
Abstract
Water creates special problems for prebiotic chemistry, as it is thermodynamically favorable for amide and phosphodiester bonds to hydrolyze. The availability of alternative solvents with more favorable properties for the formation of prebiotic molecules on the early Earth may have helped bypass this so-called “water paradox”. Formamide (FA) is one such solvent, and can serve as a nucleobase precursor, but it is difficult to envision how FA could have been generated in large quantities or accumulated in terrestrial surface environments. We report here the conversion of aqueous acetonitrile (ACN) via hydrogen cyanide (HCN) as an intermediate into FA by γ-irradiation under conditions mimicking exposure to radioactive minerals. We estimate that a radioactive placer deposit could produce 0.1‒0.8 mol FA km−2 year−1. A uraninite fission zone comparable to the Oklo reactors in Gabon can produce 0.1‒1 mol m−2 year−1, orders of magnitude greater than other scenarios of FA production or delivery for which reaching sizeable concentrations of FA are problematic. Radioactive mineral deposits may be favorable settings for prebiotic compound formation through emergent geologic processes and FA-mediated organic chemistry.
Similar content being viewed by others


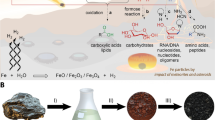
Introduction
Abiotically produced nucleic and amino acid polymers are widely viewed as key chemical intermediates that link a lifeless Earth with the universal ancestor of all known life1. Laboratory studies, however, have revealed several problems with the abiotic synthesis of these polymers prior to life’s emergence. Specifically, though the prebiotic synthesis of RNA monomers has recently been reported from relatively simple molecules2,3,4,5, only a few geochemical scenarios that can facilitate these synthesis reactions have been experimentally investigated6,7. A frequent criticism of many model prebiotic reactions is that it is unlikely the needed precursor ingredients would ever spontaneously reach the high concentrations typically employed, giving rise to a puzzle known as the Concentration Problem8. But perhaps the most deleterious barrier to the emergence of life arises from the most critical ingredient of life itself ‒ water9. The average half-life for hydrolysis of a peptide bond is on the order of 102 years at pH 7 and 25 °C10, and the half-life of an RNA phosphodiester bond at 30 °C is estimated to be less than one year under the same conditions11. Water therefore hydrolyzes biopolymers such as nucleic acids and proteins over relatively short time-scales, but is also a necessary solvent medium for all known life. This fundamental mismatch between initial and final chemical constraints on life’s origins leads to what is known as the Water Paradox12. The Concentration Problem and the Water Paradox present formidable conceptual problems for any origins of life theory, including “RNA World” scenarios that necessitate abundant, abiotic production of polynucleotides2.
Formamide (HCONH2, FA), which is a liquid under normal terrestrial surface temperature and pressure conditions, has been advanced as an alternative solvent to water that could enable chemical complexification and a means of bypassing the Water Paradox13,14,15. FA is a polar solvent that has the advantageous properties of promoting dehydration condensation reactions16,17, solubilizing phosphate minerals18, and serving as a feedstock for several biologically relevant compounds including nucleobases19,20,21, amino acids and carboxylic acids when heated and in contact with a variety of mineral catalysts5,22,23,24,25. FA can be produced via the hydration of hydrogen cyanide (HCN) or the decomposition of ammonium formate, but the competing reaction of FA hydrolysis to formic acid constrains maximum possible aqueous concentrations to approximately 10−5 M26,27,28. These dilute amounts are insufficient to validate the use of neat concentrations of FA employed in some model prebiotic syntheses, or to serve as a reservoir for abundant nucleobase production29,30,31,32. Other direct sources of FA that bypass bodies of water such as cometary33 or meteoritic34 delivery have been invoked in conjunction with ‘desert-like’ periodically dry settings or hydrothermally-driven thermophoresis as a means of concentrating FA9,27,29,30,35, but such scenarios necessitate ideal and perhaps implausible circumstances to simultaneously produce and then concentrate FA to any appreciable reservoir size36.
A localized geochemical source of FA via radiolysis of aqueous nitriles such as HCN or acetonitrile (ACN) may reduce problems associated with reaching high concentrations of FA while simultaneously mitigating the Water Paradox. Radiolysis of water produces hydroxyl radicals that increase rates of hydration and hydrolysis above those associated with thermal activation alone37. Radioactive minerals such as monazite, uraninite, and zircon are concentrated by hydrodynamic sorting in beach and river settings called placer deposits. Placers are widely distributed but heterogeneously concentrated across the Earth’s surface38,39,40 (Fig. 1a,b). The energy from radioactive minerals create fluxes of α, β and γ particles that are orders of magnitude above background levels near the surfaces of individual mineral grains (Fig. 1c). Under exceptional circumstances, uranium-rich strata formed in the Earth’s deep past created self-sustaining neutron chain reactions with radiolytic and heat power outputs that greatly exceed those of typical placer deposits41,42,43,44, a unique energy setting with prebiotic implications explored in detail by Draganic et al.45,46 and most recently expanded upon by Maruyama and Ebisuzaki47. Here, we investigate the hypothesis that γ radiolysis of atmospherically-derived aqueous nitriles by radioactive minerals could have increased production rates of FA and generated sizeable organic solvent reservoirs on the prebiotic Earth.

Factors causing heterogeneous distribution of energy from radioactive placer minerals. (a) Global distribution of major modern heavy mineral placer deposits. (b) Concentration of heavy mineral grains (dark layers) within a typical beach placer deposit; image courtesy C. Bern, United States Geological Survey. (c) Spatial distribution of α, β and γ particles around a single radioactive mineral grain.
Results
The irradiation of ACN was initially investigated as a 30 volume % (~5.7 M) aqueous solution, which was exposed to an 800 kGy dose at 3kGy h−1. GC-MS was used to analyze the resulting mixture. FA was observed along with acetaldehyde, acetamide, acetic acid and succinonitrile as major products (Fig. 2). Hydrogen cyanide (HCN) was detected by GC-MS analysis of the headspace of the reaction vial. Using a calibration curve determined from known concentrations of an authentic standard, a concentration of 6.6 mM FA was measured (Fig. 3a, SI-1). The decrease in concentration of ACN due to conversion to other products was not detectable within experimental error, consistent with the relatively low yields of organic products detected. We then examined the effects of dose rate, total dose and initial concentration of ACN in order to constrain the production of FA under varying conditions. The measured concentrations of FA using 30% ACN aqueous solutions rapidly increase between 0 and 400 kGy, then increase in an approximately linear manner with total dose without significant variation as a function of dose rate (green lines, Fig. 3a). The same general trend was observed for acetic acid, acetamide and succinonitrile. From these data, we calculated a radiolytic yield (G) for FA formation from ACN of 0.012 molecules/100 eV. Although the radiolytic yield for FA decreases with lower starting concentrations of ACN, normalizing the G-values to initial ACN molarity reveals that the production of FA becomes more efficient at lower initial ACN concentrations (Fig. 3b). Overall, the conversion of aqueous ACN to FA is robust to varying dose rate (measured from 0.5‒2.92 kGy h−1), total dose (over the range of 72 to 3200 kGy) and initial concentration of ACN (0.5‒50 volume %). At 3200 kGy total dose, additional compounds including N-methyl-acetamide, N-methyl-formamide and pyrimidine-like compounds were produced at levels that could not be detected at lower total doses (Table SI-1). Generation of FA seems robust to oxidizing conditions; replacing N2 with a standard atmosphere or adding oxygen-rich nitrate had only a minor effect on FA production.

GC-MS chromatogram of the most abundant identified products (annotated peaks) resulting from gamma irradiation of mixtures of 30% aqueous ACN. Indicated total doses were delivered at a dose rate of ~3 kGy hr−1.

FA production as a function of total dose and yield from ACN. (a) Concentration of compounds at 2.92, 1.0 and 0.5 kgry hr−1 γ-dose rates versus total dose. (b) FA yield (triangles, mM) and molar conversion efficiency (squares, dimensionless) versus initial acetonitrile concentration for 800 kGy total dose at ~3kGy hr−1 dose rate.
Given the observations described above, we infer a pathway of FA production that proceeds though HCN as an intermediate. The radiolytic generation of HCN from pure ACN has been previously reported, the mechanism of which is proposed to proceed by direct photolysis of ACN generating •CN and •CH3 radicals by homolytic cleavage48.
Subsequent abstraction of a proton from ACN, or in our case from a molecule of water, furnishes the HCN molecule.
Following the production of HCN, the generation of FA follows a well-understood mechanism49. Addition of a hydroxyl radical followed by rapid tautomeric rearrangement leads to the FA radical.
Protonation and electronic disproportionation with another radical species leads to the FA molecule. The fact that addition of oxygen or nitrate, i.e., electron scavengers, had only a minor effect on the yield of FA also suggests that •OH radicals are involved in the mechanism. Radiolysis of aqueous 25 mM NaCN at pH 7 at 2.92 kGy h−1 over a range of dose rates revealed FA as a major product detectable by GC-MS (G = 0.08 molecules/100 eV), corroborating the proposed mechanism of FA formation through an HCN intermediate.
Discussion
Radiolysis of aqueous ACN and HCN produces amide and nitrile solvents at rates that exceed hydrolytic decomposition. These experiments simulate terrestrial synthesis pathways that produce FA without invoking extraterrestrial influx or thermally mediated hydrolysis of HCN. Experiments have shown that radiolysis of N2 and CH4 yield HCN and ACN as two major products50,51; though HCN appears to be the crucial intermediate to FA production, it is also a more volatile and readily reactive compound, which poses a challenge to having it accumulate to significant concentrations. ACN, on the other hand, is much less volatile, affording it the potential to settle from the atmosphere and to collect in surface water reservoirs along with other, less abundant nitrile species52. Nitriles produced by gamma radiolysis of atmospheric N2 and CH4 proximal to radioactive minerals could have been supplemented by more distributed energy sources such as solar UV, galactic cosmic rays (GCRs) or coronal mass ejections associated with solar flare events53 throughout the atmospheric column, which would have been introduced into the system during periodic influx of external water. The result would have been robust, long-term production of FA in a fixed setting (Fig. 4).

Proposed formamide synthesis and concentration process, depicting localized production and concentration of formamide (HCONH2) near radioactive mineral deposits on terrestrial surface environments. Note that chemical reactions are not balanced for simplicity.
Repetitive wet/dry and heat/cooling cycles, occurring over a long duration of time, seem to be a prerequisite for FA-assisted prebiotic scenarios27. Heat dissipation associated with fissioning mineral seams could have assisted in the rapid concentration of FA. Laboratory FA-based prebiotic synthesis experiments employ concentrated FA or relatively high temperatures (>100 °C)26,31,54,55. Temperature fluctuations caused by water- or organic-moderated fission of uranium-bearing minerals would periodically raise temperatures above 100 °C, while also permitting influx of new sources of dissolved HCN or ACN from external sources that could be radiolyzed to produce FA. To determine if the concentration of FA is possible in the presence of the other radiolysis products observed (Fig. SI2)30, we carried out a simple concentration experiment by heating a 450 mL solution containing 250 mM each of FA, acetic acid, formic acid, acetamide and succinonitrile in an oven above a temperature of 100 °C. The concentrations of these molecules were evaluated as a function of time using 13C NMR spectroscopy (Fig. SI3). After approximately one day, all of the water had evaporated leaving behind a desiccated mixture in approximately the same molar proportions of starting materials.
The radiolysis yields determined herein enable estimation of the geochemical production parameters of FA within a radioactive mineral deposit. The G values describing the production of FA from ambient ACN and HCN are derived from this study. A typical sandstone sediment has pores that make up about 20% of the total volume56, wherein radiolytic chemistry may occur; calculated productivity is therefore reduced by approximately this proportion. The radioactivity associated with heavy mineral placer sands38,39 yields about 0.1–0.8 mol FA km−2 yr−1. This is over an order of magnitude greater than area- and time-averaged delivery from extraterrestrial sources33,34, with the added advantage that FA is produced within a relatively small volume proximal to the radioactive deposit (thus reducing reliance upon an external concentration mechanism). A fissioning mineral seam of uranium, with dimensions comparable to a typical fission zone described from the Paleoproterozoic Oklo deposit, has a combined (radiolysis and heat) power output of about 10 kilowatts42,46,57,58. About 13% of this power is composed of γ or β rays that can penetrate substantial distances beyond their host minerals59. These parameters yield an estimated FA production rate of 0.1–1 moles FA m−2 yr−1, which is over 6 orders of magnitude larger than the estimated FA production from placer minerals or delivery from cometary material per unit area34. The highest area-normalized production rate of about 1 mole FA m−2 yr−1 would require conversion of approximately 10% per day of the HCN present within the penetration range of the mineral seam’s γ radiation, and with HCN at the highest estimated concentration of oceanic reservoirs; a smaller conversion fraction per day would be required for shallow lake reservoirs with higher HCN concentrations26. This estimate does not include direct HCN or ACN production from irradiation of atmospheric gases by the deposit itself, nor does it include diffusion of HCN or ACN from adjacent volumes each day. Within this envelope of parameters, the theoretical upper limit of FA production in shallow terrestrial settings would be limited by efficiency of radiolytic conversion from HCN and ACN feedstocks rather than by the amount of nitrile feedstock abundance. An individual fission zone comparable to those at Oklo could produce an upper limit of 230,000 kg of FA over an average fission zone lifespan of about 500,000 years and within a radius of about 10 meters.
The maximum possible area-normalized reservoir capacities for each mechanism of FA input can be estimated based on production figures and energy input associated with these settings (Fig. 5). If prevailing surface conditions are assumed to induce an FA degradation rate of about 0.7% per year on all potential reservoirs (i.e., FA degradation in water at 30 °C and pH 726) then each maximum area-normalized reservoir is calculated as the ratio of normalized FA input to the degradation rate (Fig. SI4). Isolated comet impacts have a maximum reservoir size of approximately zero, because input is a step function rather than a continuous flux, and therefore FA degradation after the impact event cannot be counterbalanced except through some combination of the other input processes. Steady-state production and degradation of FA in oceanic and shallow lake reservoirs are not capable of reaching neat FA concentrations required to undergo reactions of prebiotic interest26. Among all considered FA sources that have been reported, the radiolysis of ACN and HCN within uranium fission zones have the highest area-normalized FA production rate and reservoir size, and the smallest characteristic source areas.

Formamide (FA) maximum reservoir size for different FA delivery or production mechanisms. FA sources include global comet influx34, a single comet event33, ideal pH and temperature for oceanic and lake reservoirs from HCN hydrolysis26, and radiolysis of ACN and HCN within radioactive placers and uraninite fission zones. Marker size indicates characteristic area of reservoir, with smaller markers indicating more localized processes. Heat map indicates maximum possible FA concentration of the reservoir.
Radioactive mineral deposits capable of producing abundant FA would place this compound in contact with placer minerals relevant to prebiotic chemistry. Common placer sediments include monazite ((Sm, Gd, Ce, Th)PO4), rutile (TiO2), pyrite (FeS2) and apatite (Ca10(PO4)6(OH)2)40, some of which promote condensation of FA into nucleobases14. We observed herein that radiolysis of aqueous ACN produces significant amounts of four different amide solvents (FA, acetamide, N-methyl-acetamide and N-methyl-formamide), all of which are known to support the phosphorylation of nucleosides and nucleotides with solid phase apatite60, yielding multiple pathways and abundant sources of two of three nucleic acid monomer components: nucleobases and phosphate.
Radioactive mineral deposits are unique geochemical environments that can produce FA far more quickly than it is degraded. FA is a demonstrated nucleobase precursor and water-alternative solvent with favorable properties for phosphorylation and polymerization. In conjunction with localized production of abundant FA, these geochemical settings may offer one possible means of mitigating both the Water Paradox and the Concentration Problem, at least with respect to the production of nucleotide components. Periodic drying of placer deposits has the potential to concentrate FA, other high boiling-point solvents, and solutes to a degree which renders condensation reactions favorable. Radioactive mineral deposits open the intriguing possibility that water-alternative solvents could have assisted or even enabled the emergence of polymeric chemical systems that preceded water-based life as we know it.
Methods
Reagents (including ACN, NaCN, FA, formic acid, acetamide and succinonitrile) were purchased from Sigma-Aldrich, Japan and were of 99% purity or higher and used without further purification. Purified 18 MΩ water produced by EMD Millipore’s Milli-Q® water purification system was loaded into glass vials which were pre-ashed at 500 °C for three hours to remove organic contaminants. In the case of ACN, the vials were frozen, vacuum purged and back filled with N2 three times. For HCN experiments, the solutions were prepared from NaCN and water which had been degassed with N2. The pH of the NaCN solutions were adjusted to 7 using concentrated HCl, after which the reaction vials were sealed and the solutions were subjected to further purging with N2 for a period of about five minutes to remove any residual O2. For each experiment, two sets of vials were created. One set was exposed to varying doses and dose rates of γ-radiation from a 60Co source (10‒30 cm distances, with dose rates varying from approximately 3.0‒0.5 kGry hr−1, respectively) at the Tokyo Institute of Technology’s 60Co Gamma Radiation Facility. These dose rates are roughly 75 times greater than that estimated from natural fission cores46. Another set of vials, which served as controls were kept in a darkened box at room temperature.
Volatiles in radiolysis samples were analyzed with a Shimadzu GCMS-QP2010 Ultra gas chromatrograph-mass spectrometer (GC-MS). Direct injection of liquid samples and head-space sampling were carried out using a PAL (prep and load solution) RTC autosampler. Data processing was conducted using the LabSolution_GCMSsolution version 4.11 software package. The injection method used an inlet temperature of 250 °C and a source temperature of 200 °C. The scan range was set to m/z 20–500 Da. Chromatography was conducted using an Rtx-WAX® column (60 m, 0.3 mm I.D., 0.5 mm df); helium was used as the carrier gas at a flow rate of 40 cm sec−1. The oven temperature program was: 2 min hold at 70 °C, from 70 to 240 °C at 10 °C min−1 and then the temperature was maintained at 240 °C for 5 min. Compounds were identified by comparison of their electron impact fragmentation spectra with standards from the NIST standard reference library, and by verification of spectra with off-the-shelf standard reagents. Concentrations were determined by establishing a calibration curve determined from known concentrations of authentic standards. FA heating and concentration experiment mixtures were analyzed in 10% D2O/90% H2O using a Bruker Avance III nuclear magnetic resonance (NMR) spectrometer tuned to measure 13C spectra at 100 MHz and 303 K.
Data Availability
The datasets generated during the current study are available from the corresponding author on reasonable request.
References
Miller, S. L. & Orgel, L. E. The Origin of Life on the Earth. (Prentice-Hall, Inc., 1974).
Powner, M. W., Gerland, B. & Sutherland, J. D. Synthesis of activated pyrimidine ribonucleotides in prebiotically plausible conditions. Nature 459, 239–242 (2009).
Becker, S. et al. A high-yielding, strictly regioselective prebiotic purine nucleoside formation pathway. Science 352, 833–836 (2016).
Kim, H.-J. & Benner, S. A. Prebiotic stereoselective synthesis of purine and noncanonical pyrimidine nucleotide from nucleobases and phosphorylated carbohydrates. Proceedings of the National Academy of Sciences 114, 11315–11320 (2017).
Saladino, R. et al. Meteorite-catalyzed syntheses of nucleosides and of other prebiotic compounds from formamide under proton irradiation. Proceedings of the National Academy of Sciences 112, E2746–E2755 (2015).
Saladino, R., Botta, G., Bizzarri, B. M., Di Mauro, E. & Garcia Ruiz, J. M. A global scale scenario for prebiotic chemistry: Silica-based self-assembled mineral structures and formamide. Biochemistry 55, 2806–2811 (2016).
Mulkidjanian, A. Y., Bychkov, A. Y., Dibrova, D. V., Galperin, M. Y. & Koonin, E. V. Origin of first cells at terrestrial, anoxic geothermal fields. Proceedings of the National Academy of Sciences 109, E821–E830 (2012).
De Duve, C. Selection by differential molecular survival: a possible mechanism of early chemical evolution. Proceedings of the National Academy of Sciences 84, 8253–8256 (1987).
Benner, S. A., Ricardo, A. & Carrigan, M. A. Is there a common chemical model for life in the universe? Current opinion in chemical biology 8, 672–689 (2004).
Radzicka, A. & Wolfenden, R. Rates of uncatalyzed peptide bond hydrolysis in neutral solution and the transition state affinities of proteases. Journal of the American Chemical Society 118, 6105–6109 (1996).
Wolfenden, R. Benchmark reaction rates, the stability of biological molecules in water, and the evolution of catalytic power in enzymes. Annual review of biochemistry 80, 645–667 (2011).
Benner, S. A. Paradoxes in the origin of life. Origins of Life and Evolution of Biospheres 44, 339 (2014).
Schoffstall, A. M. Prebiotic phosphorylation of nucleosides in formamide. Origins of life 7, 399–412 (1976).
Saladino, R., Crestini, C., Pino, S., Costanzo, G. & Di Mauro, E. Formamide and the origin of life. Physics of life reviews 9, 84–104 (2012).
Philipp, M. & Seliger, H. Spontaneous phosphorylation of nucleosides in formamide—Ammonium phosphate mixtures. Naturwissenschaften 64, 273–273 (1977).
Saladino, R., Botta, G., Pino, S., Costanzo, G. & Di Mauro, E. Genetics first or metabolism first? The formamide clue. Chemical Society Reviews 41, 5526–5565 (2012).
Schoffstall, A. M., Barto, R. J. & Ramos, D. L. Nucleoside and deoxynucleoside phosphorylation in formamide solutions. Origins of life 12, 143–151 (1982).
Saladino, R. et al. Origin of informational polymers: The concurrent roles of formamide and phosphates. ChemBioChem 7, 1707–1714 (2006).
Saladino, R., Crestini, C., Costanzo, G., Negri, R. & Di Mauro, E. A possible prebiotic synthesis of purine, adenine, cytosine, and 4 (3H)-pyrimidinone from formamide: implications for the origin of life. Bioorganic & medicinal chemistry 9, 1249–1253 (2001).
Ochiai, M., Marumoto, R., Kobayashi, S., Shimazu, H. & Morita, K. A facile one-step synthesis of adenine. Tetrahedron 24, 5731–5737 (1968).
Yamada, H. & Okamoto, T. A one-step synthesis of purine ring from formamide. Chemical and Pharmaceutical Bulletin 20, 623–624 (1972).
Saladino, R. et al. Synthesis and degradation of nucleic acid components by formamide and iron sulfur minerals. Journal of the American Chemical Society 130, 15512–15518 (2008).
Saladino, R. et al. One‐Pot TiO2‐Catalyzed Synthesis of Nucleic Bases and Acyclonucleosides from Formamide: Implications for the Origin of Life. ChemBioChem 4, 514–521 (2003).
Saitta, A. M. & Saija, F. Miller experiments in atomistic computer simulations. Proceedings of the National Academy of Sciences 111, 13768–13773 (2014).
Harada, K. Formation of amino-acids by thermal decomposition of formamide-oligomerization of hydrogen cyanide. Nature 214, 479–480 (1967).
Miyakawa, S., Cleaves, H. J. & Miller, S. L. The cold origin of life: A. Implications based on the hydrolytic stabilities of hydrogen cyanide and formamide. Origins of Life and Evolution of the Biosphere 32, 195–208 (2002).
Šponer, J. E. et al. Emergence of the First Catalytic Oligonucleotides in a Formamide‐Based Origin Scenario. Chemistry–A European Journal 22, 3572–3586 (2016).
Pietrucci, F. & Saitta, A. M. Formamide reaction network in gas phase and solution via a unified theoretical approach: Toward a reconciliation of different prebiotic scenarios. Proceedings of the National Academy of Sciences 112, 15030–15035 (2015).
Furukawa, Y., Kim, H.-J., Hutter, D. & Benner, S. A. Abiotic regioselective phosphorylation of adenosine with borate in formamide. Astrobiology 15, 259–267 (2015).
Barks, H. L. et al. Guanine, Adenine, and Hypoxanthine Production in UV‐Irradiated Formamide Solutions: Relaxation of the Requirements for Prebiotic Purine Nucleobase Formation. ChemBioChem 11, 1240–1243 (2010).
Burton, A. S. A. “Warm formamide” scenario for the origins of life might not be so hot: Comment on “Formamide and the origin of life” by E. Di Mauro et al. Physics of life reviews 9, 114–115 (2012).
Ferus, M. et al. High-energy chemistry of formamide: A unified mechanism of nucleobase formation. Proceedings of the National Academy of Sciences 112, 657–662 (2015).
Biver, N. et al. Complex organic molecules in comets C/2012 F6 (Lemmon) and C/2013 R1 (Lovejoy): Detection of ethylene glycol and formamide. Astronomy & Astrophysics 566, L5 (2014).
Adande, G. R., Woolf, N. J. & Ziurys, L. M. Observations of interstellar formamide: Availability of a prebiotic precursor in the galactic habitable zone. Astrobiology 13, 439–453 (2013).
Niether, D., Afanasenkau, D., Dhont, J. K. & Wiegand, S. Accumulation of formamide in hydrothermal pores to form prebiotic nucleobases. Proceedings of the National Academy of Sciences, 201600275 (2016).
Bada, J. L., Chalmers, J. H. & Cleaves, H. J. Is formamide a geochemically plausible prebiotic solvent? Physical Chemistry Chemical Physics (2016).
Buxton, G. V., Greenstock, C. L., Helman, W. P. & Ross, A. B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (⋅OH/⋅O−) in aqueous solution. Journal of Physical and Chemical Reference Data 17, 513–886 (1988).
Mohanty, A., Sengupta, D., Das, S., Saha, S. & Van, K. Natural radioactivity and radiation exposure in the high background area at Chhatrapur beach placer deposit of Orissa, India. Journal of Environmental Radioactivity 75, 15–33 (2004).
Mohapatra, S. et al. Proceedings of the DAE-BRNS fifth symposium on nuclear analytical chemistry. Mumbai, India, 330–331 (2014).
Acharya, B., Nayak, B. & Das, S. Mineralogy and mineral chemistry of placer deposit around Jhatipodar, Odisha. Journal of the Geological Society of India 86, 137–147 (2015).
Adam, Z. Actinides and life’s origins. Astrobiology 7, 852–872 (2007).
Adam, Z. R. Temperature oscillations near natural nuclear reactor cores and the potential for prebiotic oligomer synthesis. Origins of Life and Evolution of Biospheres 46, 171–187 (2016).
Meshik, A., Hohenberg, C. & Pravdivtseva, O. Record of cycling operation of the natural nuclear reactor in the Oklo/Okelobondo area in Gabon. Physical review letters 93, 182302 (2004).
Coogan, L. & Cullen, J. T. Did natural reactors form as a consequence of the emergence of oxygenic photosynthesis during the Archean. GSA Today 19, 5 (2009).
Draganic, I. G. & Adloff, J.-P. Radiation and radioactivity on earth and beyond. (CRC press, 1993).
Draganić, I., Draganić, Z. & Altiparmakov, D. Natural nuclear reactors and ionizing radiation in the Precambrian. Precambrian Research 20, 283–298 (1983).
Ebisuzaki, T. & Maruyama, S. Nuclear geyser model of the origin of life: Driving force to promote the synthesis of building blocks of life. Geoscience Frontiers 24, https://doi.org/10.1016/j.gsf.2016.09.005 (2016).
Ayscough, P., Drawe, H. & Kohler, P. The gamma radiolysis of acetonitrile. Radiation research 33, 263–273 (1968).
Bielski, B. & Allen, A. O. Radiation chemistry of aqueous cyanide ion. Journal of the American Chemical Society 99, 5931–5934 (1977).
Gautier, T. et al. Nitrile gas chemistry in Titan’s atmosphere. Icarus 213, 625–635 (2011).
Ramirez, S., Navarro-Gonzalez, R., Coll, P. & Raulin, F. Possible contribution of different energy sources to the production of organics in Titan’s atmosphere. Advances in Space Research 27, 261–270 (2001).
Cable, M. L. et al. Titan tholins: Simulating Titan organic chemistry in the Cassini-Huygens era. Chemical Reviews 112, 1882–1909 (2011).
Airapetian, V., Glocer, A., Gronoff, G., Hébrard, E. & Danchi, W. Prebiotic chemistry and atmospheric warming of early Earth by an active young Sun. Nature Geoscience (2016).
Cleaves, H. J. II et al. Mineral–organic interfacial processes: potential roles in the origins of life. Chemical Society Reviews 41, 5502–5525 (2012).
Rotelli, L. et al. The key role of meteorites in the formation of relevant prebiotic molecules in a formamide/water environment. Scientific reports 6 (2016).
Connell-Madore, S. & Katsube, T. Pore-size-distribution characteristics of Beaufort-Mackenzie Basin shale samples, Northwest Territories. (Natural Resources Canada, 2006).
Naudet, R. Des réacteurs nucléaires fossiles. Paris, France. le Commisariat a l’Energie Atomique, Eyrolles, 695 (1991).
Gauthier-Lafaye, F., Holliger, P. & Blanc, P.-L. Natural fission reactors in the Franceville basin, Gabon: A review of the conditions and results of a “critical event” in a geologic system. Geochimica et Cosmochimica Acta 60, 4831–4852 (1996).
DOE. Nuclear physics and reactor theory. (Department of Energy, Washington D. C, 1993).
Schoffstall, A. M. & Laing, E. M. Phosphorylation mechanisms in chemical evolution. Origins of life and evolution of the biosphere 15, 141–150 (1985).
Acknowledgements
This work was supported by JSPS KAKENHI Grant-in-Aid for Scientific Research on Innovative Areas “Hadean Bioscience” (Grant number JP26106003), and by the ELSI Origins Network (EON), which is supported by a grant from the John Templeton Foundation. The opinions expressed in this publication are those of the authors and do not necessarily reflect the views of the John Templeton Foundation. This work was supported by a grant from the Simons Foundation (494291, ZA). ZA was supported by an Agouron Institute Geobiology Fellowship. We thank Andrew Knoll and the Blue Marble Space Institute of Science for constructive feedback on early drafts of the manuscript.
Author information
Authors and Affiliations
Contributions
Z.A., H.C., I.Y., M.A., R.Y. and A.C.F. designed and carried out the experiments, Y.H. and A.C.F. completed the chemical analyses, and Z.A., H.C., M.A., Y.H. and A.C.F. contributed to writing the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Adam, Z.R., Hongo, Y., Cleaves, H.J. et al. Estimating the capacity for production of formamide by radioactive minerals on the prebiotic Earth. Sci Rep 8, 265 (2018). https://doi.org/10.1038/s41598-017-18483-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-18483-8
This article is cited by
-
The Habitability of Venus
Space Science Reviews (2023)
-
Ariel – a window to the origin of life on early earth?
Experimental Astronomy (2022)
-
Prognostic significance and oncogene function of cathepsin A in hepatocellular carcinoma
Scientific Reports (2021)
-
Radiolysis generates a complex organosynthetic chemical network
Scientific Reports (2021)
-
GNG7 and ADCY1 as diagnostic and prognostic biomarkers for pancreatic adenocarcinoma through bioinformatic-based analyses
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
