
Abstract
Sharks are charismatic predators that play a key role in most marine food webs. Their demonstrated vulnerability to exploitation has recently turned them into flagship species in ocean conservation. Yet, the assessment and monitoring of the distribution and abundance of such mobile species in marine environments remain challenging, often invasive and resource-intensive. Here we pilot a novel, rapid and non-invasive environmental DNA (eDNA) metabarcoding approach specifically targeted to infer shark presence, diversity and eDNA read abundance in tropical habitats. We identified at least 21 shark species, from both Caribbean and Pacific Coral Sea water samples, whose geographical patterns of diversity and read abundance coincide with geographical differences in levels of anthropogenic pressure and conservation effort. We demonstrate that eDNA metabarcoding can be effectively employed to study shark diversity. Further developments in this field have the potential to drastically enhance our ability to assess and monitor elusive oceanic predators, and lead to improved conservation strategies.
Similar content being viewed by others

Introduction
Oceanic ecosystems are increasingly impacted worldwide. Marine predators are under often unsustainable fishing pressure, which has resulted in several documented cases of stock collapses1,2,3. Elasmobranch (sharks and batoids) populations specifically have suffered from overexploitation and stock declines4,5,6,7,8,9. They are key species in virtually all marine trophic webs10,11 and have long been in conflict with human societies, due to their perceived competition with fishers12 or hazardous nature13,14. Owing to their relatively slow growth rate and low fecundity15, they are also particularly vulnerable to overfishing16,17,18. Only recently have elasmobranchs become the focus of conservation initiatives4,19, as the importance of these charismatic animals for the maintenance and resilience of healthy ecosystems is widely acknowledged20,21,22,23.
The development of management strategies for elasmobranchs depends on accurate population assessments in the field. Yet, currently established survey methods, such as fishing by long-lining or gill-netting, acoustic monitoring, baited remote underwater video (BRUV), underwater visual census (UVC) and fisheries-dependent population surveys, are often resource intensive, selective and dependent on taxonomic expertise, and sometimes invasive and potentially traumatogenic24,25,26. Therefore, biologists and managers worldwide are faced with considerable challenges due to the high effort and cost associated with the assessment and monitoring of elasmobranch biodiversity, abundance and distribution.
Environmental DNA (eDNA), DNA isolated directly from environmental samples such as soil or water, can be amplified, sequenced and assigned back to its species of origin through (meta)barcoding and has been suggested as an alternative to track species presence and abundance in their environment27,28. Due to its limited persistence in the water column — in seawater even small (100-bp) eDNA fragments degrade beyond detectability within days29— the detection of eDNA from a specific taxon indicates its recent presence in the environment30,31,32. Accordingly, over the past couple of years eDNA methods have increasingly been applied for the detection of rare and invasive species33,34,35. Moreover, it has been demonstrated that eDNA metabarcoding has the ability to outperform traditional survey methods for diverse taxa, including teleost fish, both in freshwater36,37,38,39 and in marine ecosystems40,41,42.
The first reported study to detect elasmobranch eDNA in natural water samples employed DNA barcoding (aiming to detect a single species in the environment), for the detection of the largetooth sawfish (Pristis pristis)25. Similarly, a species-specific approach was recently applied to amplify whale shark (Rhincodon typus) eDNA from oceanic water samples42. On the other hand, eDNA metabarcoding has the potential to simultaneously identify several taxa from an environmental sample28, which is clearly essential for community-level assessments. However, previous studies have encountered challenges concerning elasmobranch specific detection, when applying this multispecific approach43,44. And although three species of elasmobranch have recently been detected in a large-scale marine eDNA study using a primer set designed for teleosts45, we are still lacking evidence that eDNA metabarcoding can successfully be applied to describe elasmobranch diversity across a range of natural settings, for the purpose of ecosystem assessment and management.
Here, for the first time, we employ eDNA metabarcoding of natural seawater samples to specifically investigate shark communities in Atlantic and Pacific tropical ecosystems, using a previously published primer set targeting a 127 bp stretch of the mitochondrial COI region46. We assess the potential of this low-effort approach for multi-species elasmobranch detection, and specifically examine whether patterns of species diversity and eDNA read abundance, reflect the known degree of anthropogenic impact in two independent tropical marine systems.
In the greater-Caribbean, there has been a long and ongoing history of elasmobranch exploitation, and high anthropogenic pressure in coastal zones has led to the broad-scale depauperation of elasmobranchs on Caribbean reefs8,47. However, many species do still occur in populated areas where strong fishing regulations are in place or where specific shark conservation policies have been enacted8. In the Bahamas, for instance, gillnet and long-line fishing have been prohibited since 1991 and their national waters have been declared a shark sanctuary in June 2011, prohibiting directed fishing or even the retention of shark by-catch48. In the wider Indo-Pacific region, overfishing and poaching are also responsible for declines in elasmobranch populations49; nevertheless, elasmobranchs do still occur in relatively high numbers around remote, isolated locations such as coral reefs on uninhabited atolls in the northern Line Islands50 and the Chagos Archipelago51. Furthermore, although several widely-distributed elasmobranch species are in effect cosmopolitan or circum-tropical, significant biogeographical differences exist between the Caribbean and the Pacific Coral Sea, which allows also for a broad-scale eDNA comparison of community composition.
Elasmobranch species inventories and assessment of geographical distributions based on eDNA metabarcoding could potentially represent an important tool for rapid environmental monitoring and hence influence conservation management and policy decisions. This study represents the first targeted effort that demonstrates the effectiveness of an eDNA metabarcoding approach for the detection and monitoring of elasmobranch communities.
Results
eDNA detection of elasmobranchs
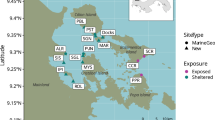
A total number of 2,972,832 reads was obtained from an Illumina MiSeq run of pooled amplicon libraries, built from 55 Caribbean and 22 New Caledonian samples (Fig. 1a,b and Supplementary Table S1). A large part of the sequenced reads (80%) originated from non-specific amplification and were shorter than the target length. After sample assignment, quality and sequence-length filtering, 284,252 reads were left; of which 21,542 could be taxonomically assigned to elasmobranchs (Supplementary Table S1). The number of elasmobranch reads per sample ranged from 0 to 5,205 (Supplementary Table S1). After the removal of singletons (MOTUs in a sample that contained only one read), taxonomic assignment from the sampled locations (Fig. 1a,b) resulted in 22 elasmobranch molecular operational taxonomic units (MOTUs), of which 12 were detected in the Caribbean, 16 in New Caledonia and 9 in both locations. Krona-like plots (Fig. 1c) display the complete taxonomic assignment for each of the sampling locations, while a Principal Component Analysis (PCA) (Fig. 1d) depicts the scattering of all the samples containing elasmobranch reads across the two biogeographic areas. Even though several MOTUs are shared between the two regions, there is still a clear spread in MOTU (species) composition between New Caledonia and the Caribbean. No elasmobranch reads were detected in the two PCR negative controls that were performed and sequenced to detect potential contamination.

Map of New Caledonian (a) and Caribbean (b) sampling locations. The intensity of dot shading in all panels indicates the level of anthropogenic impact from ‘severely impacted’ (light pink/blue) to ‘least impacted’ (dark pink/blue). The krona-like plots (c) show the complete taxonomic assignment for each of the sampling locations (with elasmobranchs in purple). The different taxonomic levels are represented by the layers of rings, starting with phylum, for the innermost layer, and subsequently class, order, family and genus radiating outwards. In the centre of each location plot, the number of elasmobranch reads compared to the total number of filtered reads, is displayed. The Principal Component Analysis (PCA) (d) depicts the scattering of the samples containing elasmobranch reads, across the two biogeographic areas. The six most discriminating taxa are labelled in full, while the rest are indicated by numbers (following alphabetical order from lines 27–50 in Supplementary Table S2), namely: 1 = C. acronotus, 2 = C. albimarginatus, 4 = C. amblyrhynchos/limbatus_Caribbean, 6 = C. brachyurus/perezii, 8 = C. melanopterus/cautus, 9 = C. leucas, 10 = C. obscurus/macloti/longimanus/galapagensis, 12 = C. perezii/falciformis_Pacific, 13 = C. plumbeus, 14 = C. plumbeus/altimus/sorrah, 15 = G. cuvier, 17 = N. brevirostris/acutidens_Pacific, 18 = R. porosus/terraenovae, 20 = S. mokarran, 21 = D. Americana, 22 = S. fasciatum. Maps made with Natural Earth. Free vector and raster map data @ naturalearthdata.com.
Using the 127-bp COI fragment, we did not find a wholly unequivocal correspondence between MOTUs and species; since some MOTUs had 100% sequence identity matches with more than one species in the BOLD database. Although this issue mostly pertained to the genus Carcharhinus, which is known to be taxonomically problematic and polyphyletic52, it also affected the less speciose Rhizoprionodon and Negaprion. Consequently, this has also resulted in MOTUs containing sequences from both Coral Sea and Caribbean species that share an identical 127 bp sequence. Thus, for a more reflective separation, these particular MOTUs have been split into their Pacific and Caribbean components (see Supplementary Table S2 for a complete data file of all reads per taxa, per sample).
Elasmobranch diversity and read abundance patterns
The Bahamas is the Caribbean sampling location least subjected to fishing pressure, as a result of its shark sanctuary status; hence, it displays the greatest elasmobranch diversity, composed of 11 different MOTUs (Fig. 2). In the samples from the locations most impacted by anthropogenic disturbances, Jamaica and Belize, only 2 and 1 elasmobranch MOTUs respectively, were detected. A similar pattern is apparent for the New Caledonian samples, where the highest diversity is found in the most remote and pristine locations, the Chesterfield Atolls (11 MOTUs) and New Caledonia North (14 MOTUs). Contrastingly, only 5 elasmobranch MOTUs were detected in the capital, Noumea, the most densely populated area of New Caledonia.

Bar plot showing the relative abundances of reads (fourth-root transformed) for every elasmobranch MOTU detected in the Caribbean and New Caledonian locations.
The violin plots of MOTU richness (Fig. 3a) and read abundances (Fig. 3b) show how the different sample values are distributed, by comparing the variable sample size distribution across the different locations. The distribution of density (number of MOTUs, Fig. 3a), and abundance of reads, (Fig. 3b) is represented by the width of the plots. For both the Caribbean (green) and the New Caledonian (blue) locations, MOTU richness (Fig. 3a) increases from left to right, following the pattern of decreasing anthropogenic disturbance. While the three least impacted locations, the Bahamas, New Caledonia North and the Chesterfield atolls, show the greatest numbers of MOTUs in one sample (6, 7 and 8 MOTUs respectively), the violin plots for these locations show that richness is more equally spread across the different samples from Chesterfield (thus more samples containing multiple MOTUs); a sample is more likely to contain only 1 or 2 MOTUs in the Bahamas whereas in Chesterfield a sample is more likely to contain 4 or 5 MOTUs. Additionally, every sample from Chesterfield contains at least 3 elasmobranch MOTUs (for detailed MOTU richness per sample, see Supplementary Table S2). The abundance of reads (Fig. 3b) per location follows the same pattern: it increases in both Caribbean and New Caledonian locations, with a decreasing level of human impact. While both New Caledonia North and Chesterfield have samples that contain more than 1000 elasmobranch sequence reads, the number of samples with more than 1000 reads is greater in the more remote Chesterfield atolls. Additionally, Chesterfield is the only location without any samples with less than 80 sequence reads.

Violin plots showing (a) elasmobranch diversity (MOTU richness) and (b) abundance of reads per sample in the different locations from the Caribbean (green) and New Caledonia (blue). The shapes indicate the density distribution of the samples, extending from the minimum to the maximum observed values. The median values are indicated by the red dots. The thick black bars are the interquartile ranges. The thin black extending lines represent the 95% confidence intervals such that the values in the wider parts of the plots are more probable than those in the narrower parts. Per region, significant differences (P < 0.05) are indicated with asterisks. Asterisk significant codes: ***P < 0.001, **P < 0.01, *P < 0.05, •P < 0.1.
Generalized Linear Model (GLM) tests (using location as categorical factor) indicate significant differences in both diversity and read abundance between locations in both the Caribbean and New Caledonia. Pair-wise comparisons (post-hoc Tukey comparisons) show that for diversity (MOTU richness, Fig. 3a), significant differences are detected between the Bahamas and Jamaica (P = 0.039) and nearly significant differences between the Bahamas and Belize (P = 0.095). While diversity in New Caledonia is significantly different between Chesterfield and Noumea (P = 0.013). For read abundances (Fig. 3b), significant differences exist between the Bahamas and the other 3 Caribbean locations (P < 0.001). Additionally, read abundances in Turks & Caicos are significantly different from Jamaica (P = 0.033) and Belize (P = 0.047). In New Caledonia, all abundance comparisons are highly significant (P < 10−10).
Species accumulation curves are plotted for each location (Fig. 4). The curves show elasmobranch diversity (MOTU richness) as a function of the number of samples in the locations from the Caribbean (A) and New Caledonia (B). Error bars indicate standard errors after 100 permutations. The results show that none of the Caribbean (Fig. 4a) or New Caledonian (Fig. 4b) samples tend to reach a plateau in MOTU richness, although, with the exception of the Bahamas, the Caribbean slopes tend to flatten after N = 10. Non-saturation of species accumulation curves suggests that increased sampling effort would be desirable for capturing total diversity in each location, particularly in the naturally more diverse tropical Pacific.

Species accumulation curves showing elasmobranch diversity (MOTU richness) as a function of the number of samples in the locations from the Caribbean (a) and New Caledonia (b). Error bars indicate standard errors after 100 permutations. Belize is absent from the plot as it contains only one elasmobranch MOTU.
Discussion
Our results demonstrate that eDNA metabarcoding can be applied to assess elasmobranch species richness and, potentially, relative abundance in natural seawater samples, the two main components of ecological communities. The derived geographical patterns of diversity and abundance of shark eDNA sequence reads may be used for monitoring purposes and ultimately to inform conservation management and policy decisions. At the global/macro-scale, the detected elasmobranch MOTUs collectively separate the Caribbean and New Caledonian regions (Fig. 1d), dominated by Carcharhinus perezii/Negaprion brevirostris and Carcharhinus amblyrhynchos / Triaenodon obesus, respectively, with the exception of the most depauperate locations (e.g. Belize, Jamaica and Noumea) which are grouped in the center of the ordination plot. Additionally, the patterns of MOTU richness and abundance of sequence reads follow the level of anthropogenic impact in each location. Remote localities such as the Chesterfield atolls and protected areas such as Bimini, Bahamas show both the highest species richness and read abundance among our samples, whereas the less remote and non-protected locations show lower values for both diversity and abundance (Figs 1c, 2 and 3).
In marine ecosystems, the impact on living resources is often framed into the Malthusian theory of human density around such ecosystems53. Several studies have shown proximity to market to be the strongest predictor of overfishing on coral reefs54,55,56. A particular case study from New Caledonia has demonstrated that travel time from the market is a strong predictor of fish biomass, predator abundance and functional diversity on coral reefs53,57. Thus, remote locations such as the Chesterfield atolls, receive de facto protection due to their isolation57. The level of elasmobranch diversity and eDNA read abundance, sheds a new light on these wilderness areas that are already known to support high levels of fish biomass51,57.
Likewise, it has previously been shown that sharks on reefs in the Greater Caribbean mostly occur in areas with low human population density or in a few places where strong fishing regulations or conservation measures have been implemented8. While none of our Caribbean sampling locations is at more than 1 hour travel away from people, The Bahamas represents one of those locations whose elasmobranch populations in particular receive protection through effective conservation measures48.
Environmental DNA metabarcoding has a number of advantages compared to classical approaches for monitoring elasmobranchs. It is a minimally invasive and resource-effective technique. The eDNA sampling and metabarcoding protocols are easy to standardize and the molecular assignment does not require taxonomic expertise. Nevertheless, our findings also reveal a number of concerns that should be addressed in future developments of shark/elasmobranch eDNA metabarcoding approaches. First the taxonomic resolution of the final dataset is strongly dependent on the choice of markers; while the use of COI as a metabarcoding marker has previously been criticized58, owing to its high sequence variability, which may impair the design of truly universal primers and complicate bioinformatics analysis - it can also be argued that COI presents two major advantages over other potential markers. First, the steadily growing international effort, headed by the Consortium for the Barcode of Life (CBOL), to develop a public DNA barcoding database with curated taxonomy, greatly facilitates taxonomic assignment. The BOLD database (http://www.boldsystems.org/)59,60 currently includes >4 million sequences belonging to over 500,000 species, curated and identified by expert taxonomists. Secondly, the high mutation rate of COI enables identification at the species level, whereas the highly conserved sequences of other markers, such as 18S, make it often impossible to distinguish at the species or genus levels; and species-level identification is crucial for studies aimed at detecting rare species, such as is often the case for sharks.
Nevertheless, using the 127 bp elasmobranch specific COI fragment46, we still recorded some ambiguity in the taxonomic assignment of some species of the genera Carcharhinus, Rhizoprionodon and Negaprion, owing to the limited sequence variability within the amplicon. Consequently, the sequences of some MOTUs are 100% identical to individuals belonging to different species in the BOLD database. In our dataset, this is presented by several MOTUs belonging to either of two or more species. Furthermore, this has resulted in 3 MOTUs having 100% sequence identity for species occurring in New Caledonia, and species occurring in the Caribbean. One particular example is the Carcharhinus amblyrhynchos/limbatus MOTU. The bulk of sequences within this MOTU are from New Caledonia, 16730 compared to 146 in the Caribbean (Supplementary Table S2). In all probability, the sequence reads from New Caledonia belong to both C. amblyrhynchos, the grey reef shark (a species abundant in this area and also the species most often visually detected during sampling operations, Supplementary Table S1) and to Carcharhinus limbatus, the blacktip shark. Since C. amblyrhynchos does not occur in the Caribbean, the 146 reads of this MOTU in the Caribbean samples most likely belong to C. limbatus. Similar considerations apply to Negaprion brevirostris/acutidens and Carcharhinus perezii/falciformis, whose MOTUs are shared between New Caledonia (365 and 206 reads respectively) and the Caribbean (91 and 1045 reads respectively). Negaprion brevirostris (lemon shark) is native to the Americas whereas N. acutidens (sicklefin lemon shark) is widely distributed throughout the Indo-Pacific. Carcharhinus falciformis, the silky shark, is circumtropical, while the distribution of Carcharhinus perezii (Caribbean reef shark) is restricted to the tropical western Atlantic Ocean.
In order to resolve the issue with closely related species, it will be essential to design alternative primers that are able to amplify a longer fragment of the gene region, in addition to an improved reference database. In relation to this, it is clear that certain elasmobranch species present in the environment at the time of sampling could not be detected using the selected primer set, as their non-degenerate sequences contain mismatches with the binding regions of several species. This is epitomized by the case of the nurse shark, Ginglymostoma cirratum, which is an abundant species in the Caribbean and some individuals were visually observed at the time of sampling. Yet, eDNA sequence reads of G. cirratum were not detected in any of our Caribbean samples. Comparing the available G. cirratum COI sequences in public repositories with our primer sequences, it becomes apparent that two mismatches with the primer 3′ end are most likely responsible for the prevention of amplification of this species’ DNA from environmental samples. In silico mismatch statistics, comparing the 3′ half of the primers with full mitochondrial genome sequences available, are listed in Supplementary Table S4, for all elasmobranch Orders. This table shows that the primers are particularly suitable for amplifying Carcharhiniformes, which contains more than half of all shark species, including those of most ecological relevance for this study. However, all the elasmobranch orders may be amplified. Potential mismatch issues may be resolved by decreasing the specificity of the primer set by incorporating degenerate bases or inosine nucleotides. However, this approach may have the undesired effect of an increase in the number of reads belonging to non-target taxa getting amplified. Clearly, this trade-off between taxonomic resolution and non-target amplification needs to be well balanced prior to applying the eDNA metabarcoding approach for the purpose of informing conservation and management decisions. Here, we focused on large-scale differences and overall patterns in relation to anthropogenic influence, but in alternative contexts it may be necessary to attain greater taxonomic accuracy (e.g. endangered or invasive species); a challenge also faced by currently established ribosomal amplicon-based analysis of other vertebrates45,61.
Quantification of eDNA relating to species abundance could provide clues to habitat use and preference, thus identifying spatial conservation priorities such as home ranges and dispersal and migration corridors62. However, whether eDNA metabarcoding can provide quantitative estimates, particularly in the case of community-level abundance, remains a controversial issue. Although amplicon sequencing produces read counts that may contain valuable information about target species abundances40,63, the interpretation of the results of amplicon studies, in the context of quantitative ecology, is not straightforward and remains ambiguous64. This is in part because shedding rates between communities, species and individuals may differ. But also because the precise relationship between amplicon abundance and taxon abundance remains unknown and likely varies among taxa63,65, as it is argued that PCR products are not fully proportional to real abundances due to the fact that primer efficiency may vary among species templates (primer bias)65,66. While previous studies have shown positive rank correlations between species abundance and read abundance38,43,63,67, as of yet, no studies have been published, revealing evidence of a relationship between relative abundance of species within a community and their respective eDNA read abundances. And while currently no experiments have been performed to empirically verify the relationship between read abundance and community biomass for elasmobranchs in particular, our data show that read abundances are higher in the more pristine/remote sampling locations and that these patterns of read abundance are coherent with expectations that can be inferred from the contrasting levels of human impact/remoteness of the different locations, for both the Caribbean and New Caledonia (Fig. 3b). While for example the species diversities of New Caledonia North and Chesterfield are very similar (Fig. 2), eDNA read abundance is significantly higher in Chesterfield (Fig. 3b), suggesting that read abundance may be correlated with remoteness. As the relationships between eDNA and species abundance become clearer, the role of eDNA in estimating species abundance in both freshwater and marine environments is likely to become more valuable, increasing the potential of future eDNA applications in research and conservation.
Conservation and management of elasmobranch diversity relies on the effective monitoring of species across large oceanic areas. While direct observation and identification of individuals are often complicated, we have demonstrated that, despite the seemingly daunting task of probing vast stretches of ocean by collecting water samples, eDNA metabarcoding has great potential for developing into an objective and powerful elasmobranch assessment tool, applicable to a wide range of ecological goals, from the mapping of diversity gradients in response to environmental variation, to the monitoring of the effectiveness of spatial protection measures.
Material and Methods
Experimental Design
Aqueous eDNA samples were collected with interoceanic replication, to test for spatial marine protection as a predictor for elasmobranch diversity. During February and March of 2015 (Supplementary Table S1), samples were collected from four Caribbean locations impacted by various levels of anthropogenic pressures (Fig. 1b). Jamaica is known to have one of the most depauperate fish populations in the Caribbean and a severely extirpated elasmobranch fauna68; thus it was expected that Jamaica would sit at the lower end of the elasmobranch diversity range and read abundance. In Belize sampling took place around the partially submerged Glover’s Reef atoll, which is part of the Mesoamerican Barrier Reef. Even though this region has a relatively large number of marine reserves, including the ‘Glovers Reef Marine Reserve’, shark sightings in the Caribbean are quite rare and relatively few shark sightings occurred in the Mesoamerican Barrier Reef area during a previously conducted survey8,69.
In the Turks & Caicos Islands, where sampling took place around South Caicos, the establishment of a shark sanctuary is under consideration; however, the islands are currently still experiencing high fishing pressure47, which tends to disproportionally reduce densities of longer-lived, larger-bodied individuals50. At the other end, the nation of Bahamas is a designated shark sanctuary48 and as such, an area characterised by consolidated shark protection. The sampling was conducted around the islands of Bimini, which consequently boast an abundant and diverse elasmobranch fauna70,71.
In the tropical Pacific, eDNA samples were collected from three locations in New Caledonia (Fig. 1a), during September, October and November of 2015 (Supplementary Table S1). New Caledonia has a unique anthropogenic impact gradient from nearly pristine to significant levels of anthropogenic disturbance57. The most heavily impacted site is represented by the capital Noumea, the most densely populated area in New Caledonia53. However, most reefs near Noumea are no-take reserves and shark fishing is historically non-existent in New Caledonia so that near Noumea, shark populations may be healthier compared to many other impacted areas. Sampling sites north of the main island Grande Terre, ‘New Caledonia North’, represent the intermediate level of anthropogenic impact, being between 70–120 km removed from the nearest human settlement and a minimum of 500 km/15 hours travel time, from the Noumea fish market. The isolated Chesterfield atolls, 550 km northwest of Grande Terre (~35 h travel time from the Noumea fish market), are the most remote of all our sampling locations. These samples were expected to show the highest levels of elasmobranch diversity and eDNA read abundance. Within all 7 (Caribbean and New Caledonian) locations, samples were collected from between 6 and 20 different sites covering a variety of habitats (Supplementary Table S1 contains coordinates per site). A total of 55 samples from the Caribbean and 22 samples from New Caledonia were collected and analysed. Each sample consisted of 4 litres of sea water, collected by either a Kemmerer type water sampler or directly with a plastic collection bottle.
Sample processing and DNA extraction
After collection, the water samples were individually covered and stored, in the dark and on ice, during transport to the local laboratory facilities. Vacuum filtration was carried out within two hours after collection. When it was not feasible to carry out filtration within two hours after collection, due to travel time to laboratory facilities, the samples were directly frozen after collection, until further processing. The sterile mixed cellulose esters (MCE) filters (Merck Millipore; 47 mm diameter; 0.45 µm pore size) containing sample filtrates were stored in 2.0 ml screw-cap microcentrifuge tubes containing silica beads. The silica beads function as a desiccator, drying out the filters and hence preventing the DNA from degrading. The sample filters were then stored at −20 °C until extraction. DNA was extracted from the filters with the Mo-Bio PowerSoil DNA Isolation Kit (www.mobio.com), following the manufacturers’ protocol. Purified extracts were assessed for DNA concentration in a Qubit fluorometer (Thermo Fisher Scientific).
Contamination Control
Contamination of samples may occur anywhere from preparing sampling equipment and collecting the samples in the field (target DNA being carried unintentionally from one locality to another), to every subsequent step of sample preparation, extraction and analysis in the laboratory. Hence, strict adherence to contamination control was followed at all field and laboratory stages in order to prevent the occurrence of contamination, including the use of disposable gloves and single use-sterile collection bottles and filtration equipment, and the bleaching (50% bleach) of sampling devices and laboratory equipment and surfaces. Additionally, a dedicated controlled eDNA lab at the University of Salford, with separate rooms designated for the physical separation of eDNA extraction, pre-PCR preparations and post-PCR procedures, was used for all laboratory work. Moreover, to identify potential contamination, DNA extraction blanks (elution buffer from extraction kit) and PCR blanks were included.
Library preparation and sequencing
For the amplification of eDNA metabarcoding markers, an elasmobranch specific COI primer set was used. This previously published primer set consisted of a novel reverse primer ‘Shark COI-MINIR’ 5′-AAGATTACAAAAGCGTGGGC-3′46 and two universal fish barcoding forward primers FishF2 5′-TCGACTAATCATAAAGATATCGGCAC-3′ and VF2 5′-TCAACCAACCACAAAGACATTGGCAC-3′72, yielding an amplicon of 127 bp46. For the multiplex Illumina sequencing run, we used 4 sets of 24 primers with attached 8-base sample-specific oligo-tags differing in at least 3 bases73. In order to increase variability of the amplicon sequences, a variable number (2, 3 or 4) of fully degenerate positions (Ns) was added at the beginning of each primer74. The full, sequenced PCR product, consisted then of 195 bp, including the amplicon, primers, sample tags and leading N’s.
For PCR amplification, a single step protocol was used, directly attaching the 8-base tagged primers. The PCR mix recipe was as follows: a total volume of 20 µl included 2 µl 10x buffer (BioLine), 0.6 µl 50 mM MgCl (BioLine), 0.5 µl of each of the 5 μM forward primers (Eurofins), 1 µl of the 5 µM reverse primer, 0.2 µl 10 mM dNTP mix (BioLine), 0.2 µl BioTaq DNA polymerase (5 u/μl, BioLine), a standardised amount (10 ng) of the filter-extracted eDNA template, and 13 µl sterile water. The PCR profile included an initial denaturing step of 95 °C for 15 min, 35 cycles of 94 °C 1 min, 52 °C 1 min and 72 °C 1 min and a final extension step of 72 °C for 5 minutes. The quality of all amplifications was assessed by electrophoresis, running the products through a 1.5% agarose gel stained with Gel Red (Cambridge Bioscience) and visualized on a UV light platform. All PCR products (including one replicate per sample and 2 PCR negative controls) were pooled into 4 multiplexed sample pools (each composed of 24 individually-tagged samples) and purified using MinElute columns (Qiagen). Four Illumina libraries were subsequently built from the four pools, using the NextFlex PCR-free library preparation kit (BIOO Scientific). The libraries were quantified using the NEBNext qPCR quantification kit (New England Biolabs) and pooled in equimolar concentrations along with 1% PhiX (v3, Illumina) serving as a positive sequencing quality control. The libraries with a final molarity of 8 pM were sequenced on an Illumina MiSeq platform in a single MiSeq flow cell using v2 chemistry (2 × 150 bp paired-ends).
Bioinformatic and Statistical Analyses
The bioinformatic analysis was based on the OBITools metabarcoding software suite75. The pipeline used for data analysis is summarized in Supplementary Methods S3. Quality of the reads was assessed using FastQC. Paired-end reads were aligned using illuminapairedend and alignments with quality score >40 were kept. The aligned dataset was demultiplexed using ngsfilter. The length distribution of the demultiplexed reads showed a large percentage of short fragments (<95 bp), originating from non-specific amplifications and primer-dimer artefacts, which were not removed during the size selection step of library preparation. Thus, a length filter (obigrep) was applied to the aligned reads (120–135 bp) in order to select only the fragments with the correct target size. Reads containing ambiguous bases were also removed. The reads were subsequently dereplicated using obiuniq and a chimera removal step was performed using the uchime-denovo algorithm76 implemented in vsearch77. The MOTUs were delimited using the sumaclust algorithm75 with a constant similarity threshold of 99%. Taxonomic assignment of the representative sequences for each Molecular Operational Taxonomic Unit (MOTU) was performed using the ecotag algorithm75. We built a bespoke elasmobranch reference database using a custom R script for retrieving all COI elasmobranch sequences available from the BOLD database60, and subsequently selecting those that included our 127 bp target fragment. In order to add homologous sequences from other, non-elasmobranch taxa, an in silico PCR was performed against release R117 of the EMBL-EBI database using ecoPCR78. Subsequently, the obtained reference sequences were added to the elasmobranch sequences obtained from BOLD. These additional reference sequences were added to our elasmobranch database in order to avoid the incorrect assignment of amplified sequences, belonging to other taxa, to elasmobranchs. This combined reference database is available from http://github.com/metabarpark/Reference-databases. The final refining of the dataset included taxonomy clustering of MOTUs assigned to the same species.
Due to its high sensitivity, an additional challenge associated with eDNA metabarcoding, is the risk of contamination79,80, and hence the possibility of introducing false positive results. While it is certainly possible to detect a species present in a sample, represented by a single sequence read, it is not possible to completely exclude contamination (or sequencing error) as the potential cause of MOTUs containing only a single read, i.e. to dismiss single reads as potential false positives. Accordingly, we have opted for a more conservative approach and have removed all single read MOTUs from our samples.
All statistical analyses were performed in R v 3.3.0 (https://www.R-project.org/). The vegan package v. 2.4–081 was used for the calculation of sample-based species accumulation curves. A generalized linear model approach was used for testing differences in MOTU richness and read abundances (square-root transformed) as a function of location, using the glm2 package v. 1.1.282. The Poisson distribution family function was used for modelling the residuals and package multcomp83 was used for post-hoc Tukey comparisons. All custom R scripts are publicly available from http://github.com/metabarpark.
Data and materials availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Original DNA sequences (after initial quality filtering, paired-end alignment and length filtering) can be found at: https://data.mendeley.com/datasets/489gckxbz3/1.
References
Jackson, J. B. et al. Historical overfishing and the recent collapse of coastal ecosystems. Science 293, 629–637 (2001).
Mullon, C., Freon, P. & Cury, P. The dynamcis of collapse in world fisheries. Fish Fish. 6, 111–120 (2005).
Myers, R. A. & Worm, B. Rapid worldwide depletion of predatory fish communities. Nature 423, 280–283 (2003).
Camhi, M. D., Valenti, S. V., Fordham, S. V., Fowler, S. L. & Gibson, C. The conservation status of pelagic sharks and rays, IUCN Species Survival Commission’s Shark Specialist Group. 19–23, doi:978-0-9561063-1-5 (2009).
Robbins, W. D., Hisano, M., Connolly, S. R. & Choat, J. H. Ongoing Collapse of Coral-Reef Shark Populations. Curr. Biol. 16, 2314–2319 (2006).
Simpfendorfer, C. A., Hueter, R. E., Bergman, U. & Connett, S. M. H. Results of a fishery-independent survey for pelagic sharks in the western NorthAtlantic, 1977–1994. Fish. Res. 55, 175–192 (2002).
Spaet, J. L. Y. & Berumen, M. L. Fish market surveys indicate unsustainable elasmobranch fisheries in the Saudi Arabian Red Sea. Fish. Res. 161, 356–364 (2015).
Ward-Paige, C. A. et al. Large-Scale Absence of Sharks on Reefs in the Greater-Caribbean: A Footprint of Human Pressures. PLoS One 5, e11968 (2010).
Worm, B. et al. Global catches, exploitation rates, and rebuilding options for sharks. Mar. Policy 40, 194–204 (2013).
Heupel, M. R., Knip, D. M., Simpfendorfer, C. A. & Dulvy, N. K. Sizing up the ecological role of sharks as predators. Mar. Ecol. Prog. Ser. 495, 291–298 (2014).
Navia Andrés, F., Mejía-Falla Paola, A., López-García, J. & Giraldo Alan, C.-E. V. H. How many trophic roles can elasmobranchs play in a marine tropical network? Mar. Freshw. Res. https://doi.org/10.1071/MF16161 (2017).
Gilman, E. et al. Shark interactions in pelagic longline fisheries. Mar. Policy 32, 1–18 (2008).
Muter, B. A., Gore, M. L., Gledhill, K. S., Lamont, C. & Huveneers, C. Australian and U.S. news media portrayal of sharks and their conservation. Conserv. Biol. 27, 187–196 (2013).
Simpfendorfer, C. A., Heupel, M. R., White, W. T. & Dulvy, N. K. The importance of research and public opinion to conservation management of sharks and rays: Asynthesis. Mar. Freshw. Res. 62, 518–527 (2011).
Stevens, J. The effects of fishing on sharks, rays, and chimaeras (chondrichthyans), and the implications for marine ecosystems. ICES J. Mar. Sci. 57, 476–494 (2000).
Bonfil, R. Overview of world elasmobranch fisheries. FAO. Fisheries Technical Paper341. (1994).
Garcia, V. B., Lucifora, L. O. & Myers, R. A. The importance of habitat and life history to extinction risk in sharks, skates, rays and chimaeras. Proc Biol Sci 275, 83–89 (2008).
Musick, J. A., Burgess, G., Cailliet, G., Camhi, M. & Fordham, S. Management of Sharks and Their Relatives (Elasmobranchii). Fisheries 25, 9–11 (2000).
Dulvy, N. K. et al. Extinction risk and conservation of the world’s sharks and rays. Elife 3, e00590 (2014).
Baum, J. K. & Worm, B. Cascading top-down effects of changing oceanic predator abundances. J. Anim. Ecol. 78, 699–714 (2009).
Estes, J. A. et al. Trophic Downgrading of Planet Earth. Science 333, 301–306 (2011).
Ferretti, F., Worm, B., Britten, G. L., Heithaus, M. R. & Lotze, H. K. Patterns and ecosystem consequences of shark declines in the ocean. Ecol. Lett. 13, 1055–1071 (2010).
Heithaus, M. R., Wirsing, A. J. & Dill, L. M. The ecological importance of intact top-predator populations: a synthesis of 15 years of research in a seagrass ecosystem. Mar. Freshw. Res. 63, 1039–1050 (2012).
Lodge, D. M. et al. Conservation in a cup of water: Estimating biodiversity and population abundance from environmental DNA. Mol. Ecol. 21, 2555–2558 (2012).
Simpfendorfer, C. A. et al. Environmental DNA detects Critically Endangered largetooth sawfish in the wild. Endanger. Species Res. 30, 109–116 (2016).
Wheeler, Q. D. Taxonomy: Impediment or Expedient? Science 303, 285–285 (2004).
Ji, Y. et al. Reliable, verifiable and efficient monitoring of biodiversity via metabarcoding. Ecol. Lett. 16, 1245–1257 (2013).
Taberlet, P., Coissac, E., Hajibabaei, M. & Rieseberg, L. H. Environmental DNA. Mol. Ecol. 21, 1789–1793 (2012).
Thomsen, P. F. et al. Detection of a Diverse Marine Fish Fauna Using Environmental DNA from Seawater Samples. PLoS One 7, e41732 (2012).
Barnes, M. A. et al. Environmental Conditions Influence eDNA Persistence in Aquatic Systems. Environ. Sci. Technol. 48, 1819–1827 (2014).
Jerde, C. L., Mahon, A. R., Chadderton, W. L. & Lodge, D. M. ‘Sight-unseen’ detection of rare aquatic species using environmental DNA. Conserv. Lett. 4, 150–157 (2011).
Pilliod, D. S., Goldberg, C. S., Arkle, R. S. & Waits, L. P. Factors influencing detection of eDNA from a stream-dwelling amphibian. Mol. Ecol. Resour. 14, 109–116 (2014).
Takahara, T., Minamoto, T. & Doi, H. Using Environmental DNA to Estimate the Distribution of an Invasive Fish Species in Ponds. PLoS One 8, e56584 (2013).
Turner, C. R., Uy, K. L. & Everhart, R. C. Fish environmental DNA is more concentrated in aquatic sediments than surface water. Biol. Conserv. 183, 93–102 (2015).
Wilcox, T. M. et al. Robust Detection of Rare Species Using Environmental DNA: The Importance of Primer Specificity. PLoS One 8, e59520 (2013).
Civade, R. et al. Spatial Representativeness of Environmental DNA Metabarcoding Signal for Fish Biodiversity Assessment in a Natural Freshwater System. PLoS One 11, e0157366 (2016).
Deiner, K., Fronhofer, E. A., Mächler, E., Walser, J.-C. & Altermatt, F. Environmental DNA reveals that rivers are conveyer belts of biodiversity information. Nat. Commun. 7, 12544 (2016).
Hänfling, B. et al. Environmental DNA metabarcoding of lake fish communities reflects long-term data from established survey methods. Mol. Ecol. 25, 3101–3119 (2016).
Valentini, A. et al. Next-generation monitoring of aquatic biodiversity using environmental DNA metabarcoding. Mol. Ecol. 25, 929–942 (2016).
Port, J. A. et al. Assessing vertebrate biodiversity in a kelp forest ecosystem using environmental DNA. Mol. Ecol. 25, 527–541 (2016).
Yamamoto, S. et al. Environmental DNA metabarcoding reveals local fish communities in a species-rich coastal sea. Sci. Rep. 7, 40368 (2017).
Sigsgaard, E. E. et al. Population characteristics of a large whale shark aggregation inferred from seawater environmental DNA. Nat. Publ. Gr. 1, 1–4 (2016).
Kelly, R. P., Port, J. A., Yamahara, K. M. & Crowder, L. B. Using Environmental DNA to Census Marine Fishes in a Large Mesocosm. PLoS One 9, e86175 (2014).
Miya, M. et al. MiFish, a set of universal PCR primers for metabarcoding environmental DNA from fishes: detection of more than 230 subtropical marine species. R. Soc. Open Sci. 2, 150088 (2015).
Thomsen, P. F. et al. Environmental DNA from Seawater Samples Correlate with Trawl Catches of Subarctic, Deepwater Fishes. PLoS One 11, e0165252 (2016).
Fields, A. T., Abercrombie, D. L., Eng, R., Feldheim, K. & Chapman, D. D. A Novel Mini-DNA Barcoding Assay to Identify Processed Fins from Internationally Protected Shark Species. PLoS One 10, e0114844 (2015).
Newton, K., Côté, I. M., Pilling, G. M., Jennings, S. & Dulvy, N. K. Current and Future Sustainability of Island Coral Reef Fisheries. Curr. Biol. 17, 655–658 (2007).
Chapman, D. D. et al. Give Shark Sanctuaries a Chance. Science 339, 757–757 (2013).
Werry, J. M. et al. Reef-Fidelity and Migration of Tiger Sharks, Galeocerdo cuvier, across the Coral Sea. PLoS One 9, e83249 (2014).
Sandin, S. A. et al. Baselines and Degradation of Coral Reefs in the Northern Line Islands. PLoS One 3, e1548 (2008).
Graham, Na. J. & Mcclanahan, T. R. The Last Call for Marine Wilderness? Bioscience 63, 397–402 (2013).
Sorenson, L., Santini, F. & Alfaro, M. E. The effect of habitat on modern shark diversification. J. Evol. Biol. 27, 1536–1548 (2014).
Maire, E. et al. How accessible are coral reefs to people? A global assessment based on travel time. Ecol. Lett. 19, 351–360 (2016).
Cinner, J. E. et al. Comanagement of coral reef social-ecological systems. Proc. Natl. Acad. Sci. 109, 5219–5222 (2012).
Cinner, J. E. et al. Evaluating Social and Ecological Vulnerability of Coral Reef Fisheries to Climate Change. PLoS One 8, e74321 (2013).
Cinner, J. E. & McClanahan, T. R. Socioeconomic factors that lead to overfishing in small-scale coral reef fisheries of Papua New Guinea. Environ. Conserv. 33, 73–80 (2006).
D’agata, S. et al. Marine reserves lag behind wilderness in the conservation of key functional roles. Nat. Commun. 7, 12000 (2016).
Deagle, B. E. et al. DNA metabarcoding and the cytochrome c oxidase subunit I marker: not a perfect match. Biol. Lett. 10, 1789–1793 (2014).
Hebert, P. D. N., Cywinska, A., Ball, S. L. & DeWaard, J. R. Biological identifications through DNA barcodes. Proc R Soc B 270 (2003).
Ratnasingham, S. & Hebert, P. D. N. bold: The Barcode of Life Data System. Mol. Ecol. Notes 7, 355–364, (http://www.barcodinglife.org) (2007).
Kelly, R. P. et al. Genetic signatures of ecological diversity along an urbanization gradient. PeerJ 4, e2444 (2016).
Barnes, M. A. & Turner, C. R. The ecology of environmental DNA and implications for conservation genetics. Conserv. Genet. 17, 1–17 (2016).
Evans, N. T. et al. Quantification of mesocosm fish and amphibian species diversity via environmental DNA metabarcoding. Mol. Ecol. Resour. 16, 29–41 (2016).
Kelly, R. P. Making environmental DNA count. Mol. Ecol. Resour. 16, 10–12 (2016).
Elbrecht, V. & Leese, F. Can DNA-based ecosystem assessments quantify species abundance? Testing primer bias and biomass-sequence relationships with an innovative metabarcoding protocol. PLoS One 10, 1–16 (2015).
Clarke, L. J., Soubrier, J., Weyrich, L. S. & Cooper, A. Environmental metabarcodes for insects: in silicoPCR reveals potential for taxonomic bias. Mol. Ecol. Resour. 14, 1160–1170 (2014).
Klobucar, S. L., Rodgers, T. W. & Budy, P. At the forefront: evidence of the applicability of using environmental DNA to quantify the abundance of fish populations in natural lentic waters with additional sampling considerations. Can. J. Fish. Aquat. Sci. 1–5, https://doi.org/10.1139/cjfas-2017-0114 (2017).
Hawkins, J. P. & Roberts, C. M. Effects of artisanal fishing on Caribbean coral reefs. Conserv. Biol. 18, 215–226 (2004).
Bond, M. E. et al. Reef Sharks Exhibit Site-Fidelity and Higher Relative Abundance in Marine Reserves on the Mesoamerican Barrier Reef. PLoS One 7, e32983 (2012).
Guttridge, T. L. et al. Philopatry and Regional Connectivity of the Great Hammerhead Shark, Sphyrna mokarran in the U.S. and Bahamas. Front. Mar. Sci. 4, 3 (2017).
Jennings, D. E. et al. Assessment of the aquatic biodiversity of a threatened coastal lagoon at Bimini, Bahamas. J. Coast. Conserv. 16, 405–428 (2012).
Ward, R. D., Zemlak, T. S., Innes, B. H., Last, P. R. & Hebert, P. D. N. DNA barcoding Australia’s fish species. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 360, 1847–1857 (2005).
Guardiola, M. et al. Deep-Sea, Deep-Sequencing: Metabarcoding Extracellular DNA from Sediments of Marine Canyons. PLoS One 10, e0139633 (2015).
Wangensteen, O. S. & Turon, X. Metabarcoding Techniques for Assessing Biodiversity of Marine Animal Forests. Marine Animal Forests 1–29, https://doi.org/10.1007/978-3-319-17001-5_53-1 (2016).
Boyer, F. et al. obitools: A unix-inspired software package for DNA metabarcoding. Mol. Ecol. Resour. 16, 176–182 (2016).
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C. & Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200 (2011).
Rognes, T., Flouri, T., Nichols, B., Quince, C. & Mahé, F. VSEARCH: a versatile open source tool for metagenomics. PeerJ 4, e2584 (2016).
Ficetola, G. F. et al. An In silico approach for the evaluation of DNA barcodes. BMC Genomics 11, 434 (2010).
Goldberg, C. S. et al. Critical considerations for the application of environmental DNA methods to detect aquatic species. Methods Ecol. Evol. 1299–1307, https://doi.org/10.1111/2041-210X.12595 (2016).
Thomsen, P. F. & Willerslev, E. Environmental DNA - An emerging tool in conservation for monitoring past and present biodiversity. Biol. Conserv. 183, 4–18 (2015).
Oksanen, J. et al. Community Ecology Package. R package (2016).
Marschner, I. Fitting Generalized Linear Models, Package ‘glm2’ (2015).
Hothorn, T., Bretz, F. & Westfall, P. Simultaneous Inference in General Parametric Models, Package ‘multcomp’. Cran (2016).
Acknowledgements
We thank all staff, students and volunteers from Glover’s Reef Research Station, Discovery Bay Marine Laboratory and Field Station, Montego Bay Marine Park, Bimini Biological Field Station (Sharklab), the SFS Center for Marine Studies, and the crew of the research vessel Amborella, for their assistance with sample collection. We also thank Alexandre De Menezes and Ian Goodhead for technical discussions and Andjin Siegenthaler for assistance with our sampling area maps. We are also grateful to Kathryn Matthews and Angelo Villagomez for logistic support. This work was co-funded by the Pew Charitable Trusts, the University of Salford R&E strategy funding, the Total Foundation and the Government of New Caledonia
Author information
Authors and Affiliations
Contributions
S.M. and J.B. conceived, designed and coordinated the study; J.B., S.M., D.D.C., G.B., D.B., T.L.G., H.H., D.M. and L.V. contributed to fieldwork and sample collection; Laboratory experiments and data analyses were conducted by J.B. and O.S.W.; J.B. wrote the manuscript; all authors read and commented on the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bakker, J., Wangensteen, O.S., Chapman, D.D. et al. Environmental DNA reveals tropical shark diversity in contrasting levels of anthropogenic impact. Sci Rep 7, 16886 (2017). https://doi.org/10.1038/s41598-017-17150-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-17150-2
This article is cited by
-
Environmental DNA biomonitoring reveals seasonal patterns in coral reef fish community structure
Environmental Biology of Fishes (2022)
-
Current status and future perspectives of Neotropical freshwater stingrays (Potamotrygoninae, Myliobatiformes) genetics
Environmental Biology of Fishes (2022)
-
Seeing through sedimented waters: environmental DNA reduces the phantom diversity of sharks and rays in turbid marine habitats
BMC Ecology and Evolution (2021)
-
Environmental DNA reveals the fine-grained and hierarchical spatial structure of kelp forest fish communities
Scientific Reports (2021)
-
Environmental biomonitoring of reef fish community structure with eDNA metabarcoding in the Coral Triangle
Environmental Biology of Fishes (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
