methane (CH4) from the soil, and potentially generate a positive feedback to climate warming7.
Despite prolonged subzero temperature and low water availability, a diverse group of microorganisms inhabit permafrost soils8,9,10,11. In a broader biogeography study, the Arctic tundra surface soils showed similar levels of bacterial diversity with soils of other biomes12. Microbial communities were also compared between active and permafrost soil layers and both layers displayed highly similar phylogenetic and functional community composition13. The majority of the microorganisms present in permafrost soils are cold-adapted heterotrophs belonging to the bacterial phyla such as Proteobacteria, Actinobacteria, Acidobacteria, Firmicutes, Bacteroidetes, and Chloroflexi, while others belong to archaeal phyla Euryarchaeota, Crenarchaeota, and Thaumarchaeota 14. However, it is still not well understood how this complex active and permafrost soil layer microbial community is influenced by various environmental and ecological factors.
Understanding the factors influencing the assembly of soil microorganisms along the permafrost thaw gradient is critical to predict the potential consequences of climate change1. In recent years, several studies have reported the depth specific profile of microbial communities along permafrost thaw gradient and revealed that microbial community compositions are influenced by a number of abiotic factors such as soil pH15, moisture16, conductivity17, soil organic matter quality18, and nutrient availability19,20,21. However, it is still not clear how the microbial community composition and diversity would vary along the permafrost thaw gradient sampled across different ecosystems.
Deterministic and stochastic processes are known to influence the assembly of microbial species in a local community22,23,24. Recent studies have suggested that rather than acting independently, both deterministic and stochastic processes interact to shape microbial community assembly25,26,27. The relative influence of deterministic and stochastic processes in shaping microbial community assembly is examined in various environments25,26,28,29,30, except in terrestrial cryoenvironments, which have been relatively poorly studied31. Thus, there is need to study the microbial community assembly and underlying ecological processes along the permafrost thaw gradient from different ecosystems.
We sampled four soil cores with different vegetation covers in Alaska to examine bacterial and archaeal communities along active layer and permafrost thaw gradient. This study was set out to answer the following questions:
-
(1)
How do the abundance, composition and diversity of prokaryotic community vary along the depth in permafrost soils?
-
(2)
Are there any bacterial or archaeal taxa specific to sampled sites or soil layers of the cores? Are those taxa are associated with unique geochemistry or trapped gases of the cores?
-
(3)
What are the major environmental factors that influence the assembly of prokaryotic community along the depth?
-
(4)
How does the relative influence of deterministic and stochastic processes vary along the depth to influence the assembly of prokaryotic community?
Materials and Methods
Site description and soil core sampling
Four different sites were selected for this study located along the trans-Alaskan pipeline (Fig. 1). These sites were one in tundra near Sag River (TS), two were in black spruce boreal forest towards north and south of Yukon River (NY and SY), and the other one was also in a black spruce forest in interior Alaska dominated by Sphagnum moss at Poker Flat Research Range (PF). Except TS which was located in continuous permafrost zone, the other three sites were located within discontinuous permafrost zone (see32 for detailed site descriptions).

Soil core sampling site locations along the haul road of Alaska. The map was generated in R software version 3.2.351 using package ‘mapdata’78 (https://cran.rproject.org/web/packages/mapdata/index.html). TS = tundra near Sag River; NY = north of Yukon River; SY = south of Yukon River; PF = Poker Flat Research Range.
Field sampling was carried out during the early spring in the months of April to May 2013. Soil cores were collected using a SIPRE coring auger (3-inches in diameter with PVC liners fitted in the auger). The TS, NY and SY sites were completely frozen from surface to bottom during the time of drilling, whereas the top 20–30 cm soil layer of PF site was thawed. All the four cores were sealed in PVC liners (90 cm long and 7.6 cm in diameter) and shipped to the laboratory in frozen state (−20 °C). A total of 42 samples were collected from four cores for further analysis, with 8–13 samples taken along the depth from each core.
Soil properties and trapped greenhouse gas analysis
Soil pH and electrical conductivity (EC) were measured in a soil-water suspension (1:5 ratio, w/v) using a pH/EC benchtop meter (Orion Star A215, Thermo Scientific, Waltham, MA, USA). Gravimetric water content (GWC) was measured by weighing the soils before and after drying at 105 °C for 12 h. Total organic carbon (TOC) and total nitrogen (TN) contents were measured using an elemental analyzer (FlashEA 1112 Thermo Electron corporation, Waltham, Massachusetts, USA) at the National Instrumentation Center for Environmental Management (NICEM, Seoul, Korea).
Soil core temperature was measured from boreholes drilled into the surface at 80 to 90 cm depths since 2013 to now using custom-made temperature profilers, consisting of 12 temperature sensors (TMC20-HD, Onset Comp., USA) and three data loggers (U12-008, Onset Comp., USA). The trapped CH4 and CO2 in frozen soil samples were analyzed using a headspace gas extraction method33. The frozen soil samples collected along the depth of each core were taken in a 40 ml sampling vials and filled with 10 ml 6 M NaCl solution. The sampling vials were sealed and heated at 65 °C for 30 minutes to extract the headspace air. The CH4 and CO2 concentrations of headspace air were measured by injecting 1 ml gas into an Agilent Technologies 7890 A gas chromatograph with flame ionization (FID) and thermal conductivity detectors (TCD) (Agilent Tech, Santa Clara, CA, USA) at the Korea Institute of Geoscience and Mineral Resources (KIGAM, Daejeon, Korea). The methane and carbon dioxide mixing ratios were calculated from peak areas by comparing with several standard gases (10.2, 100, 1050 ppm, 1%, 10% for CH4, and 1%, 10% for CO2). Total uncertainties for the whole procedure were better than 6.9% for CH4 and 6.2% for CO2. All the measurements of soil properties and trapped greenhouse gases were performed previously by Byun et al.32.
DNA extraction, PCR amplification and sequencing
For each sample, a single DNA extraction was performed on 0.50 g of soil using FastDNATM SPIN Kit for Soil (MP Biomedicals, Santa Ana, CA, USA) according to the manufacturer’s protocol. The primer pair Bakt_341F (5′-CCTACGGGNGGCWGCAG-3′) and Bakt_805R (5′-GACTACHVGGGTATCTAATCC-3′) was used to amplify the V3 and V4 region of bacterial 16 S rRNA gene34. Whereas, the V3 region of archaeal 16 S rRNA gene was amplified using the primer pair S-D-Arch-0349-a-S-17 (5′-GYGCASCAGKCGMGAAW-3′) and S-D-Arch-0519-a-A-16 (5′-TTACCGCGGCKGCTG-3′)35. The resulting amplicons were sequenced using paired-end (2 × 300 bp) Illumina MiSeq system (Illumina,USA) at Macrogen Incorporation (Seoul, Korea).
Sequence Processing
The pair end sequences were trimmed based on quality scores with Sickle (https://github.com/najoshi/sickle) followed by error correction with BayesHammer36.
The resulting quality trimmed and error corrected paired-end sequences were assembled using PANDAseq software37. This approach has been shown to reduce the substitution error rates by ~93% in Illumina MiSeq sequence data38. Further sequence processing steps were performed on mothur pipeline39. The assembled sequences were aligned against a SILVA alignment (http://www.arb-silva.de/), and subsequently denoised using ‘pre.cluster’ command implemented in mothur. Chimeric sequences were removed using mothur’s command ‘chimera.uchime’ in de novo mode40. The bacterial and archaeal 16 S rRNA gene sequences were taxonomically classified against EzTaxon-extended database41, using the naïve Bayesian classifier implemented in mothur (at 80% bootstrap cutoff with 1000 iterations). The sequences were further clustered to operational taxonomic units (OTUs) at 97% sequence similarity level using the average neighbor algorithm implemented in mothur. The entire singleton OTUs were removed prior to statistical analysis.
Despite using Archaea-specific primer set, we detected a lot of bacterial reads in each sample, which resulted in highly variable archaeal sequence reads across samples (120–6495 reads). Therefore, we only performed community composition analyses on archaeal sequences. For bacterial sequences, we analyzed the composition, diversity and assembly processes with rarified sequence data (2,453 reads per sample). All the sequence data used in this study are deposited to the Sequence Read Archive (SRA) at NCBI under the accession number SRP124819.
Quantitative PCR analysis
We performed quantitative PCR (qPCR) using CFX96 qPCR System (Bio-Rad, Hercules, CA) with SYBR Green as reporter dye (Bio-Rad, USA) to estimate the 16 S rRNA gene abundance of bacterial and archaea. Partial 16 S rRNA genes were amplified using lineage-specific primer pairs targeting bacteria (341 F: 5′-CCTACGGGAGGCAGCAG-3′)/797 R: 5′-GGACTACCAGGGTCTAATCCTGTT-3′42,43,) and archaea (Arch349F: 5′-GYGCASCAGKCGMGAAW-3′/Arch806R(5′-GGACTACVSGGGTATCTAAT-3′35,). For each sample, the qPCR reaction was carried out in duplicates for 40 cycles with denaturation at 94 °C for 25 s, primer annealing for bacteria at 50 °C and for archaea at 50 °C for 25 s, and extension at 72 °C, each for 25 s. A tenfold dilution series of plasmids containing the bacterial and archaeal 16 S rRNA gene from soil samples were used as the standard curves.
Phylogenetic analysis
A maximum-likelihood tree was constructed with aligned bacterial 16 S rRNA gene sequences using FastTree program with default settings44. Standardized effect size measure of mean nearest taxon distance (SES.MNTD) was calculated using the null model ‘taxa.labels’ (999 randomization) in Picante R package45. The lower magnitude of SES.MNTD values (<0) indicate phylogenetic clustering, whereas higher magnitude of SES.MNTD values (>0) indicate phylogenetic over dispersion46. Turnover in phylogenetic community composition was quantified using β-mean nearest taxon distance (βMNTD) in Picante R package (‘comdistnt’ function), which is between-community analog of MNTD. Furthermore, to infer the relative influence of community assembly processes, we calculated β-nearest taxon index (βNTI) which is referred as difference between observed and mean of null distribution of βMNTD normalized by its standard deviation. The pairwise comparisons with βNTI values <−2 and >+2 indicate deterministic assembly with less than (homogenous selection) and more than (variable selection) expected phylogenetic turnover28, respectively.
Statistical analysis
The correlation matrix of soil properties data was used to perform principal components analysis (PCA) in Canoco 5.0 (Biometrics, Wageningen, The Netherlands). Permutational multivariate analysis of variance (PerMANOVA, ‘adonis’ function in vegan R package47) was used to test the effect of sampling site on soil properties data (Euclidean distance matrix of normalized data) with 9999 random permutations.
Non-metric multi-dimensional scaling (NMDS) ordinations were used to visualize the pairwise Bray–Curtis dissimilarity matrices. We used PerMANOVA to evaluate the effect of sampling site on Bray–Curtis dissimilarity matrices (9999 random permutations). To examine the association between community structure and soil properties, the vectors of significant soil properties were fitted onto NMDS ordination space by using ‘envfit’ function of vegan R package. Redundancy analysis (RDA)-based variation partitioning analysis35 was performed to assess the relative influence of soil depth, sampling site, and soil chemical properties on composition of bacterial and archaeal community. We used Hellinger-transformed OTU abundance table as response variable for variation partitioning analysis.
We performed linear regression analysis to analyze the relationship between alpha-diversity (Shannon index) and depth. Furthermore, liner mixed effects model was used to investigate the effect of soil properties on Shannon diversity index using ‘lme’ function in nlme R package48. Soil properties were treated as fixed effects, whereas sampling site was considered as a random effect.
To evaluate the effect of depth and soil properties on relative influence of bacterial community assembly processes, the pairwise comparisons of βNTI values were regressed against Euclidean distance matrices of depth and soil properties. The statistical significance of the resulting comparisons was determined by Mantel tests (‘mantel’ function in Ecodist R package49). Furthermore, we used multiple regression on distance matrices (MRM) approach50 to evaluate the relative importance of each of the soil properties on bacterial phylogenetic community assembly (βNTI). All the statistical analyses were performed using R software version 3.2.351.
Results and Discussion
Soil properties
PCA ordination indicated the in terms of measured soil properties all the soil cores were distinct, except NY and SY cores (Fig. S1). A total of 93.7% of the variance was explained by the first two axes of PCA, with axis 1 and 2 explaining 54% and 39.7% of the total variance, respectively. PerMANOVA analysis further indicated a statistically significant effect of soil core sampling site on soil properties (R2 = 0.59, P < 0.001). Soil pH and electrical conductivity linearly increased along depth profile in each core (Fig. S2), with lowest pH and EC values being observed in PF core. These trends are in accordance with previous studies showing increase in soil pH15,18,52 and EC31 along depth profiles. The thick peat layer of PF core has resulted in detection of highest TOC, TN and GWC contents per unit soil mass in PF core, while other cores (TS, NY and SY) had relatively small amount of TOC, TN and GWC contents. Except pH and EC, the other parameters did not show consistent trend along depth profile in each core (Fig. S2).
Quantitative PCR and dominant bacterial and archaeal taxa
Vertical trends in the bacterial and archaeal abundances detected using qPCR varied by sampling site but they shifted in a similar manner with soil depth in TS, NY, and SY cores except for PF core (Fig. 2). In TS core, both bacterial and archaeal abundances declined with increasing depth with an abundance peak at surface soils. The high abundance of bacteria and archaea in surface soils compared to deeper soils has already been reported in previous studies13,52. The TS core has thinner organic layer and this might be the reason for observed strong negative correlation between bacterial and archaeal abundances and soil depth in TS core. In NY and SY cores, both bacterial and archaeal abundances were overall relatively uniform throughout the soil layers with a range of abundance fluctuations found at certain depths (Fig. 2). In PF core, bacterial abundance remained relatively constant across the depth, whereas archaeal abundance dramatically declined at a depth of 60 cm and maintained the abundance level down to the 80 cm. It is not clear why there is marked diminution in archaeal abundance at these depths. Regarding that methane concentration started increasing from a depth of 60 cm in PF core, the replacement of archaeal populations by methanogenic archaea at methane-rich soil layers may result in reduction of entire archaeal abundance.

Bacterial and archaeal 16 S rRNA gene copies quantification along depth profiles of (a) TS, (b) NY, (c) SY, and (d) PF cores.
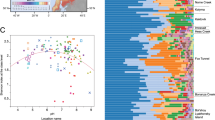
A total of 469,028 good quality 16 S rRNA gene sequences were obtained from 43 samples, and of which 369,582 and 99,446 sequences were of bacteria and archaea, respectively. The most abundant bacterial phylum across all samples was Actinobacteria (23.9% on average) followed by Proteobacteria (15.3%), Acidobacteria (14.7%), Chloroflexi (12%), Firmicutes (5%), Bacteroidetes (4.9%), Verrucomicrobia (3.6%), Gemmatimonadetes (2.9%), Planctomycetes (1.7%), Caldiserica (1.7%), AD3 (1.1%), OD1 (1.0%) and TM7 (0.9%) (Fig. 3a). The relative abundance of dominant bacterial phyla showed consistent depth related trend across all cores. Of these the relative abundance of Proteobacteria, Acidobacteria, Bacteroidetes, Verrucomicrobia, Plactomycetes and Cyanobacteria maintained at similar levels or decreased with depth (Fig. 3a). Our results are consistent with the findings of Kim et al.18, who also observed similar depth related trends in most these taxa in soil cores collected in Alaska. The decrease in relative abundance of Proteobacteria and Bacteroidetes with depth could be related to their copiotrophic life strategies because nutrient levels are normally greatest in top soils and deeper layers contain relatively less amounts of nutrients53. Verrucomicrobia and Plactomycetes were also shown earlier to dominate surface peat soils compared to deeper mineral soils54. The decrease in relative abundance of Acidobacteria could be related to increase in soil pH with depth, as several previous studies have showed that the relative abundance of Acidobacteria had a strong negative correlation with soil pH55,56,57,58. The decrease in relative abundance of photosynthetic bacterial phylum Cyanobacteria with depth could be related to reduced sunlight exposure with depth. Furthermore, consistent with prior studies, the relative abundance of Actinobacteria and Firmicutes increased with depth (Fig. 3a), which were commonly dominated in deeper mineral soils15,31. It has been also suggested that Actinobacteria and Firmicutes play important roles in anaerobic degradation of soil organic carbon at deeper depth levels54. Interestingly, Chloroflexi, which was shown earlier to dominate deeper soil layers15,18,31, showed contrasting trend with depth between cores (Fig. 3a). In TS core, there was a large proportion of Chloroflexi found throughout the active layer and its relative abundance became sharply reduced below 58 cm depth. In contrast, Chloroflexi was almost absent down to 55 cm depth in the active layer of PF core and became dominant in permafrost layer below a depth of 72 cm. These contrasting patterns suggest that some other unmeasured factors, either biotic or abiotic, may be involved in driving the relative abundance of Chloroflexi. Interestingly, an unknown Chloroflexi lineage (GQ396871_f) dominated deeper layers of PF core, whereas the majority of Chloroflexi in upper layers of TS core consist of family Anaerolineaceae, which are frequently found in petroleum-enriched areas and have potential of hydrocarbon degradation in anoxic conditions59,60. When representative sequences of major Anaerolineaceae OTUs were blast-searched against nr/nt database, they matched most closely to bacterial clone sequences recovered from benzene-contaminated river sediment, hydrocarbon-contaminated aquifer and oil reservoir (>99.0% identity with DQ463268, AF050569 and JQ668598, respectively). The greater relative abundance of phylum Caldiserica in permafrost layers of TS core also supports this because an unknown genus (EU266853_g) comprised the majority of this phylum and members belonging to this genus are often found in hydrocarbon-rich environments61,62. Another interesting finding was that the candidate bacterial phylum AD3 was dominant (~10%) in deeper permafrost layer of only PF core (Fig. 3a). The dominance of AD3 in deeper subsurface soils has been reported earlier18,21,63. Members of AD3 are known to be more abundant in deeper layers of mineral soil with low nutrient contents63. It corresponds to our results that SOC and TN contents dramatically declined below around permafrost table (80 cm depth) of PF core and soil texture also shifted from organic to mineral layers at the equivalent depth.

Relative abundance of dominant (a) bacterial (at phylum level) and (b) archaeal (at class level) taxa along depth profiles of soil cores. Permafrost table was represented by red dashed lines in TS and PF cores.
The most dominant archaeal taxa (at the class level) detected were different in each core, except in NY and SY cores (Fig. 3b). Methanomicrobia (61%) was the most dominant taxon detected in TS core (Fig. 3b), whereas FFSB (44%) was dominant in PF core (Fig. 3b). Both NY and SY cores showed similar taxonomic distribution with dominance of group 1.1b archaeal class (NY: 97% and SY: 88%) (Fig. 3b). The major depth related trends in the relative abundance of dominant archaeal taxa were observed only in TS and PF cores (Fig. 3b). The relative abundance of Methanobacteria decreased along depth profile in TS cores. Members of the Methanobacteria are hydrogenotrophic methanogens and more commonly found in upper soils15,64,65. The relative abundance of FFSB, which was the most dominant taxa in PF core, decreased with depth (Fig. 3b). The most obvious explanation for this finding is the increase in soil pH (4.34 to 5.62) along depth in PF core, as it is well documented that FFSB dominate acidic soils66,67,68, and their abundance decline quickly with increasing pH69,70. Furthermore, the relative abundance of Methanomicrobia increased with depth in PF core. Our findings support those of Deng et al.15, who found that the relative abundance of Methanomicrobia was higher in mineral permafrost layers.
The average methane concentrations of TS and PF core were much higher compared to other two cores with sharp increase in methane concentration at some depths (TS: at 27 and 81 cm depth; PF: at 78 cm depth) (Fig. S2). Stable isotope analysis revealed that methane at methane concentration peaks in these two cores is of microbial origin (see Fig. 3, Byun et al.32). Therefore, we further tested if there is any possible association between trapped methane and methanogenic archaeal taxa. We found significant correlations between methane concentrations and relative abundance of the dominant known methanogenic archaeal genera in both TS and PF cores (Fig. 4). In TS core, methane concentrations were significantly correlated with the relative abundance of hydrogenotrophic Methanobacterium (Fig. 4a), indicating that this hydrogenotrophic genus play important roles in subsurface methane generation under anaerobic conditions. The members of hydrogenotrophic methanogens were also found dominant in other studies on Arctic tundra soils15,71. It remains a moot point why methane and Methanobacterium relative abundance peak at both active and permafrost layer depths. Regarding the predominance of bacterial lineages associated with hydrocarbon degradation throughout the active layer, a methane peak at this depth may be ascribed to increased methanogenic degradation of hydrocarbons under poorly drained conditions formed at active layer of the TS core.

Correlation (Pearson) between the methane concentration and relative abundance of dominant methanogenic archaeal genera in (a) TS and (b) PF cores. Isotopic signatures of carbon and hydrogen (δ13CCH4 and δDCH4) at methane concentration peaks were retrieved from Byun et al.32.
In PF core, the methane concentrations were strongly correlated with the relative abundance of acetoclastic Methanosarcina (Fig. 4b). This observation suggests that the methane peak in deeper permafrost layers of PF core was mainly formed due to acetoclastic methanogenesis. This is in agreement with the findings of other studies reporting the dominance of acetoclastic methanogenesis over hydrogenotrophic methanogenesis in peat soils72,73,74.
Community composition
The NMDS ordination plot showed that the bacterial and archaeal community composition of soil cores was strongly clustered by sampling site (Fig. 5). The PerMANOVA analyses revealed that sampling site explained 48.7 and 54.5% of the variations in bacterial and archaeal community composition, respectively (P < 0.001, 9999 permutations). Moreover, certain dominant bacterial and archaeal OTUs also exhibited site-specific patterns (Fig. S3). Furthermore, we partitioned the explained proportion of the variation in bacterial and archaeal communities between soil properties, depth and geographical distance (Fig. S4). The largest fraction of the variation was explained by the geographically structured soil properties (35.6% for bacteria and 47.2% for archaea), of which the unique component of soil properties explained 13.4% and 10.8% of the variation in bacterial and archaeal communities, respectively. In comparison to soil properties, the unique component of geographical distance explained less variation - about 4.3% and 6.1% in bacterial and archaeal communities, respectively. The unique proportion of the variation explained by soil depth was very small and was only significant in explaining the variation in bacterial communities (Fig. S4). A large proportion of the variation (about 43.5% for bacteria and 35.4% for archaea) remained unexplained. We did not find significant influence of soil depth on bacterial and archaeal community composition (P > 0.05). This result is in contrast with the findings of Deng et al.15, who found that the largest fraction of variation in soil core bacterial community was explained by depth. However, in agreement with our result, Hu et al.31 found that the vertical distribution of bacterial community in permafrost soil core was primarily determined by physicochemical conditions rather than depth.

Non-metric multi-dimensional scaling plot of pairwise Bray–Curtis dissimilarities of (a) bacterial (b) archaeal communities. The vectors of significantly (P < 0.05) correlated variables are shown on the plot.
The environmental vector fitting analysis indicated that soil parameters such as pH, EC, GWC, and TOC significantly influenced the community composition of both bacteria and archaea (Fig. 5). It is well known that pH is one of the most important determinants of soil microbial community composition across various scales12,55,56,57. Soil pH has also been reported to influence the microbial community structure along the depth profile in permafrost soils15,18,20. Electrical conductivity31, GWC21 and total organic carbon20 are also reported earlier to influence the composition of the microbial communities with depth in permafrost soils. We also observed significant association between CO2 and CH4 concentrations and archaeal and bacterial community composition (Fig. 5). One of the possible reasons of this association could be that variations in soil physicochemical parameters along depth profile leads to change in community structure of bacteria and archaea, which might involve in key metabolic processes (such as acetoclastic methanogenesis or syntrophic acetate oxidation75) and influence the concentrations of CO2 and CH4 along the depth of soil cores.
Diversity and assembly processes
Bacterial diversity (Shannon index) decreased significantly with depth across all cores (Adj. R2 = 0.27, P < 0.001), and also influenced by soil properties (Table 1). The decrease in bacterial diversity along depth profile is commonly observed in several previous studies15,18,52. The change in environmental conditions with depth generate a strong ecological filter and this lead to decrease the bacterial diversity along depth profile, as many of the bacterial taxa are less likely to survive in deeper soil environment. The SES.MNTD values obtained using the null model were less than zero in all soil cores (Fig. 6a), indicating that the bacterial assemblages were phylogenetically clustered in each core along all depth. Our results are consistent with the findings of several previous studies in a wide range of environments31,76,77, which indicated that bacterial communities had a tendency to be phylogenetically clustered than expected by chance. The SES.MNTD values were positively correlated with depth across all cores (Fig. 6b). This indicates that bacterial community was shifted from phylogenetically more clustered assembly to less clustered assembly with increasing soil depth. It has been observed recently that bacterial lineages are more likely to be clustered in surface soils than subsurface soils77. Phylogenetically less clustered bacterial lineages in deeper soil layers could be resulted due to higher relative influence of stochastic processes in deeper soil layers than in surface soils.

(a) Box plot showing variation in standardized effect size measure of mean nearest taxon distance (SES.MNTD) values of bacterial community along depth profiles of each soil core. (b) Relationship between depth and SES.MNTD across soil cores.
We also calculated βNTI metric, which provides important insights about the community assembly processes. The turnover in βNTI metric among depth profiles across cores was significantly (Mantel P < 0.05) influenced with environmental distance, but not soil depth (Fig. 7). Among the soil properties measured, the turnover in βNTI metric was influenced by EC, CO2, GWC, pH, CH4 and carbon to nitrogen ratio (C:N) (Table 2). These results indicate that with progressive change in environmental distance, the community assembly processes shift from homogenous selection, to stochasticity, to variable selection. Overall, these results suggest that both deterministic and stochastic processes are important in governing the assembly of bacterial community along depth profiles and their relative influence vary with change in environment.

The relationships between β-nearest taxon index (βNTI) and (a) change in depth, and (b) environmental distances (Euclidean) between samples across soil cores. The statistically significant (Mantel test, 9999 permutations, P < 0.05) relationship is shown with solid red line.
Due to logistical limitations on sampling, we took only one soil core from each sampling site; however, this is insufficient to make any generalization about the depth profile of microbial communities in Alaskan soils. Therefore, the present study should be considered as a preliminary attempt for more in-depth future studies to provide a comprehensive understanding of depth profile of the microbial communities in Alaskan soils.
Conclusions
In conclusion, our study demonstrates that the taxonomic composition of bacterial and archaeal communities of Alaskan soil cores is influenced by local environmental conditions (primarily soil pH, EC, GWC and TOC) rather than depth. However, bacterial and archaeal abundance and diversity were decreased along depth profile. The relative abundance of known methanogenic archaeal genera (Methanobacterium and Methanosarcina) was significantly correlated with the methane peaks detected at various depth levels in two soils cores. Bacterial phylogenetic community structure was shifted from more clustered to less clustered assembly with depth. Both deterministic and stochastic processes were involved in governing the assembly of bacterial community, and their relative influence varied strongly with change in environment rather than depth. Further studies on taxonomy and phylogeny of soil microbial communities along permafrost thaw gradient are necessary in order to have comprehensive understanding of the microbial ecology of permafrost soils.
Data availability statement
All the 16 S rRNA gene sequence data used in this study are deposited to the Sequence Read Archive (SRA) at NCBI under the accession number SRP124819.
References
Graham, D. E. et al. Microbes in thawing permafrost: the unknown variable in the climate change equation. ISME J 6, 709–712 (2012).
Schuur, E. A. G. et al. Vulnerability of permafrost carbon to climate change: implications for the global carbon cycle. Bioscience 58, 701–714 (2008).
Tarnocai, C. et al. Soil organic carbon pools in the northern circumpolar permafrost region. Global Biogeochem Cycles 23, GB2023 (2009).
Romanovsky, V. E., Smith, S. L. & Christiansen, H. H. Permafrost thermal state in the polar Northern Hemisphere during the international polar year 2007–2009: a synthesis. Permafr Periglac Process 21, 106–116 (2010).
Schuur, E. A. G. et al. The effect of permafrost thaw on old carbon release and net carbon exchange from tundra. Nature 459, 556–559 (2009).
Pautler, B. G., Simpson, A. J., Mcnally, D. J., Lamoureux, S. F. & Simpson, M. J. Arctic permafrost active layer detachments stimulate microbial activity and degradation of soil organic matter. Environ Sci Technol 44, 4076–4082 (2010).
Schuur, E. A. G. et al. Climate change and the permafrost carbon feedback. Nature 520, 171–179 (2015).
Bakermans, C., Skidmore, M. L., Douglas, S. & McKay, C. P. Molecular characterization of bacteria from permafrost of the Taylor Valley, Antarctica. FEMS Microbiol Ecol 89, 331–346 (2014).
Frank-Fahle, B. A., Yergeau, É., Greer, C. W., Lantuit, H. & Wagner, D. Microbial functional potential and community composition in permafrost-affected soils of the NW Canadian Arctic. PLoS ONE 9, e84761 (2014).
Ganzert, L., Bajerski, F. & Wagner, D. Bacterial community composition and diversity of five different permafrost-affected soils of Northeast Greenland. FEMS Microbiol Ecol 89, 426–441 (2014).
Coolen, M. J., van de Giessen, J., Zhu, E. Y. & Wuchter, C. Bioavailability of soil organic matter and microbial community dynamics upon permafrost thaw. Environ Microbiol 13, 2299–2314 (2011).
Chu, H. et al. Soil bacterial diversity in the Arctic is not fundamentally different from that found in other biomes. Environ Microbiol 12, 2998–3006 (2010).
Yergeau, E., Hogues, H., Whyte, L. G. & Greer, C. W. The functional potential of high Arctic permafrost revealed by metagenomic sequencing, qPCR and microarray analyses. ISME J 4, 1206–1214 (2010).
Jansson, J. K. & Taş, N. The microbial ecology of permafrost. Nat Rev Microbiol 12, 414–425 (2014).
Deng, J. et al. Shifts of tundra bacterial and archaeal communities along a permafrost thaw gradient in Alaska. Mol Ecol 24, 222–234 (2015).
Zhang, X. et al. Soil moisture effect on bacterial and fungal community in Beilu River (Tibetan Plateau) permafrost soils with different vegetation types. J Appl Microbiol 114, 1054–1065 (2013).
Hu, W. et al. Diversity and community structure of fungi through a permafrost core profile from the Qinghai‐Tibet Plateau of China. J Basic Microbiol 54, 1331–1341 (2014).
Kim, H. M. et al. Vertical distribution of bacterial community is associated with the degree of soil organic matter decomposition in the active layer of moist acidic tundra. J Microbiol 54, 713–723 (2016).
Koyama, A., Wallenstein, M. D., Simpson, R. T. & Moore, J. C. Soil bacterial community composition altered by increased nutrient availability in Arctic tundra soils. Front Microbiol 5, 516 (2014).
Gittel, A. et al. Distinct microbial communities associated with buried soils in the Siberian tundra. ISME J 8, 841–853 (2014).
Taş, N. et al. Impact of fire on active layer and permafrost microbial communities and metagenomes in an upland Alaskan boreal forest. ISME J 8, 1904–1919 (2014).
Torsvik, V., Ovreas, L. & Thingstad, T. F. Prokaryotic diversity - Magnitude, dynamics, and controlling factors. Science 296, 1064–1066 (2002).
Baas-Becking, L. G. M. Geobiologie of inleiding tot de milieukunde. (eds Van Stockum, W. P. & Zoon, N. V., Den Haag [Netherlands], 1934).
Peay, K. G., Garbelotto, M. & Bruns, T. D. Evidence of dispersal limitation in soil microorganisms: isolation reduces species richness on mycorrhizal tree islands. Ecology 91, 3631–3640 (2010).
Wang, J. et al. Phylogenetic beta diversity in bacterial assemblages across ecosystems: deterministic versus stochastic processes. ISME J 7, 1310–1321 (2013).
Stegen, J. C., Lin, X., Konopka, A. E. & Fredrickson, J. K. Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J 6, 1653–1664 (2012).
Stegen, J. C. et al. Quantifying community assembly processes and identifying features that impose them. ISME J 7, 2069–2079 (2013).
Dini-Andreote, F., Stegen, J. C., van Elsas, J. D. & Salles, J. F. Disentangling mechanisms that mediate the balance between stochastic and deterministic processes in microbial succession. Proc Natl Acad Sci USA 112, E1326–E1332 (2015).
Martínez, I. et al. The gut microbiota of rural Papua New Guineans: composition, diversity patterns, and ecological processes. Cell Rep 11, 527–538 (2015).
Stegen, J. C. et al. Groundwater-surface water mixing shifts ecological assembly processes and stimulates organic carbon turnover. Nat Commun 7, 11237 (2016).
Hu, W. et al. Relative roles of deterministic and stochastic processes in driving the vertical distribution of bacterial communities in a permafrost core from the Qinghai-Tibet Plateau, China. PLoS ONE 10, e0145747 (2015).
Byun, E., Yang, J. W., Kim, Y. & Ahn, J. Trapped greenhouse gases in the permafrost active layer: preliminary results for methane peaks in vertical profiles of frozen Alaskan soil cores. Permafr Periglac Process 28, 477–484 (2016).
Kim, J. H. et al. Inferences on gas transport based on molecular and isotopic signatures of gases at acoustic chimneys and background sites in the Ulleung Basin. Org Geochem 43, 26–38 (2012).
Herlemann, D. P. et al. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J 5, 1571–1579 (2011).
Klindworth, A. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res 41, e1 (2013).
Nikolenko, S. I., Korobeynikov, A. I. & Alekseyev, M. A. BayesHammer: Bayesian clustering for error correction in single-cell sequencing. BMC Genomics 14, S7 (2013).
Masella, A. P., Bartram, A. K., Truszkowski, J. M., Brown, D. G. & Neufeld, J. D. PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics 13, 31 (2012).
Schirmer, M. et al. Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic Acids Res 43, e37 (2015).
Schloss, P. D. et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75, 7537–7541 (2009).
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C. & Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200 (2011).
Kim, O. S. et al. Introducing EzTaxon-e: a prokaryotic 16S rRNA gene sequence database with phylotypes that represent uncultured species. Int J Syst Evol Microbiol 62, 716–721 (2012).
Lane, D. J. 16S/23S rRNA sequencing. In: Nucleic acid techniques in bacterial systematics (eds Stackebrandt, E. & Goodfellow, M.) 115–175 (John Wiley & Sons, 1991).
Nadkarni, M. A., Martin, F. E., Jacques, N. A. & Hunter, N. Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology 148, 257–266 (2002).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2-approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490 (2010).
Kembel, S. W. et al. Picante: R tools for integrating phylogenies and ecology. Bioinformatics 26, 1463–1464 (2010).
Webb, C. O., Ackerly, D. D., McPeek, M. A. & Donoghue, M. J. Phylogenies and community ecology. Annu Rev Ecol Syst 33, 475–505 (2002).
Oksanen, J. et al. The vegan package: Community Ecology Package. R package version 2.3-5, Available at: CRAN.R-project.org/package = vegan (2007).
Pinheiro, J., Bates, D., DebRoy, S. & Sarkar, D. R Core Team, nlme: Linear and Nonlinear Mixed Effects Models. R package version 3.1-120, Available at: CRAN.R-project.org/package = nlme (2015).
Goslee, S. C. & Urban, D. L. The ecodist package for dissimilarity-based analysis of ecological data. J Stat Softw 22, 1–19 (2007).
Legendre, P., Lapointe, F. J. & Casgrain, P. Modeling brain evolution from behavior - a permutational regression approach. Evolution 48, 1487–1499 (1994).
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing (https://www.R-project.org), Vienna, Austria (2015).
Eilers, K. G., Debenport, S., Anderson, S. & Fierer, N. Digging deeper to find unique microbial communities: the strong effect of depth on the structure of bacterial and archaeal communities in soil. Soil Biol Biochem 50, 58–65 (2012).
Fierer, N., Bradford, M. A. & Jackson, R. B. Toward an ecological classification of soil bacteria. Ecology 88, 1354–1364 (2007).
Tveit, A., Schwacke, R., Svenning, M. M. & Urich, T. Organic carbon transformations in high-Arctic peat soils: key functions and microorganisms. ISME J 7, 299–311 (2013).
Lauber, C. L., Hamady, M., Knight, R. & Fierer, N. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl Environ Microbiol 75, 5111–5120 (2009).
Tripathi, B. M. et al. Tropical soil bacterial communities in Malaysia: pH dominates in the equatorial tropics too. Microbial Ecol 64, 474–484 (2012).
Rousk, J. et al. Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J 4, 1340–1351 (2010).
Jones, R. T. et al. A comprehensive survey of soil acidobacterial diversity using pyrosequencing and clone library analyses. ISME J 3, 442–453 (2009).
Liang, B. et al. High frequency of Thermodesulfovibrio spp. and Anaerolineaceae in association with Methanoculleus spp. in a long-term incubation of n-alkanes-degrading methanogenic enrichment culture. Front Microbiol 7, 1431 (2016).
Sherry, A. et al. Anaerobic biodegradation of crude oil under sulphate-reducing conditions leads to only modest enrichment of recognized sulphate-reducing taxa. Int Biodeter Biodegr 81, 105–113 (2013).
Winderl, C., Anneser, B., Griebler, C., Meckenstock, R. U. & Lueders, T. Depth-resolved quantification of anaerobic toluene degraders and aquifer microbial community patterns in distinct redox zones of a tar oil contaminant plume. Appl Environ Microbiol 74, 792–801 (2008).
Tischer, K. et al. Microbial communities along biogeochemical gradients in a hydrocarbon‐contaminated aquifer. Environ Microbiol 15, 2603–2615 (2013).
Costello, E. K. Molecular Phylogenetic Characterization of High Altitude Soil Microbial Communities and Novel, Uncultivated Bacterial Lineages (ProQuest, 2007).
Thauer, R. K. Biochemistry of methanogenesis: a tribute to Marjory Stephenson: 1998 Marjory Stephenson Prize Lecture. Microbiology 144, 2377–2406 (1998).
Conrad, R., Schütz, H. & Babbel, M. Temperature limitation of hydrogen turnover and methanogenesis in anoxic paddy soil. FEMS Microbiol Ecol 3, 281–289 (1987).
Nicol, G. W., Tscherko, D., Embley, T. M. & Prosser, J. I. Primary succession of soil Crenarchaeota across a receding glacier foreland. Environ Microbiol 7, 337–347 (2005).
Nicol, G. W., Campbell, C. D., Chapman, S. J. & Prosser, J. I. Afforestation of moorland leads to changes in crenarchaeal community structure. FEMS Microbiol Ecol 60, 51–59 (2007).
Kemnitz, D., Kolb, S. & Conrad, R. High abundance of Crenarchaeota in a temperate acidic forest soil. FEMS Microbiol Ecol 60, 442–448 (2007).
Lehtovirta, L. E., Prosser, J. I. & Nicol, G. W. Soil pH regulates the abundance and diversity of Group 1.1c Crenarchaeota. FEMS Microbiol Ecol 70, 367–376 (2009).
Tripathi, B. M. et al. pH dominates variation in tropical soil archaeal diversity and community structure. FEMS Microbiol Ecol 86, 303–311 (2013).
Barbier, B. A. et al. Methane-cycling communities in a permafrost-affected soil on Herschel Island, Western Canadian Arctic: active layer profiling of mcrA and pmoA genes. FEMS Microbiol Ecol 82, 287–302 (2012).
Schmidt, O., Hink, L., Horn, M. A. & Drake, H. L. Peat: home to novel syntrophic species that feed acetate-and hydrogen-scavenging methanogens. ISME J 10, 1954–1966 (2016).
Tveit, A. T., Urich, T., Frenzel, P. & Svenning, M. M. Metabolic and trophic interactions modulate methane production by Arctic peat microbiota in response to warming. Proc Natl Acad Sci USA 112, E2507–E2516 (2015).
Metje, M. & Frenzel, P. Methanogenesis and methanogenic pathways in a peat from subarctic permafrost. Environ Microbiol 9, 954–964 (2007).
Kato, S. & Watanabe, K. Ecological and evolutionary interactions in syntrophic methanogenic consortia. Microbes Environ 25, 145–151 (2010).
Horner-Devine, M. C. & Bohannan, B. J. Phylogenetic clustering and overdispersion in bacterial communities. Ecology 87, S100–108 (2006).
Chu, H. et al. Bacterial community dissimilarity between the surface and subsurface soils equals horizontal differences over several kilometers in the western Tibetan Plateau. Environ Microbiol 18, 1523–1533 (2016).
Brownrigg, R., Minka, T., Becker, R. & Wilks, A. Mapdata: Extra map databases. R package version:2.2-2. Available at: CRAN.R-project.org/package = mapdata (2013).
Acknowledgements
This work was supported by the Korea Research Fellowship Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (NRF-2015H1D3A1066568 (PN16030, KOPRI) and NRF-2015R1A2A2A01003888). This study was also supported by a National Research Foundation of Korea Grant from the Korean Government (MSIP) (NRF-2011-0021063, 0021067 (PN15082, KOPRI)), (NRF-2016M1A5A1901769), (NRF-2016M1A5A1901770 (KOPRI-PN17082)) (Title: Circum Arctic Permafrost Environment Change Monitoring, Future Prediction and Development Techniques of Useful Biomaterials (CAPEC Project)), and KOPRI’s basic research project (PE17010). This study was also conducted under the JAMSTEC-IARC Collaboration Study (JICS project), with funding provided by the Japan Agency for Marine-Earth Science and Technology (JAMSTEC) under a grant to the International Arctic Research Center (IARC). Finally, this manuscript is dedicated to the late Dr. Rikie Suzuki of the JAMSTEC.
Author information
Authors and Affiliations
Contributions
B.M.T., M.K. and Y.K.L. conceived of the study. Y.K., J.W.Y. and J.A. performed field sampling. M.K., E.B. and J.W.Y. contributed to lab experiments. B.M.T. and M.K. analyzed the data. B.M.T. and M.K. wrote the main manuscript text and all authors approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tripathi, B.M., Kim, M., Kim, Y. et al. Variations in bacterial and archaeal communities along depth profiles of Alaskan soil cores. Sci Rep 8, 504 (2018). https://doi.org/10.1038/s41598-017-18777-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-18777-x
This article is cited by
-
Microbial life in 25-m-deep boreholes in ancient permafrost illuminated by metagenomics
Environmental Microbiome (2023)
-
Warming changes the composition and diversity of fungal communities in permafrost
Annals of Microbiology (2023)
-
Distinct taxonomic and functional profiles of high Arctic and alpine permafrost-affected soil microbiomes
Environmental Microbiome (2023)
-
Permafrost microbial communities and functional genes are structured by latitudinal and soil geochemical gradients
The ISME Journal (2023)
-
Vertical Distribution of Soil Bacterial Communities in Different Forest Types Along an Elevation Gradient
Microbial Ecology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
