
Abstract
The Mycobacterium tuberculosis complex (MTBC) is the collective term given to the group of bacteria that cause tuberculosis (TB) in mammals. It has been reported that M. tuberculosis H37Rv, a standard reference MTBC strain, is attenuated in cattle compared to Mycobacterium bovis. However, as M. tuberculosis H37Rv was isolated in the early 1930s, and genetic variants are known to exist, we sought to revisit this question of attenuation of M. tuberculosis for cattle by performing a bovine experimental infection with a recent M. tuberculosis isolate. Here we report infection of cattle using M. bovis AF2122/97, M. tuberculosis H37Rv, and M. tuberculosis BTB1558, the latter isolated in 2008 during a TB surveillance project in Ethiopian cattle. We show that both M. tuberculosis strains caused reduced gross pathology and histopathology in cattle compared to M. bovis. Using M. tuberculosis H37Rv and M. bovis AF2122/97 as the extremes in terms of infection outcome, we used RNA-Seq analysis to explore differences in the peripheral response to infection as a route to identify biomarkers of progressive disease in contrast to a more quiescent, latent infection. Our work shows the attenuation of M. tuberculosis strains for cattle, and emphasizes the potential of the bovine model as a ‘One Health’ approach to inform human TB biomarker development and post-exposure vaccine development.
Similar content being viewed by others


Introduction
The Mycobacterium tuberculosis complex (MTBC), the group of pathogens that cause tuberculosis (TB) in mammals, show distinct host preference. Mycobacterium tuberculosis is the hallmark member of the MTBC and the most deadly human pathogen globally, with close to 2 billion people infected worldwide1. The animal-adapted members of the MTBC are named after the host of initial/most frequent isolation, and comprise: the exemplar animal pathogen and predominant agent of bovine TB M. bovis2; M. microti3; the ‘Dassie bacillus’4,5; M. caprae6; M. pinnipedii7; M. orygis8, and M. mungi9. A caveat is that these species designations do not define host exclusivity; MTBC members can infect a range of mammals to greater or lesser degrees. The central feature of host adaptation is the ability to sustain within a host population. Thus, M. bovis can infect and cause disease in humans; however, the capacity of M. bovis to transmit between immunologically competent humans is severely limited compared to M. tuberculosis10,11. Similarly, reports suggest that M. tuberculosis appears attenuated in a bovine host compared to M. bovis12,13,14.
The advent of systems for mutagenesis of mycobacteria allied with (largely) murine screens for phenotype has provided enormous insight into the virulence genes and pathogenic strategies of mycobacteria. Yet the basis for host preference across the MTBC is largely unknown. Members of the MTBC share >99% nucleotide identity across their genomes, with for example ~2,400 SNPs between M. bovis AF2122/97 and M. tuberculosis H37Rv15,16,17. Regions of difference (RD), deleted loci absent from one MTBC member relative to another, serve as unique markers to differentiate species with some having had functional roles ascribed. The RD1 locus of M. tuberculosis encodes a type VII secretion system whose role in virulence of M. tuberculosis has been convincingly explored using a range of in vitro and model systems18. However, RD1-like deletions from M. microti and the ‘dassie’ bacillus indicate that discrete evolutionary scenarios have moulded the virulence strategies and genomes of the MTBC bacilli19,20. Similarly, functional impacts of single nucleotide polymorphisms (SNPs) between the various MTBC and their potential role in host-pathogen interaction have been described21. Much work remains in describing the precise host and pathogen molecular factors involved in host preference22.
An essential step to defining host tropic factors is defining tractable experimental models; given the range of wild and domesticated mammals that are susceptible to infection by MTBC members, this is no small task. Nevertheless, with the aim of exploring MTBC host preference, we have previously explored the comparative virulence of M. tuberculosis and M. bovis in a bovine experimental infection model, and showed that M. tuberculosis H37Rv was attenuated compared to M. bovis AF2122/9713. However, while M. tuberculosis H37Rv is the reference strain of the MTBC, its isolation was first reported 193523 and it has been maintained in multiple laboratories globally, with separate extant ‘versions’ of M. tuberculosis H37Rv24. The possibility remained that other M. tuberculosis clinical isolates would show a different phenotype in a bovine infection. Indeed, there have been reports of the isolation of M. tuberculosis from cattle25,26,27, including our own work where we previously isolated M. tuberculosis strains from lesions identified in Ethiopian cattle28,29. This would suggest that either there exist strains of M. tuberculosis with virulence characteristics that allow them to infect and cause disease in cattle, or that the cattle from which these M. tuberculosis strains were isolated had greater susceptibility to infection because of being immune comprised, co-infections, age, malnutrition, or other predisposing factors such as being in an environment of continuous exposure to M. tuberculosis.
We therefore set out to evaluate the virulence of M. tuberculosis in the bovine host, using both M. tuberculosis H37Rv and a recent M. tuberculosis bovine isolate as the comparator. For the latter we chose to use an M. tuberculosis strain isolated from an Ethiopian Zebu bull, M. tuberculosis BTB1558, and compared its virulence in an experimental bovine infection to the M. tuberculosis H37Rv and M. bovis AF2122/97 reference strains.
Material and Methods
Ethical permission
Ethical permission was obtained from the APHA Animal Use Ethics Committee (UK Home Office PCD number 70/6905), AHRI/ALERT Ethics Review Committee (Ethiopia) and the French Research and Education Ministry, via the Val de Loire Ethical Committee (CEEAVDL, #19) for INRA (France), with all experiments on live animals performed in accordance with relevant guidelines and regulations.
Mycobacterial strains and culturing protocols
Three strains were used for this bovine challenge experiment: M. bovis AF2122/97 is a field strain isolated from a cow in Great Britain in 199716. M. tuberculosis H37Rv was from the APHA culture stocks. The virulence of both the M. bovis AF2122/97 and M. tuberculosis H37Rv stocks has been confirmed via inoculation of guinea pigs13, with both seed stocks clearly virulent in this model. M. tuberculosis BTB1558 was isolated in 2008 from the cranial mediastinal lymph node of a Zebu bull (Bos indicus) at the Ghimbi abattoir, Ethiopia. The lesion from which the strain was isolated was classed as localised, and was not caseous or calcified; no nasal secretions were present at the ante-mortem investigation. An M. tuberculosis strain of the same spoligotype as BTB1558 (SIT 764) was isolated from a human pulmonary TB patient in Ethiopia30; both the cattle and the human isolate have been confirmed as members of the Euro-American lineage, also known as Lineage 431,32.
M. bovis AF2122/97 and M. tuberculosis BTB1558 had been passaged a maximum of five times prior to the challenge experiment. Seed stocks had been cultured to mid-log phase in Middlebrook 7H9 media (Difco, UK) supplemented with 10% (v/v) Middlebrook acid-albumin-dextrose-catalase enrichment (Difco), 4.16 g/l sodium pyruvate (Sigma-Aldrich, UK) and 0.05% (v/v) Tween 80 (Sigma-Aldrich) and stocks stored frozen at −80 °C. The colony forming units (CFU)/ml of bacterial stocks, infection inoculum, and homogenised tissue was determined by bacterial enumeration of a serial dilution cultured on modified Middlebrook 7H11 agar33. Inoculated plates were incubated at 37 °C for four weeks (six weeks for tissue samples) prior to enumeration of individual colonies on the agar plates. All seed stocks were confirmed with a viability of approximately 2 × 107 CFU/ml prior to further use.
Preparation of M. bovis and M. tuberculosis infection inoculum
Frozen seed stocks were thawed and diluted in 7H9 medium to a final concentration of approximately 5 × 103 CFU/ml. For each animal, 2 ml of this infection inoculum were drawn into a 5 ml Luer-lock syringe. An aliquot of the M. bovis AF2122/97, M. tuberculosis H37Rv, and M. tuberculosis BTB1558 inocula was retained to determine, retrospectively, the concentration of bacilli used in each inoculum. Heat-inactivated samples of each strain were used to confirm the identify of the strains by large sequence polymorphism29,34 and spoligotyping35.
Cattle infection
All animal infections were carried out within the INRA ‘Platform for experimentation on infectious diseases’ biocontainment level 3 suites. Twelve female Limousin x Simmenthal cattle of approximately six months of age raised from birth within INRA’s animal facility were divided into three groups of four. An infective dose of 1 × 104 CFU was targeted for each strain; inocula were delivered endobronchially in 2 ml of 7H9 medium as described previously36. In brief, animals were sedated with xylazine (Rompun® 2%, Bayer, France) according to the manufacturer’s instructions (0.2 mL/100 kg, IV route) prior to the insertion of an endoscope through the nasal cavity into the trachea for delivery of the inoculum through a 1.8 mm internal diameter cannula (Veterinary Endoscopy Services, U.K.) above the bronchial opening to the cardiac lobe and the main bifurcation between left and right lobes. Two ml of PBS were used to rinse any remains of the inoculum into the trachea and then cannula and endoscope were withdrawn. The canal through which the cannula was inserted into the endoscope was rinsed with 20 ml of PBS and the outside of the endoscope was wiped with sterilizing wipes (Medichem International, U.K.) prior to infection of the next animal. Retrospective counting of the inocula revealed infection with 1.66 × 104 CFU M. tuberculosis H37Rv; 2.2 × 104 CFU M. tuberculosis BTB1558; and 1.12 × 104 CFU M. bovis AF2122/97.
Monitoring of infection by the IFN-γ release Assay (IGRA)
Blood was collected from animals one day prior to the infectious challenge (−1) and every two weeks after infection until the animals were culled. Heparinized whole blood (250 μl) was incubated with a selection of mycobacterial antigens: PPD-Avium (PPD-A) or PPD-Bovine (PPD-B) (Prionics) respectively at 25 IU and 30 IU final; or peptide pools covering ESAT6/CFP10, Rv3873 or Rv3879c added in a volume of 25 μl to a final concentration of 10 μg/ml. Pokeweed mitogen was used as the positive control at 10 µg/ml, and a media-only negative control also included. After 24 h in 5% (v/v) CO2 atmosphere at 37 °C stimulated bloods were centrifuged (400 × g for 5 min); 120 µl of supernatant was removed and stored at −80 °C for subsequent IFN-γ quantification using the Bovigam kit (Prionics) in accordance with the manufacturer’s instructions.
For RNA extractions, 4 ml of heparinized whole blood was incubated with PBS or PPD-B in the same condition as described above. After 24 hr 3 ml of stimulated blood were transferred to TempusTM Blood RNA tubes (Life Technologies) and stored at −80 °C. The remaining 1 ml was centrifuged and the supernatant stored at −80 °C for cytokine analyses.
Multiplex ELISA analyses
PPD-B stimulated whole blood supernatants were assayed for cytokine levels using a custom-designed bovine Meso Scale Discovery® (MSD) multiplex protein analysis platform (Meso Scale Discovery®, Gaithersburg, MD, USA). The bovine cytokines analysed were: IL-1β, IL-6, IL-10, IL-12, and TNF-α. Multiplex 96 well plates (supplied with target capture antibodies spotted onto separate carbon electrodes in each well) were blocked with MSD® assay buffer for 30 min at room temperature before the addition of 25 µl samples or MSD® standards (prepared according to manufacturer’s instructions). Following 2 h sample incubation at room temperature, plates were washed and incubated for a further 2 h with a combined cocktail of biotinylated detection antibodies for each cytokine and MSD® SULFO-TAG™-labelled Streptavidin (according to the manufacturers’ instructions). After a final wash, plates were coated with MSD® Buffer-T and luminescence (OD455 nm) was measured on a SECTOR® Imager 6000 instrument (MSD). IL-6, IL-10 and IL-12 responses are reported as U/ml while IL-1β and TNF-α responses are reported in ng/ml as interpolated from the standard curves for each cytokine included on each plate.
Gross pathology and histopathology
Ten weeks after infection animals inoculated with M. tuberculosis were killed and subjected to post-mortem analysis as indicated elsewhere37; animals inoculated with M. bovis were sacrificed six weeks after infection for post-mortem analysis as above. The presence of gross pathological TB-like lesions was scored as previously described (37). For histology, a cross-sectional slice of the lymph node was collected into a 100 ml pot containing buffered formalin. Collected tissue samples were shipped overnight from INRA to APHA Weybridge for subsequent processing.
Tissues evaluated for gross pathology included the following lymph nodes: left and right parotid, lateral retropharyngeal, medial retropharyngeal, submandibular, caudal, cranial mediastinal and cranial tracheobronchial and pulmonary lymph nodes; lung tissue samples were also taken. Tissue samples were preserved in 10% phosphate buffered formalin for 7 days before being embedded in paraffin wax. Four-micron sections were cut and stained with hematoxylin and eosin (H&E); Ziehl-Neelsen staining was carried out for the detection of acid-fast bacilli (AFB). For histopathology, a full section of thoracic (caudal mediastinal, cranial mediastinal, cranial tracheobronchial, left and right bronchial) and extrathoracic (left and right parotid, left and right medial retropharyngeal, left and right lateral retropharyngeal, left and right mandibular) lymph nodes, left and right tonsils and lung were stained with heamoatxilin and eosin (H&E) for examination by light microscopy (at x100 magnification) to assess the number, developmental stage and distribution of each granuloma (I-IV) as well as assessing the quantity and location of AFB as previously described38,39.
Evaluation of tissue bacterial load
For bacteriology, up to 20 g of tissue was collected into 25 ml sterile containers and frozen at −80 °C on the day of collection. Frozen tissues were shipped at +4 °C to APHA Weybridge and immediately upon arrival were homogenised using a Seward Stomacher Paddle Blender with bacterial enumeration undertaken as previously described37. Macerates were plated on modified 7H11 agar plates containing 10% (vol/vol) Middlebrook oleic acid-albumin-dextrose-catalase enrichment33. Plates were seeded with 500 µl, 50 µl and 5 µl of macerate; 450 µl and 500 µl of PBS was added to the plates containing 50 µl and 5 µl respectively to help distribute the macerate on the whole plate. Using this method the limit of detection was 2 CFU/ml.
RNA extraction and library preparation
Thirty-two strand-specific RNA-Seq libraries were prepared from whole blood from M. bovis AF2122/7 and M. tuberculosis H37Rv infected animals (n = 4) at day 14 and day 42 that were either stimulated or not with PPD-B. Total RNA including miRNA was extracted from the Tempus™ Blood RNA Tubes using the Preserved Blood RNA Purification Kit I (Norgen Biotek Corp, Canada) according to the manufacturer’s instructions. Twelve random samples were chosen to assess RNA integrity using the RNA 6000 Nano Kit (Agilent) in conjunction with the Agilent 2100 Bioanalyzer. RNA Integrity Numbers (RINs) ranged from 8 to 9.1 (8.6 average). RNA was quantified using the NanoDrop™ ND-1000 Spectrophotometer (Thermo Fisher Scientific) and 200 ng of total RNA was subjected to two rounds of Poly(A) mRNA purification using the Dynabeads® mRNA DIRECT™ Micro Kit (Invitrogen™) according to the manufacturer’s recommendations. The samples were prepared for sequencing using the ScriptSeq™ v2 RNA-Seq Library Preparation Kit, Index PCR Primers and the FailSafe™ PCR enzyme system according to manufacturer’s specifications (Illumina® Inc., Madison, WI, USA). The Agencourt® AMPure® XP system (Beckman Coulter Genomics, Danvers, MA, USA) was utilised to purify the resulting RNA-Seq libraries. Libraries were quantified using the Quant-iT dsDNA Assay Kit and subsequently pooled in equimolar concentrations (Thermo Fisher Scientific). The 32 sequencing libraries were pooled and sequenced over three lanes of an Illumina HiSeq 2500 Rapid Run flow cell (v1) in paired end (2 × 100 bp) format by Michigan State University Research Technology Support Facility, Michigan, USA. Base-calling and demultiplexing was performed with Illumina Real Time Analysis [v1.17.21.3] and Illumina Bcl2Fasta [v1.8.4] respectively. The RNA-Seq data has been deposited in the European Nucleotide Archive, accession number PRJEB22247.
RNA-Seq pipeline
The pipelines used for the analysis of the RNA-Seq data are available on GitHub (https://github.com/kerrimalone). Computational analyses were performed on a 32-node Compute Server with Linux Ubuntu [version 12.04.2]. Briefly, pooled libraries were deconvoluted, adapter sequence contamination and paired-end reads of poor quality were removed using cutadapt [v1.13] (Phred > 28)40 and the filterbytile.sh script from the BBMap package41. At each step, read quality was assessed with FastQC [v0.11.5]42. Paired-end reads were aligned to the Bos taurus reference genome (B. taurus UMD 3.1.1) with the STAR software43. Read counts for each gene were calculated using featureCounts, set to unambiguously assign uniquely aligned paired-end reads in a stranded manner to gene exon annotation (B. taurus UMD 3.1.1 GCF_000003055.6)44. Differential gene expression analysis was performed using the edgeR Bioconductor package that was customised to filter out all bovine ribosomal RNA (rRNA) genes, genes displaying expression levels below one count per million (CPM) in at least four individual libraries and identify differentially expressed (DE) genes correcting for multiple testing using the Benjamini-Hochberg method with a log2 fold change (log2FC) greater than 1/less than -1 and a False-Discovery Rate (FDR) threshold of significance ≤0.0545. DE gene expression was evaluated between M. bovis and M. tuberculosis infected animals for unstimulated blood samples (unpaired statistics) for each time point in addition to between unstimulated and PPD-B-stimulated blood samples for the M. bovis and M. tuberculosis infected animals independently at each time point (paired statistics). Cellular functions and pathways over-represented in DE gene lists were assessed using the SIGORA R package while graphical representation of data results was achieved using the R packages ggplot2, VennDiagram and related supporting packages46,47,48.
miRNA RT-qPCR
MicroRNA miR-155 was selected for analysis based on its suggested role in immune response to M. bovis infection49. As the human and bovine sequences are identical, hsa-miR-155–5p primers were purchased from Exiqon miRCURY UniRT miRNA primers (catalogue number 204308).
Results
Immune response in infected cattle
Three groups of four cattle each were infected endobronchially with 1.66 × 104 CFU M. tuberculosis H37Rv, 2.2 × 104 CFU M. tuberculosis BTB1558, or 1.12 × 104 CFU M. bovis AF21222/97, respectively. Because of the need to restrict the total time the experiment would run in the containment facility, animals infected with M. tuberculosis strains H37Rv or BTB1558 were maintained for 10 weeks, while M. bovis AF2122/97-infected animals were maintained for 6 weeks, after which all animals underwent post-mortem examination. Antigen specific IFN-γ responses were detected against both PPD-B (data not shown) and a cocktail of ESAT-6/CFP-10 peptides (Fig. 1) two to three weeks after infection, with no significant difference in responses between groups over the course of the infections.

Infection of cattle with M. tuberculosis H37Rv, M. tuberculosis BTB1558 or M. bovis AF2122/97 induces similar peripheral immune responses. Blood was collected at regular intervals from cattle prior to and after experimental infection with M. tuberculosis H37Rv (n = 4), M. tuberculosis BTB1558 (n = 4), or M. bovis AF2122/97 (n = 4). Whole blood was isolated and stimulated with a cocktail of peptides derived from ESAT-6 and CFP-10. The responses in the infected cattle are shown: M. tuberculosis H37Rv (squares); M. tuberculosis BTB1558 (triangles); M. bovis AF2122/97 (circles). M. bovis infected animals were maintained for 6 weeks, M. tuberculosis groups for 10 weeks. Data for each time point is presented as the mean response + SEM.
Supernatants from PPD-B stimulated samples were also checked for IL-1β, IL-6, IL-10, IL-12, and TNF-α production over the course of infection using the MSD multiplex platform (Fig. 2). Over the first 6 weeks after infection, all animals showed similar responses to all strains, although M. tuberculosis BTB1558 generated higher IL-1β responses at day 28 compared to responses induced by M. tuberculosis H37Rv or M. bovis AF2122/97. M. tuberculosis BTB1558 induced higher production of IL-10, IL-12, and TNF-α than M. tuberculosis H37Rv at the later time points (days 56 and 70) due to responses waning in the M. tuberculosis H37Rv group from day 42 onwards. Less than 1 U/mL of IL-6 were detected in both infection groups (data not shown).

Cytokine analysis of stimulated whole blood from M. tuberculosis H37Rv, M. tuberculosis BTB1558 or M. bovis AF2122/97 infected cattle. PPD-B stimulated-whole blood supernatants were assayed for IL-1β, IL-10, IL-12 (D), and TNF-α cytokine levels using a custom-designed bovine MSD. The response in the M. tuberculosis H37Rv infected cattle is shown as squares; M. tuberculosis BTB1558 is shown as triangles; and M. bovis AF2122/97 shown as circles; all groups contained 4 animals. IL-10 and IL-12 responses are reported as U/ml while IL-1β and TNF-α responses are reported in ng/ml as interpolated from the standard curves for each cytokine included on each plate. Data for each time point is presented as the mean response + SEM.
Gross pathology
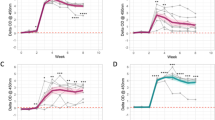
As described above, animals infected with M. bovis AF2122/97 were culled 6 weeks post-infection, while M. tuberculosis groups were culled after 10 weeks. Lungs and lymph nodes (thoracic and extrathoracic) were examined for gross lesions. Typical gross lesions were isolated granulomas or coalescing clusters of granulomas of variable size ranging from 5 to 10 mm in diameter. Gross pathology scores are summarised in Fig. 3(A–D). Statistically significant differences were observed in lung gross lesions of animals infected with M. bovis compared to animals infected with M. tuberculosis H37Rv or M. tuberculosis BTB1558; no significant differences were observed comparing the lung lesions of the two sets of M. tuberculosis infected animals. Lung gross lesions were only observed in animals infected with M. bovis AF2122/97 with scores ranging from 5 to 10 and scores of 0 (no visible lesions) in animals infected with M. tuberculosis H37Rv or BTB1558. Thoracic lymph nodes (cranial mediastinal, caudal mediastinal, right bronchial, left bronchial and cranial tracheobronchial) showed scores of between 4 and 14 in animals infected with M. bovis AF2122/97; animals infected with M. tuberculosis H37Rv had a score of 1; animals infected with M. tuberculosis BTB1558 had scores ranging between 0 and 2. Extra-thoracic lymph nodes from the head and neck region (right and left lateral retropharyngeal, right and left medial retropharyngeal, right and left parotid and right and left submandibular) showed scores of between 0 and 7 in animals infected with M. bovis AF2122/97; scores of between 0 and 2 in animals infected with M. tuberculosis BTB1558; animals infected with M. tuberculosis H37Rv did not show any gross visible lesions in extra-thoracic lymph nodes. No statistically significant differences were observed between the three groups of infected animals in the lesions observed in these nodes. No lesions were found in the tonsils in any group. All animals showed TB-like gross lesions in at least one organ.

Pathology scores across M. tuberculosis H37Rv, M. tuberculosis BTB1558 or M. bovis AF2122/97 infected cattle. Pathology scores in the thoracic (A), extra thoracic lymph nodes (LNs) (B), and lungs (C) of animals infected with M. tuberculosis H37Rv (squares); M. tuberculosis BTB1558 (triangles), or M. bovis AF2122/97 (circles). Total gross pathology score is shown in (D). Data for each time point is presented as the mean + SEM. The difference in pathology scores was statistically significant in the lungs (ρ 0.0052) and in the total pathology score (ρ 0.0105).
Culture of M. bovis and M. tuberculosis from processed samples
The presence of bacteria in the harvested tissue samples was investigated by semi-quantitative culture (Table 1). It was possible to culture the respective infecting strain from at least one organ from all experimental animals. However, the number of CFU/ml was usually low for animals infected with either of the two M. tuberculosis strains, with zero bacterial counts recorded in the lungs for all eight animals infected with either strain of M. tuberculosis. Respiratory lymph nodes were mostly affected, with all 12 animals having bacterial counts in this organ system (Table 1).
Histopathology
In H&E stained sections, four stages of granulomas were identified as previously described39,50. Briefly, Stage I (initial) granulomas comprised clusters of epithelioid macrophages, low numbers of neutrophils and occasional Langhans’ multinucleated giant cells (MNGCs). Stage II (solid) granulomas were more regular in shape and surrounded by a thin and incomplete capsule. The cellular composition was primarily epithelioid macrophages, with Langhans’ MNGCs present and some infiltration of lymphocytes and neutrophils. Necrosis was minimal or not present. Stage III (necrotic) granulomas were all fully encapsulated with central areas of necrosis. The necrotic centres were surrounded by epithelioid macrophages and Langhans’ MNGCs, and a peripheral zone of macrophages, clustered lymphocytes and isolated neutrophils extended to the fibrotic capsule. Stage IV (mineralised) granulomas were completely surrounded by a thick fibrous capsule and displayed central areas of caseous necrosis with extensive mineralization. The central necrosis was surrounded by epithelioid macrophages and Langhans’ MNGCs cells with a peripheral zone of macrophages and dense clusters of lymphocytes just inside the fibrous capsule. Granulomas were frequently multicentric, with several granulomas coalescing.
The number of granulomas of each stage in each tissue was variable (Table 2). Most of the histopathological lesions were observed in the thoracic lymph nodes with only two animals from BTB1558 and two others from the M. bovis AF2122/97 group showing granulomas in extrathoracic lymph nodes. The number of granulomas observed in tissues from animals infected with M. bovis AF2122/97 was significantly higher than the small number of granulomas observed in animals infected with either strain of M. tuberculosis. Moreover, the majority of granulomas observed in both H37Rv and BTB1558 infected animals were classed as stage I, with a few stage II granulomas, while M. bovis AF2122/97 infected animals showed granulomas of all development stages (I-IV) (Table 2). AFBs were identified in at least one ZN stained tissue section from every animal (data not shown).
Transcriptome analysis of M. bovis AF2122/97 and M. tuberculosis H37Rv infected animals
As the peripheral immune responses were broadly similar across all groups, yet pathological examination revealed significant differences, we used transcriptomic analysis of stimulated and non-stimulated whole blood samples as an unbiased tool to identifying global peripheral blood markers that would correlate with the pathological outcomes. We chose to analyse just the M. bovis AF2122/97 and M. tuberculosis H37Rv groups as they presented the extremes in terms of immune responses and pathological presentations. Blood samples cultured with medium alone (negative control) or with PPD-B from M. bovis AF2122/97 (n = 4) and M. tuberculosis H37Rv (n = 4) infection groups at days 14 and 42 were selected for analysis. Strand-specific RNA-Seq libraries (n = 32) were prepared from these blood samples and after sequencing on an Illumina HiSeq 2500, quality-control and filtering of sequencing reads yielded a mean of 14,981,780 paired-reads per individual library (2 × 100 nucleotides); these data satisfy previously defined criteria for RNA-Seq experiments with respect to sequencing depth51,52,53. Alignment of filtered paired-end reads to the B. taurus reference genome UMD3.1.1 yielded a mean of ~13.5 million read pairs (~90.5%) mapping to unique locations per library. Gene count summarisation resulted in an average of 59% of read pairs being assigned to B. taurus reference genome annotations based on strict sense strand and counting specifications. Gene filtering resulted in 17,663 sense genes (57.5% of all B. taurus reference genes in the RefSeq annotations) that were suitable for differential expression analysis. Multidimensional scaling analysis amongst the 32 sequencing libraries using the filtered and normalised gene counts (n = 17,663 genes) revealed that PPD-B stimulation was the largest discriminator that placed the samples into two distinct groups (Figure S1).
Differential gene expression in unstimulated whole blood from M. bovis AF2122/97 and M. tuberculosis H37Rv infected animals
Pairwise analysis of the transcriptome from unstimulated bovine whole blood samples at day 42 versus day 14 revealed increases in the number of differentially expressed (DE) genes at day 42 post infection with either M. bovis AF2122/97 or M. tuberculosis H37Rv (Fig. 4A), with a greater number of genes found differentially expressed in M. tuberculosis H37Rv infected animals at day 42, respectively (344 vs. 70 genes).

Transcriptome analysis of unstimulated whole blood after M. bovis AF2122/97 or M. tuberculosis H37Rv infection. (A) The number of differentially expressed genes in unstimulated whole blood for M. bovis- (blue) and M. tuberculosis- (purple) infected animals at day 42 -v- day 14 post infection (−1 > log2FC < 1, FDR < 0.05). The green Venn diagram depicts the overlap of differentially expressed genes from the direct comparison of unstimulated whole blood samples from M. bovis and M. tuberculosis infected animals at day 14 and day 42 post infection (−1 > log2FC < 1, FDR < 0.05). (B) The top 10 differentially expressed genes between M. bovis- (blue) and M. tuberculosis- (purple) infected animals at day 14- and day 42-post infection (FDR < 0.05). The graph depicts positive relative log2 fold change values where a gene that shows increased expression in M. bovis infected animals is relative to its expression in M. tuberculosis infected animals and vice versa. (C) Pathway enrichment analysis results for the list of 523 differentially expressed genes between M. bovis and M. tuberculosis infected animals at day 14-post infection. The graph depicts the enrichment of each pathway in the differentially expressed gene list based on the SIGORA successes metric (circle size) and the number of genes annotated within the pathway (“#genes in pathway”) while the colour bar depicts the significance of the association (Bonferroni < 0.05). (D) The relative expression (transcripts per million, TPM, “log(TPM +1)”) and the relative change in expression (log2 fold change (“Log2FC”)) of the genes belonging to the Antigen activation of B cell Receptor (R-HSA-983695), Collagen degradation (R-HSA-1442490) and Extracellular matrix (“ECM”) organization (R-HSA-1474244) pathways enriched for the comparison of M. bovis and M. tuberculosis infected animals at day 14 post infection. Genes that pass multiple hypothesis testing are denoted with an asterisk (Benjamini-Hochberg, FDR < 0.05) (*).
Direct comparison of unstimulated whole blood samples between M. bovis AF2122/97 and M. tuberculosis H37Rv infected animals at day 14 and again at day 42 revealed 523 and 76 DE gene respectively, with 27 of these identified as DE at both time-points (Fig. 4A, green) between the two infection models. Amongst the top 10 DE genes upregulated in M. bovis AF2122/97 infected animals (or conversely, downregulated in M. tuberculosis H37Rv animals) included those encoding: the macrophage restricted cell surface receptor SIGLEC1 that is known to be involved in pathogen uptake, antigen presentation and lymphocyte proliferation54; the CD4-coreceptor and fractalkine receptor CX3CR1, which has been linked to tuberculosis susceptibility and the impairment of macrophage and dendritic cell migration55,56; and Interferon-Regulated Resistance GTP-Binding Protein (MX1) at day 14 (Fig. 4B). At day 42 the expression of the following genes was at a relatively higher level in M. bovis AF2122/97 infected animals than in M. tuberculosis H37Rv infected animals (Fig. 4B): CXCL9, a previously described potential TB biomarker50,57; mediator of mycobacterial-induced cytokine production in macrophages CD18058; and the chemokine CXCL11.
T-cell chemotactic factor CCL17 had the highest log-fold change in M. tuberculosis H37Rv infected samples in comparison to M. bovis AF2122/97 infections at day 14. Genes also expressed to a higher level in M. tuberculosis infected animals at day 14 were: the defensin DEFB10; platelet aggregation inducing factor PDPN, which is expressed on macrophages and epithelioid cells within the tuberculous granuloma59; macrophage lipid export complex member ABCG1; and IL6 (in contrast with the ELISA data from the same time point, Fig. 2). At day 42 post infection the fractalkine receptor CX3CR1, a marker of Th1 stage differentiation during tuberculosis60, and the V-ATPase subunit gene ATP6V0D2 were higher in M. tuberculosis H37Rv infected samples (Fig. 4B).
Pathway enrichment analysis revealed pathways such as Extracellular matrix organization (R-HSA-1474244), Collagen degradation (R-HSA-1442490), Interferon alpha/beta signaling (R-HSA-909733) and B cell receptor second messenger signaling (R-HSA-983695) being significantly associated with the 523 DE genes between M. tuberculosis and M. bovis AF2122/97 infected animals at day 14 (Bonferroni < 0.005) (Fig. 4C,D). Further investigation revealed higher expression of 25/44 genes belonging to the antigen activation of B cell receptor signaling pathway (R-HSA-983695) in M. bovis AF2122/97 infected animals at day 14, such as membrane associated CD19, CD80, TREM2 and second messengers LYN, SYK, BTK, BLNK, PLCG2 along with B-cell receptor encoding genes CD79a and CD79b (Fig. 4D). The enrichment of collagen degradation and extracellular matrix organization pathways in the 523 DE gene list highlighted genes of the matrix metallopeptidase (MMP) family such as MMP1, MMP3, MMP12 and MMP14 (Fig. 4C). MMP proteins have been linked to leukocyte migration and the progression of granuloma formation during tuberculosis; MMP1 and MMP14 gene products are key for the destruction of collagen and alveolar destruction with increased expression of MMP14 found in the sputum of tuberculosis patients61,62. Gene expression of the matrix-associated cytokine SPP1 (i.e. Osteopontin/OPN) was upregulated in M. bovis infected animals at day 14. SPP1 enhances IFN-γ and IL-12 production, and increased levels of SPP1 have been reported in the blood of TB patients versus controls63.
Pathway enrichment analysis with the 76 common DE genes at day 42-post infection revealed no significantly associated pathways.
Differential gene expression in PPD-B-stimulated whole blood from M. bovis AF2122/97 and M. tuberculosis H37Rv infected animals
To assess antigen-stimulated alteration in the whole blood transcriptome, blood samples were stimulated with PPD-B overnight and subsequently compared to unstimulated blood samples. PPD-B stimulation resulted in the differential expression of 2,622 and 1,586 genes at day 14 and 1,446 and 2,107 at day 42 in M. bovis AF2122/97 and M. tuberculosis H37Rv infected animals respectively in comparison to control samples (Fig. 5A).

Comparative transcriptome analysis of unstimulated vs. PPD-B stimulated whole blood from M. bovis AF2122/97 or M. tuberculosis H37Rv infected cattle. (A) The overlap of differentially expressed genes from the comparison of unstimulated whole blood samples and PPD-B stimulated whole blood samples for M. bovis AF2122/97 infected animals at day 14 and day 42 (blue) and M. tuberculosis H37Rv infected animals at day 14 and day 42 (purple) (−1 > log2FC < 1, FDR < 0.05). (B) The relative expression of Interferon stimulated genes (R-HSA-877300) from the comparison of unstimulated whole blood samples and PPD-B stimulated whole blood samples for M. bovis AF2122/97 infected animals at day 14 and day 42 and M. tuberculosis H37Rv infected animals at day 14 and day 42. Genes that pass multiple hypothesis testing are denoted with an asterisk (Benjamini-Hochberg, FDR < 0.05) (*). (C) Pathway enrichment analysis results for 658 genes that are significantly differentially expressed (−1 > log2FC < 1, FDR < 0.05) in PPD-B stimulated whole blood samples from both M. bovis AF2122/97- and M. tuberculosis H37Rv- infected animals at both 14 and 42 days post infection in comparison to unstimulated whole blood samples. The graph depicts the association of each pathway with the differentially expressed gene list based on the SIGORA successes metric (circle size) and the number of genes annotated within the pathway (“#genes in pathway”) while the colour bar depicts the significance of the association (Bonferroni < 0.05). (D) The relative expression (transcripts per million, TPM, “log(TPM +1)”) and the relative change in expression (log2 fold change (“Log2FC”)) of 658 genes that are significantly differentially expressed (−1 > log2FC < 1, FDR < 0.05) in PPD-B stimulated whole blood samples from both M. bovis AF2122/97- and M. tuberculosis H37Rv- infected animals at both 14 and 42 days post infection in comparison to unstimulated whole blood samples. The genes are associated with pathways related to TNF signalling (R-HSA-5668541, R-HSA-5676594, R-HSA-5357786, R-HSA-5669034), Antigen processing-Cross presentation (R-HSA-1236975) and Chemokines and complement activation (R-HSA-380108, R-HSA-173736). Genes that pass multiple hypothesis testing are denoted with an asterisk (Benjamini-Hochberg, FDR < 0.05) (*).
Firstly, a “core” response to PPD-B stimulation amounting to 658 genes was identified, representing genes that were consistently DE regardless of infectious agent or time post infection (Fig. 5A). As expected, IFNG was strongly upregulated in stimulated blood samples at both time points in both M. bovis AF2122/97 and M. tuberculosis H37Rv infected animals (Fig. 5B). Furthermore, 47/152 genes from the Interferon gamma signaling pathway (R-HSA-877300) were significantly differentially expressed (−0.75 < log2FC > 0.75) across the comparative groups and time points (Fig. 5B). Pathway enrichment analysis of the 658 core response genes revealed significantly associated pathways such as those involved in TNF-signaling (R-HSA-5668541, R-HSA-5676594, R-HSA-5357786, R-HSA-5669034), Chemokine receptors bind chemokines (R-HSA-380108), Antigen processing-Cross presentation (R-HSA-1236975) and Initial triggering of complement (R-HSA-173736) (Fig. 5C). The change in expression of the genes within these pathways can be seen in Fig. 5D; an overall downwards trend in genes encoding complement related factors was found (e.g. C1Q1, C1QA, CFD and CFP) along with strong upregulation of TNF-signaling related factors (e.g. TNF, NFKB2, TNFAIP3 and lymphotoxins alpha and beta LTA/LTB) upon PPD-B stimulation of whole blood samples (Fig. 5D).
Second, there are 159 and 179 DE genes in either M. bovis AF2122/97 or M. tuberculosis H37Rv infected animals at both time points (Fig. 5A). These 338 genes represent a divergence in response to PPD-B stimulation between the two infection groups and the top ranking genes amongst them are presented in Figure S2 (log2FC ratio). The increased expression of CCL17, DEFB10 and matrix metalloproteinase MMP12 with the decreased expression of bactericidal permeability increasing protein BPI, CD164 and IL5 receptor IL5RA can differentiate M. bovis AF2122/97 infected animals from M. tuberculosis H37Rv infected animals at both day 14 and day 42 post infection upon PPD-B stimulation. Conversely, the increased expression of interferon inducible dyamin MX2 at day 14 and cytokine receptors IL22RA2 and XCR1 at day 42 post infection, and decreased expression of T-cell regulator TNFSF18 at day 14- and TLR5, defensin DEFB5 and V-ATPase subunit gene ATP6V0D2 at day 42- post infection, distinguished M. tuberculosis H37Rv infected from M. bovis AF2122/97 infected animals upon PPD-B stimulation.
miR-155 analysis
miR-155 has been identified as a potential biomarker of disease development and/or severity in cattle infected with M. bovis49. To explore its utility in this study, we analysed the abundance of miR-155 in PPD-B stimulated vs. unstimulated whole blood from M. bovis AF2122/97 and M. tuberculosis H37Rv infected cattle over the infection time course using RT-qPCR (Figure S3). The animals infected with M. tuberculosis H37Rv showed a steady increase in expression of miR-155 after PPD-B stimulation over the course of infection, while M. bovis infected animals showed a greater baseline expression prior to infection and hence a more modest increase over the time course. While a single miRNA will lack specificity as a biomarker, these results support the potential of miR-155 as a part of a biomarker panel to assess infection with tubercle bacilli.
Discussion
This work set out to explore whether a recent isolate of M. tuberculosis, recovered from a TB lesion identified in an Ethiopian bull, would trigger a similar immunological and pathological response to the M. tuberculosis H37Rv reference strain when used to experimentally infect cattle. This work sought to build on our previous study that had shown M. tuberculosis H37Rv to be attenuated in the bovine host by using a recent clinical isolate to address issues around possible laboratory-adaptation of the H37Rv strain that may have reduced its virulence in the bovine model. Furthermore we wished to see whether the application of transcriptomics would reveal potential biomarkers to differentiate between the initial stages of a progressive, active M. bovis infection and a more quiescent, latent infection with M. tuberculosis.
Our results showed that both M. tuberculosis BTB1558 and M. tuberculosis H37Rv were considerably attenuated in the bovine host when compared to M. bovis AF2122/97. This is despite the fact that animals infected with either M. tuberculosis strain were left to progress for 10 weeks, while the M. bovis infected animals were culled 6 weeks after infection. M. bovis induced a greater level of pathology than either of the M. tuberculosis strains. Although M. tuberculosis BTB1558 appeared to induce a slightly greater level of pathology in head lymph nodes than M. tuberculosis H37Rv, this difference was not significant; no difference was detected in the level of pathology induced by either of the M. tuberculosis strains in respiratory lymph nodes or lungs. The apparent arrest of M. tuberculosis granulomas in stages I and II, compared to infection with M. bovis that produced lesions from stages I-IV is another striking difference in infection outcome. The bacteriological culture results showed that both strains of M. tuberculosis persisted in cattle, at least over the 10 weeks of the infection time course. Interestingly, Bezos and colleagues also reported reduced pathological lesion scores for goats infected with M. tuberculosis as compared to infections with M. bovis or M. caprae64.
The kinetics and magnitude of bovine peripheral blood immune responses across all three infection groups were broadly similar over the initial 6 week phase of infection. In previous work13 we had seen that while stimulation of whole blood with the antigen Rv3879c triggered IFN-γ production in M. bovis-infected cattle, M. tuberculosis H37Rv-infected cattle showed no responses. In this current work Rv3879c stimulation of whole blood provided less definitive outcomes, as while Rv3879c triggered minimal IFN-γ responses in blood from M. tuberculosis H37Rv or BTB1558 infected animals, the baseline responses to Rv3879c stimulation in M. bovis AF2122/97 infected cattle were high prior to infection, and showed no increase in response over the course of infection (data not shown). As analysis of antigen-induced peripheral blood cytokine responses failed to reflect the distinct pathological presentations between the M. bovis AF2122/97 and M. tuberculosis groups, we applied RNA-Seq transcriptomics of whole blood in an attempt to identify biomarkers that would better distinguish the groups, focusing on the M. bovis AF2122/97 and M. tuberculosis H37Rv groups. This analysis revealed discrete genes and pathways that distinguished the groups and were indicative of accelerated disease development in the M. bovis AF2122/97 infected animals, and included previously described biomarkers of disease progression or susceptibility such as MMPs, CX3CR1, CXCL9 and SSP1/OPN50,57,60,62,63. These changes in peripheral gene expression over the course of infection provide biomarker candidates of disease progression for validation in new studies.
Classic experiments from the late 19th century by Smith, Koch and von Behring showed that bovine and human tubercle bacilli showed distinct virulence in animal models, and in particular that the human bacillus was attenuated when used to infect cattle. Our work has recapitulated these findings, using both the standard M. tuberculosis H37Rv strain as well as an M. tuberculosis isolate (BTB1558) recovered from a bovine lesion. The attenuation shown by the M. tuberculosis BTB1558 strain in our experimental model compared to that seen in the field situation in Ethiopia appears likely due to increased susceptibility of the original affected animal. It should be noted however that from a human and animal health perspective, it is possible that cattle infected with M. tuberculosis may still be transmissible and shed bacilli (e.g. through nasal secretions, aerosol from the lungs or through milk) albeit at low levels. Such scenarios are more probable in countries without statutory control of bovine TB, where the case rates of active human TB are high, interaction between humans and cattle are frequent, and consumption of raw milk common.
While the outcome of infection with M. tuberculosis in humans is more a spectrum than a bipolar, active vs. latent, state65,66, infection with M. tuberculosis in cattle could be indicative of a latent infection being established, as compared to an active disease status upon M. bovis infection. The bovine infection model may therefore offer a tractable experimental system in which to explore the reactivation of M. tuberculosis infection and to define prognostic biomarkers of the development of active disease. Furthermore, infection of cattle with M. tuberculosis to establish latent infection may provide a tractable outbred model in which to explore post-exposure vaccination strategies.
In conclusion, our work has shown that M. tuberculosis isolates, whether the H37Rv type strain or a recent isolate, are attenuated for virulence in a bovine infection model as compared to M. bovis. This work contributes further evidence as to the distinct host preference of tubercle bacilli, provides a basis to disect molecular mechanisms of virulence in M. bovis as compared to M. tuberculosis, and offers a model in which to explore latent TB infection.
References
WHO. http://www.who.int/tb/publications/global_report/en/ (2017).
Karlson, A. G. & Lessel, E. F. Mycobacterium bovis nom. nov. Int J Syst Bacteriol 20, 273–282 (1970).
Wells, A. G. The murine type of tubercle bacillus (The vole acid-fast bacillus). MRC Spec. Rep. Ser. med. Res. Coun., Lond (1946).
Cousins, D. V., Peet, R. L., Gaynor, W. T., Williams, S. N. & Gow, B. L. Tuberculosis in imported hyrax (Procavia capensis) caused by an unusual variant belonging to the Mycobacterium tuberculosis complex. Veterinary microbiology 42, 135–145 (1994).
Smith, N. The ‘Dassie’ bacillus. Tubercle 41, 203–212 (1960).
Aranaz, A. et al. Mycobacterium tuberculosis subsp. caprae subsp. nov.: a taxonomic study of a new member of the Mycobacterium tuberculosis complex isolated from goats in Spain. Int J Syst Bacteriol 49(Pt 3), 1263–1273 (1999).
Cousins, D. V. et al. Tuberculosis in seals caused by a novel member of the Mycobacterium tuberculosis complex: Mycobacterium pinnipedii sp. nov. International journal of systematic and evolutionary microbiology 53, 1305–1314 (2003).
van Ingen, J. et al. Characterization of Mycobacterium orygis as M. tuberculosis complex subspecies. Emerging infectious diseases 18, 653–655, https://doi.org/10.3201/eid1804.110888 (2012).
Alexander, K. A. et al. Novel Mycobacterium tuberculosis complex pathogen, M. mungi. Emerging infectious diseases 16, 1296–1299, https://doi.org/10.3201/eid1608.100314 (2010).
Francis, J. Control of infection with the bovine tubercle bacillus. Lancet 1, 34–39 (1950).
Magnus, K. Epidemiological Basis of Tuberculosis Eradication 3. Risk of Pulmonary Tuberculosis after Human and Bovine Infection. Bull World Health Organ. 35, 483–508 (1966).
Koch, R. An Address on the Fight against Tuberculosis in the Light of the Experience that has been Gained in the Successful Combat of other Infectious Diseases. Br Med J 2, 189–193 (1901).
Whelan, A. O. et al. Revisiting Host Preference in the Mycobacterium tuberculosis Complex: Experimental Infection Shows M. tuberculosis H37Rv to Be Avirulent in Cattle. Plos One 5, https://doi.org/10.1371/journal.pone.0008527 (2010).
von Berhing, E. In Nobel Lectures, Physiology or Medicine 1901–1921 (Elsevier Publishing Company, 1901).
Cole, S. T. et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544, https://doi.org/10.1038/31159 (1998).
Garnier, T. et al. The complete genome sequence of Mycobacterium bovis. Proc Natl Acad Sci USA 100, 7877–7882, https://doi.org/10.1073/pnas.1130426100 (2003).
Malone, K. M. et al. Updated Reference Genome Sequence and Annotation of Mycobacterium bovis AF2122/97. Genome Announc 5, https://doi.org/10.1128/genomeA.00157-17 (2017).
Groschel, M. I., Sayes, F., Simeone, R., Majlessi, L. & Brosch, R. ESX secretion systems: mycobacterial evolution to counter host immunity. Nat Rev Microbiol 14, 677–691, https://doi.org/10.1038/nrmicro.2016.131 (2016).
Brodin, P. et al. Bacterial artificial chromosome-based comparative genomic analysis identifies Mycobacterium microti as a natural ESAT-6 deletion mutant. Infect Immun 70, 5568–5578 (2002).
Mostowy, S., Cousins, D. & Behr, M. A. Genomic interrogation of the dassie bacillus reveals it as a unique RD1 mutant within the Mycobacterium tuberculosis complex. J Bacteriol 186, 104–109 (2004).
Gonzalo-Asensio, J. et al. Evolutionary history of tuberculosis shaped by conserved mutations in the PhoPR virulence regulator. Proc Natl Acad Sci USA 111, 11491–11496, https://doi.org/10.1073/pnas.1406693111 (2014).
Behr, M. A. & Gordon, S. V. Why doesn't Mycobacterium tuberculosis spread in animals? Trends Microbiol 23, 1–2, https://doi.org/10.1016/j.tim.2014.11.001 (2015).
Steenken, W., Oatway, W. H. & Petroff, S. A. Biological Studies of the Tubercle Bacillus: Iii. Dissociation and Pathogenicity of the R and S Variants of the Human Tubercle Bacillus (H(37). J Exp Med 60, 515–540 (1934).
Ioerger, T. R. et al. Variation among genome sequences of H37Rv strains of Mycobacterium tuberculosis from multiple laboratories. J Bacteriol 192, 3645–3653, https://doi.org/10.1128/JB.00166-10 (2010).
Cadmus, S. et al. Molecular analysis of human and bovine tubercle bacilli from a local setting in Nigeria. J Clin Microbiol 44, 29–34, https://doi.org/10.1128/JCM.44.1.29-34.2006 (2006).
Chen, Y. et al. Potential challenges to the Stop TB Plan for humans in China; cattle maintain M. bovis and M. tuberculosis. Tuberculosis (Edinb) 89, 95–100, https://doi.org/10.1016/j.tube.2008.07.003 (2009).
Ocepek, M., Pate, M., Zolnir-Dovc, M. & Poljak, M. Transmission of Mycobacterium tuberculosis from human to cattle. J Clin Microbiol 43, 3555–3557, https://doi.org/10.1128/JCM.43.7.3555-3557.2005 (2005).
Ameni, G. et al. Mycobacterium tuberculosis infection in grazing cattle in central Ethiopia. Vet J 188, 359–361, https://doi.org/10.1016/j.tvjl.2010.05.005 (2011).
Berg, S. et al. The burden of mycobacterial disease in ethiopian cattle: implications for public health. PLoS One 4, e5068, https://doi.org/10.1371/journal.pone.0005068 (2009).
Firdessa, R. et al. Mycobacterial lineages causing pulmonary and extrapulmonary tuberculosis, Ethiopia. Emerging infectious diseases 19, 460–463, https://doi.org/10.3201/eid1903.120256 (2013).
Comas, I., Homolka, S., Niemann, S. & Gagneux, S. Genotyping of genetically monomorphic bacteria: DNA sequencing in Mycobacterium tuberculosis highlights the limitations of current methodologies. PLoS One 4, e7815, https://doi.org/10.1371/journal.pone.0007815 (2009).
Stucki, D. et al. Mycobacterium tuberculosis lineage 4 comprises globally distributed and geographically restricted sublineages. Nat Genet 48, 1535–1543, https://doi.org/10.1038/ng.3704 (2016).
Gallagher, J. & Horwill, D. M. A selective oleic acid albumin agar medium for the cultivation of Mycobacterium bovis. J Hyg (Lond) 79, 155–160 (1977).
Gordon, S. V. et al. Identification of variable regions in the genomes of tubercle bacilli using bacterial artificial chromosome arrays. Mol Microbiol 32, 643–655 (1999).
Kamerbeek, J. et al. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J Clin Microbiol 35, 907–914 (1997).
Whelan, A. et al. Immunogenicity comparison of the intradermal or endobronchial boosting of BCG vaccinates with Ad5-85A. Vaccine 30, 6294–6300, https://doi.org/10.1016/j.vaccine.2012.07.086 (2012).
Vordermeier, H. M. et al. Correlation of ESAT-6-specific gamma interferon production with pathology in cattle following Mycobacterium bovis BCG vaccination against experimental bovine tuberculosis. Infect Immun 70, 3026–3032 (2002).
Vordermeier, H. M. et al. Viral booster vaccines improve Mycobacterium bovis BCG-induced protection against bovine tuberculosis. Infect Immun 77, 3364–3373, https://doi.org/10.1128/IAI.00287-09 (2009).
Wangoo, A. et al. Advanced granulomatous lesions in Mycobacterium bovis-infected cattle are associated with increased expression of type I procollagen, gammadelta (WC1+) T cells and CD 68+ cells. J Comp Pathol 133, 223–234, https://doi.org/10.1016/j.jcpa.2005.05.001 (2005).
Martin, M. Cutadapt Removes Adapter Sequences From High-Throughput Sequencing Reads. EMBnet.journal http://journal.embnet.org/index.php/embnetjournal/article/view/200%3E (2011).
Bushnell, B. BBMap short read aligner, and other bioinformatic tools, sourceforge.net/projects/bbmap/ (2017).
Andrew, S. FastQC, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2017).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, https://doi.org/10.1093/bioinformatics/bts635 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930, https://doi.org/10.1093/bioinformatics/btt656 (2014).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140, https://doi.org/10.1093/bioinformatics/btp616 (2010).
Foroushani, A. B., Brinkman, F. S. & Lynn, D. J. Pathway-GPS and SIGORA: identifying relevant pathways based on the over-representation of their gene-pair signatures. PeerJ 1, e229, https://doi.org/10.7717/peerj.229 (2013).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis. (Springer-Verlag, 2009).
Chen, H. VennDiagram: Generate High-Resolution Venn and Euler Plots, https://cran.r-project.org/web/packages/VennDiagram/index.html (2016).
Golby, P., Villarreal-Ramos, B., Dean, G., Jones, G. J. & Vordermeier, M. MicroRNA expression profiling of PPD-B stimulated PBMC from M. bovis-challenged unvaccinated and BCG vaccinated cattle. Vaccine 32, 5839–5844, https://doi.org/10.1016/j.vaccine.2014.07.034 (2014).
Aranday-Cortes, E. et al. Upregulation of IL-17A, CXCL9 and CXCL10 in early-stage granulomas induced by Mycobacterium bovis in cattle. Transbound Emerg Dis 60, 525–537, https://doi.org/10.1111/j.1865-1682.2012.01370.x (2013).
Castel, S. E., Levy-Moonshine, A., Mohammadi, P., Banks, E. & Lappalainen, T. Tools and best practices for data processing in allelic expression analysis. Genome Biol 16, 195, https://doi.org/10.1186/s13059-015-0762-6 (2015).
Tarazona, S., Garcia-Alcalde, F., Dopazo, J., Ferrer, A. & Conesa, A. Differential expression in RNA-seq: a matter of depth. Genome Res 21, 2213–2223, https://doi.org/10.1101/gr.124321.111 (2011).
Wang, Y. et al. Evaluation of the coverage and depth of transcriptome by RNA-Seq in chickens. BMC Bioinformatics 12(Suppl 10), S5, https://doi.org/10.1186/1471-2105-12-S10-S5 (2011).
O'Neill, A. S., van den Berg, T. K. & Mullen, G. E. Sialoadhesin - a macrophage-restricted marker of immunoregulation and inflammation. Immunology 138, 198–207, https://doi.org/10.1111/imm.12042 (2013).
Hall, J. D. et al. The impact of chemokine receptor CX3CR1 deficiency during respiratory infections with Mycobacterium tuberculosis or Francisella tularensis. Clin Exp Immunol 156, 278–284, https://doi.org/10.1111/j.1365-2249.2009.03882.x (2009).
Sakai, S. et al. Cutting edge: control of Mycobacterium tuberculosis infection by a subset of lung parenchyma-homing CD4 T cells. J Immunol 192, 2965–2969, https://doi.org/10.4049/jimmunol.1400019 (2014).
Aranday-Cortes, E. et al. Transcriptional profiling of disease-induced host responses in bovine tuberculosis and the identification of potential diagnostic biomarkers. PLoS One 7, e30626, https://doi.org/10.1371/journal.pone.0030626 (2012).
Yu, C. H. et al. RP105 Engages Phosphatidylinositol 3-Kinase p110delta To Facilitate the Trafficking and Secretion of Cytokines in Macrophages during Mycobacterial Infection. J Immunol 195, 3890–3900, https://doi.org/10.4049/jimmunol.1500017 (2015).
Feng, Y. et al. Platelets direct monocyte differentiation into epithelioid-like multinucleated giant foam cells with suppressive capacity upon mycobacterial stimulation. J Infect Dis 210, 1700–1710, https://doi.org/10.1093/infdis/jiu355 (2014).
Sallin, M. A. et al. Th1 Differentiation Drives the Accumulation of Intravascular, Non-protective CD4 T Cells during Tuberculosis. Cell Rep 18, 3091–3104, https://doi.org/10.1016/j.celrep.2017.03.007 (2017).
Salgame, P. MMPs in tuberculosis: granuloma creators and tissue destroyers. J Clin Invest 121, 1686–1688, https://doi.org/10.1172/JCI57423 (2011).
Sathyamoorthy, T. et al. Membrane Type 1 Matrix Metalloproteinase Regulates Monocyte Migration and Collagen Destruction in Tuberculosis. J Immunol 195, 882–891, https://doi.org/10.4049/jimmunol.1403110 (2015).
Koguchi, Y. et al. High plasma osteopontin level and its relationship with interleukin-12-mediated type 1 T helper cell response in tuberculosis. Am J Respir Crit Care Med 167, 1355–1359, https://doi.org/10.1164/rccm.200209-1113OC (2003).
Bezos, J. et al. Goats challenged with different members of the Mycobacterium tuberculosis complex display different clinical pictures. Vet Immunol Immunopathol 167, 185–189, https://doi.org/10.1016/j.vetimm.2015.07.009 (2015).
Barry, C. E. III et al. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat Rev Microbiol 7, 845–855, https://doi.org/10.1038/nrmicro2236 (2009).
Esmail, H. et al. Characterization of progressive HIV-associated tuberculosis using 2-deoxy-2-[18F]fluoro-D-glucose positron emission and computed tomography. Nat Med 22, 1090–1093, https://doi.org/10.1038/nm.4161 (2016).
Acknowledgements
The authors are grateful to the members of the scientific and animal staff of the Plate-Forme d’Infectiologie Expérimentale, PFIE, INRA, 37380, Nouzilly, France, especially to the study managers Céline Barc and Philippe Bernardet and animal technicians Olivier Boulesteix and Joël Moreau. We gratefully acknowledge funding from: the European Commission’s Seventh Framework Programme (FP7, 2007–2013) Research Infrastructures Action under the grant agreement No. FP7-228394 (NADIR project); EC H2020 program grant number 643381 (TBVAC2020); Wellcome Trust grant number 075833/A/04/Z under their “Animal health in the developing world” initiative; Science Foundation Ireland Investigator Award 08/IN.1/B2038; Wellcome Trust PhD awards 097429/Z/11/Z (K.R-A.) and 102395/Z/13/Z (A.S.).
Author information
Authors and Affiliations
Contributions
B.V.R., S.B., P.S., N.W., M.V., S.V.G. conceived the project; B.V.R., S.B., A.O.W., S.H., F.C., F.J.S., B.K., R.S., A.S., P.S. performed experiments; B.V.R., A.O.W., S.H., F.C., F.J.S., R.S., A.S., K.M., K.R.A., P.S., N.W., M.V., S.V.G. analysed data; G.A., A.A. provided new regents; B.V.R., S.B., S.H., F.J.S., K.M., M.V., S.V.G. wrote the paper, and all authors agreed to the final version for submission.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Villarreal-Ramos, B., Berg, S., Whelan, A. et al. Experimental infection of cattle with Mycobacterium tuberculosis isolates shows the attenuation of the human tubercle bacillus for cattle. Sci Rep 8, 894 (2018). https://doi.org/10.1038/s41598-017-18575-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-18575-5
This article is cited by
-
Genomic insights into anthropozoonotic tuberculosis in captive sun bears (Helarctos malayanus) and an Asiatic black bear (Ursus thibetanus) in Cambodia
Scientific Reports (2024)
-
Exploring virulence in Mycobacterium bovis: clues from comparative genomics and perspectives for the future
Irish Veterinary Journal (2023)
-
Isolation and comparative genomics of Mycobacterium tuberculosis isolates from cattle and their attendants in South India
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
