
Abstract
Tuberous sclerosis complex (TSC) is a rare autosomal dominant disorder causing benign tumors in the brain and other vital organs. The genes implicated in disease development are TSC1 and TSC2. Here, we have performed mutational analysis followed by a genotype-phenotype correlation study based on the clinical characteristics of the affected individuals. Twenty unrelated probands or families from Greece have been analyzed, of whom 13 had definite TSC, whereas another 7 had a possible TSC diagnosis. Using direct sequencing, we have identified pathogenic mutations in 13 patients/families (6 in TSC1 and 7 in TSC2), 5 of which were novel. The mutation identification rate for patients with definite TSC was 85%, but only 29% for the ones with a possible TSC diagnosis. Multiplex ligation-dependent probe amplification (MLPA) did not reveal any genomic rearrangements in TSC1 and TSC2 in the samples with no mutations identified. In general, TSC2 disease was more severe than TSC1, with more subependymal giant cell astrocytomas and angiomyolipomas, higher incidence of pharmacoresistant epileptic seizures, and more severe neuropsychiatric disorders. To our knowledge, this is the first comprehensive TSC1 and TSC2 mutational analysis carried out in TSC patients in Greece.
Similar content being viewed by others
Introduction
Tuberous sclerosis complex (TSC) is a tumour suppressor syndrome of neurocutaneous origin. It exhibits an autosomal dominant mode of inheritance with an incidence varying from 1/6,000 to 1/10,000 births. Two-thirds of the cases are sporadic1. The disorder causes benign hamartomatous tumour growths in multiple organ systems, primarily in the brain, eyes, heart, kidney, skin and lungs2. Tubers growing in the brain are the main factors contributing to high morbidity and mortality in TSC. Brain involvement is often associated with the presence of epilepsy and TAND (Tuberous sclerosis Associated Neuropsychiatric Disorders)3. Clinical diagnosis of TSC is based upon identification of agreed and published major and minor diagnostic criteria4,5.
Twο genes have been identified as responsible for causing TSC: TSC1 located on chromosome 9q34 and TSC2 on 16p13.3. The TSC2 gene was identified first by positional cloning. It contains 41 exons distributed over a genomic area of approximately 40 kb, produces a 5.5 kb transcript, and codes for a 200 kD protein named tuberin6. The TSC1 gene was found to extend over a genomic region of 55 kb. It contains 23 exons, two of which are untranslated, generating an 8.5 kb transcript, from which a 130 kD protein product called hamartin is deriving7. Hamartin and tuberin were shown to be stabilized with the help of TBC1D7 and act in a multimeric complex, which suppresses cell growth by inhibiting the activation of mTOR, a master kinase that regulates multiple processes, including translation, ribosome and macromolecule biogenesis, neoangiogenesis, and autophagy8,9. In cells lacking wild type hamartin or tuberin, mTOR is dysregulated, leading to abnormal differentiation and development, and generation of enlarged cells, like the ones seen in TSC lesions10.
To date, more than 500 unique TSC1 pathogenic variants have been identified in at least 1,350 probands/families, whereas another 1,400 mutations have been detected in TSC2, in over 3,600 probands/families1. Approximately, 80% of individuals diagnosed as definite TSC sufferers have been found to harbour pathogenic variants in TSC1 or TSC2. The majority of remaining patients were shown to be mosaics or bear deep intronic mutations, if not possessing genetic changes in promoter or untranslated regions of these genes11. In this paper, we report a mutational analysis of TSC1 and TSC2 genes in 20 probands/families from Greece, of which 13 had a definite and 7 a possible clinical diagnosis of TSC. Additionally, we have assessed the distribution and type of mutations, and tried to build genotype-phenotype correlations between and within TSC1 and TSC2 cases.
Results and Discussion
Patients’ characteristics
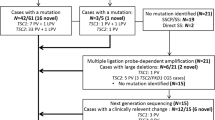
In this work, we present results from twenty unrelated probands from Greece that were referred to the laboratory of Environmental Mutagenesis and Carcinogenesis (Molecular Diagnosis of Genetic Diseases Project) for TSC genetic testing. Thirteen probands (65%) had a definite clinical diagnosis, whereas the rest (35%) presented with clinical findings meeting the criteria for possible TSC diagnosis. Clinical characteristics of affected individuals were identified by doctors of various medical specialties and were reported to us by Paediatric Neurologists or the families themselves, based on the medical records of the patients. Clinical diagnosis was based on the revised diagnostic criteria for TSC4,5.
The phenotypic data along with neurobehavioral features are presented in Table 1. Nevertheless, it should be stressed that various clinical characteristics tend to appear at different ages of the affected individuals. Therefore, the phenotype of a number of young or very young patients could change as they get older. Eighty five percent (11/13) of the patients with a definite TSC diagnosis had a TSC1 (31%) or TSC2 (54%) mutation identified, whereas in patients with possible TSC diagnosis, only TSC1 mutations have been detected in 29% of the cases (2/7). The patients in which no mutations have been identified (35%) were excluded from phenotype/genotype analysis.
Identification and characterization of mutations
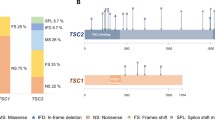
In the present study, we performed mutational analysis in the coding exons and intron/exon junctions of both TSC1 and TSC2 in a total of twenty patients/families. TSC mutation screening was carried out in the probands, but whenever possible, the presence of pathogenic sequence variants was investigated in parental DNAs and in other family members as well. In total, thirteen mutations have been identified (Table 2). Five out of the thirteen (38.5%) have never been reported elsewhere; with the exception of the pathogenic variant identified one family, all the rest were de novo mutations. The novel mutations presented in this work are c.1681_1700del20 (p.Ser561Glyfs*20) and c.2263 C > T (p.Gln755*) in TSC1; c.648 + 1 G > T, c.826delA (p.Met276CysFs*17) and c.4942 A > T (p.Ile1648Phe) in TSC2. No recurrent mutations were identified in either gene.
In more details, six disease-causing mutations were identified in TSC1 (46%), and seven in TSC2 (54%). TSC1 sequence variants included 3 nonsense mutations producing premature termination codons; 2 deletions, which caused frameshifts also resulting in the truncation of the produced protein; and 1 missense mutation. Three of these were familial mutations, and another three were de novo, while four out of the six had been previously reported in LOVD (Leiden Open Variation Database, http://chromium.lovd.nl/LOVD2/TSC/home.php). In TSC2, the seven mutations detected consisted of 1 insertion and 1 deletion (frameshifts), 2 missense, and 3 splice-site mutations. Here, only one out of the seven mutations was of familial origin, with the rest being de novo. Four of those had been reported previously, whereas 3 were novel (Table 2). The mutations identified by Sanger sequencing were not clustered on a particular exon in either of the genes, whereas no Copy Number Variants were detected in TSC1 or TSC2 using MLPA analysis. Finally, only one out of the four TSC families identified here presented with multiple (>2) members affected by TSC1 disease. This family is of interest as the affected individuals present with significant phenotypic differences in clinical characteristics (Fig. 1), suggesting that additional factors interfere with development of the disease phenotype.

Family n° 10: Mutation c.1681_1700del20 leads to different clinical characteristics in the affected family members.
Characterization of the additional variants identified
In this study, apart from the reported mutations with obvious or inferred pathogenic activity, additional variants have been identified. In total, 25 additional variants have been detected, from which 9 were found in TSC1 and 16 in TSC2, whereas 4 among them have not been reported previously (1 at the TSC1 locus and 3 at TSC2). These additional variants fell into two groups: (1) TSC1 and TSC2 variants co-existing with a TSC-causing mutation already detected in the bearers (Tables 3); and (2) TSC1 and TSC2 variants in patients with no TSC mutation identified (NMI) (Table 4).
In total, 17 out of the 25 additional variants identified were intronic. Since these could possibly have pathogenic activity due to their possible involvement in alternative splicing, we have screened LOVD and ClinVar for all additional variants, and found that in a significant percentage they have been characterized as benign or likely benign (Tables 3 and 4). As a conclusion, it is likely that most of the additional variants identified in this work that are also present in LOVD and/or ClinVar, are polymorphisms with no apparent clinical significance.
Genotype-Phenotype Correlations
Definite vs possible TSC cases
By definition, possible TSC patients present few symptoms, and very likely, a milder disease expression. Indeed, this was the case with our study, where among patients with possible TSC, displaying a generally milder disease phenotype, only 2 were found to bear a disease-causing mutation and this was detected in the TSC1 gene.
TSC1 vs TSC2 disease
Generally, definite TSC2 patients presented with a more severe phenotype compared to definite TSC1 patients. While cortical dysplasias were often seen in both TSC1 and TSC2 disease (TSC1: 5/6 or 83%; TSC2: 5/7 or 71%), development of SEGAs was a characteristic of TSC2 probands only (TSC1: 1/5 or 20%; TSC2: 5/7 or 71%). Moreover, epileptic seizures were more severe in TSC2 patients, and this was followed by more pronounced TAND characteristics. Additionally, multiple retinal hamartomas and a single case of LAM were observed in TSC2 patients only, whereas AML were present in both TSC1 and TSC2 patients in rather similar percentages (TSC1: 2/6 or 33%; TSC2: 3/7 or 43%).
Familial vs de novo TSC mutations
A significantly higher percentage of patients with de novo TSC mutations (69%) were identified compared to familial cases (31%). Of the 4 familial mutations reported here, 3 (75%) were detected in TSC1 and 1 (25%) in TSC2. In the three TSC1 families (6, 10 and 14), the pathogenic variant was passed on by the father. More specifically, all families suffered from premature termination of protein synthesis, with families 6 and 10 having one nonsense mutation each, and family 10 bearing a deletion of 20 nucleotides, which causes a frameshift. It is interesting to note that each of the 4 affected individuals in the latter family displays a different set of clinical characteristics, ranging from fibroadenomas plus SEGA with no epilepsy to SEGA with uncontrollable seizures, severe mental retardation and autism, all induced by the same TSC1 mutation (Fig. 1).
On the contrary, in TSC2 family 11, the mutation was transmitted by the mother, and it was a splice-site mutation. In this family, in order to determine whether the c.5160 + 5 G > Τ variant in intron 39 could be the cause of a splicing error, RT-PCR was performed on peripheral blood lymphocyte RNA obtained from the proband. Sequencing of the RT-PCR product revealed that this single nucleotide substitution was enough to induce skipping of TSC2 exon 39 (Fig. 2).

Family n° 11: RT-PCR analysis followed by Sanger sequencing. (a) Pedigree, (b) RT-PCR analysis, (c) Sanger sequencing of wild-type versus mutant cDNA, (d) wild-type sequence of TSC2 exons 38–40. Due to the c.5160+5 G > T splice-site mutation, exon 39 is absent from the mRNA produced by the mutant allele.
Among the 9 de novo cases, 3 TSC1 (33%) and 6 TSC2 (67%) mutations were found. In total, we have identified 4 frameshift, 3 nonsense, 3 missense and 3 splice-site mutations.
Prediction of structural consequences of TSC1 and TSC2 missense mutations
In order to assess possible pathogenicity of the Ile1648Phe (I1648F) TSC2 missense variant, which is presented in this work for the first time, we compared 3D-models of Ile1648Phe TSC2 variant versus wild-type, but also of p.Arg246Lys (R246K) TSC1 missense variant versus wild-type, as an evaluation of our prediction.
To investigate the effect of the TSC1 p.Arg246Lys missense mutation at the protein level, we first produced 3D-models of the core domain of wt hTSC1 protein and of its R246K variant, as described in Material & Methods. As shown in Fig. 3, the 3D-model of the TSC1 R246K variant is very similar to that of the wt protein. In addition, the 246 Arg/Lys side chains are involved in intra-molecular interactions in both models (Fig. 3). It is therefore unlikely that this sequence change affects either the structure or the interactions of the TSC1 protein. These observations are in line with data in the literature which show that the Arg246Lys substitution does not affect TSC1 function, and suggest that the effect of the TSC1 p.Arg246Lys mutation is rather a result of alternative splicing12.

Prediction of possible structural consequences of TSC1 R246K missense mutation: Superposition of the final 3D-models (energy minimized average MD models) of the core domain (aa: 1–262) of wt hTSC1 and of its variant, R246K. The region of the mutation is boxed and magnified (right panel) for each one of the models, for reasons of clarity.
On the contrary, as shown by mapping of the Ile1648Phe change on the 3D-model of the catalytic domain of TSC2 produced as described in Materials & Methods section (Fig. 4), substitution of the Ile residue at position 1648 by the much bulkier Phe residue, is anticipated to disrupt the structural integrity of this TSC2 domain due to steric hindrance with hydrophobic residues of the region (shown in grey sticks in Fig. 4). Therefore, it is most likely that the TSC2 p.Ile1648Phe mutation, by disrupting the structure of the catalytic domain of the TSC2 protein, impedes its GAP activity.

Prediction of possible structural consequences of TSC2 I1648F missense mutation: Details of the 3D-model of the catalytic domain of TSC2 at the Ile 1648 region for the wt (left panel) and the I1648F variant (right panel). The protein is depicted as ribbon model, including α-helices and β-strands. The amino acid 1648 is shown in ball-and-sticks and hydrophobic residues of the region are depicted as grey sticks.
Probands/families with no mutation identified (NMI)
In this work, in 7/20 probands/families, pathogenic mutations could not be identified. All 7 cases were de novo, with relatively mild symptoms of the disease. Nevertheless, since NMI refers to definite TSC patients11 and given the fact that here only 2 out of the 7 were definite TSC cases, with the remaining 5 being characterized as possible TSC, only these 2 are clear NMI, whereas the rest could be alternatively suffering from a disease other than TSC.
Generally, in NMI cases, pathogenic mutations could have been missed mainly because (a) they are found in genomic areas of TSC1 or TSC2 that are not covered during genetic analysis; or (b) the individuals are mosaics with just a small percentage of cells with a mutated TSC1 or TSC2.
In studies similar to the present one, where genetic testing is carried out in a diagnostic setting, usually, analysis of promoter regions, 5′- and 3′-UTRs and deep intronic areas of TSC1 and TSC2 genes is not included13,14,15,16. In a few studies, where TSC1 and TSC2 promoter region analysis has been performed, the levels of mutations detected were either very low or null17,18.
The main reasons for exclusion of the above mentioned genomic areas in usual TSC genetic analysis are the limitations imposed by direct Sanger sequencing, but also the fact that in the majority of the patients, pathogenic mutations are detected in the exons and the intron/exon boundaries of the TSC genes14,19,20.
Nevertheless, nowadays, there is a trend towards the use of NGS-based technologies in TSC genetic analysis, exactly due to the fact that these new methods have the ability to cover readings of the whole length of TSC1 and TSC2 genomic areas, including the promoters, UTRs, and whole intron sequences, but also because of their sensitivity, where mutations can be detected in the presence of even only 9% of the minor allele11,17,21. Of course, on the other hand, the use of NGS-based genetic analysis cannot solve the problem of the variants of unknown clinical significance (VUS) and eliminate the need for functional analysis11,17,21.
Although the major contributions on TSC mutation scanning have been based on Sanger sequencing and less than 10 NGS papers have appeared in the TSC literature until this day, one cannot ignore the advantages of NGS in TSC genetic analysis over Sanger sequencing, which is posing some inherent limitations in our study.
Conclusions
In this study, TSC1 and TSC2 disease percentages were rather similar (46% vs 54%), but detection of mutations proved more effective in patients having definite TSC (85%) than in patients having a possible TSC diagnosis (29%). Five new mutations were identified, while TSC1 disease presented with a milder phenotype, consistent with previous reports14,19,20. Most TSC2 mutations identified were de novo (86%). This was probably due to the more severe TSC2 disease phenotype, which likely prevents these individuals from having a family. In agreement with the above, most familial cases (75%) had TSC1 disease, likely due to its milder phenotype. The same was observed for all the patients with possible TSC diagnosis (100%), and the single family with multiple affected members. Nevertheless, familial cases could be slightly underrepresented in our study, since in some families only one of the parents was available for genetic testing. Finally, because TSC is one of the few rare diseases for which a targeted drug therapy is available, a more accurate genetic testing protocol should be introduced in order to help uncover the underlying molecular events in NMI individuals. A closer collaboration of scientists with TSC patient groups worldwide will probably shed more light on genotype/phenotype correlations in the near future, in the direction of improving the quality of life of patients suffering from TSC and families.
Patients and Methods
Patients
The study protocol and the informed consent forms were approved by the Bioethics Committee of NCSR “Demokritos”. All patients referred to the laboratory of Environmental Mutagenesis and Carcinogenesis, Molecular Diagnosis of Genetic Diseases Project, for genetic testing. In four cases (families 7, 8, 9 and 11) prenatal diagnosis has also been performed. Before molecular diagnosis, written informed consent forms were signed by all probands or parents, whereas after analysis, families were informed in detail on the outcome of the genetic test. Finally, the study was in agreement with the 1975 Helsinki statement, revised in 1983.
Mutation detection
Genomic DNA was extracted from peripheral blood lymphocytes according to the standard saturated salt-chloroform extraction protocol. Purity and concentration of isolated DNA were measured using a NanoDrop™ spectrophotometer, while the quality of genomic DNA was evaluated through agarose gel electrophoresis. The entire translated regions of TSC1 (exons 3 to 23) and TSC2 (exons 1 to 41) were PCR-amplified and then directly sequenced using the Sanger method. All primer sequences and PCR conditions used are shown in Tables S1 and S2 (see Supplemental Materials and Methods). Cycle sequencing reactions were performed using the v3.1 BigDye Terminator Cycle Sequencing kit (Applied Biosystems, Foster City, CA), and then analysed on an ABI Prism® Genetic Analyzer. Sequences obtained were aligned against reference sequences from the Genbank (Accession Numbers: NG_012386.1 (TSC1) and NG_005895.1 (TSC2)), and examined for the presence of variants. Family members of mutation carriers were being informed in counselling sessions, and if they consented, they were subjected to genetic analysis for the specific mutation. The origin of mutations (inherited or de novo) was inferred after testing both parents (when available). No paternity test was performed.
Confirmation of the presence of an aberrant mRNA splice variant by RT-PCR analysis followed by DNA sequencing
In order to test for possible splicing anomalies within TSC2 mRNA transcripts (family 11), we performed total RNA extraction from peripheral blood lymphocytes, using ΤRI REAGENT (Molecular Research Center Inc, Cincinnati, OH). Subsequent cDNA synthesis was carried out using M-MLV RT (Life Technologies, Carlsbad, CA). In the particular family, RT-PCR analysis was performed with the help of a forward primer on exon 38 and a reverse primer on exon 41. RT-PCR products were sequenced and analysed on an ABI Prism® Genetic Analyzer.
Multiplex ligation-dependent probe amplification (MLPA)
Possible large rearrangements in TSC1 and TSC2 were assessed by Multiplex Ligation-dependent Probe Amplification (MLPA) using the SALSA MLPA P124 and SALSA MLPA P046, respectively, following manufacturer’s instructions (MRC-Holland, Netherlands).
Molecular Modeling of missense mutations
The 3D-model of the core domain of human TSC1 (aa: 1–262) was obtained from the Swiss_Model22 and was based on the known crystal structure of the TSC1 core domain from S. pombe 23 (PDB entry: 1KK0). The initial models of both the wild-type (wt) hTSC1 core domain and of its R1246K variant were subsequently subjected to molecular dynamics (MD) simulations in explicit water, using a procedure similar to that applied in Voukkalis et al. 201624. Three independent, 50 ns long MD simulations were performed for each molecule. The 3D-model of the GAP domain of human TSC2 (aa: 1502–1756) was constructed using a combination of the Swiss-Pdb Viewer program25 and the Phyre server26. The known crystal structure of the Rap1GAP catalytic domain27 (PDB entry: 1SRQ) was used as template, for this purpose.
Data availability
Data are available upon request.
References
Northrup, H., K, M. K., Pearson, D. A. & Au, K. S. In GeneReviews(R) (eds R. A. Pagon et al.) (2015).
Leung, A. K. & Robson, W. L. Tuberous sclerosis complex: a review. J Pediatr Health Care 21, 108–114, https://doi.org/10.1016/j.pedhc.2006.05.004 (2007).
Roach, E. S. Applying the Lessons of Tuberous Sclerosis: The 2015 Hower Award Lecture. Pediatr Neurol 63, 6–22, https://doi.org/10.1016/j.pediatrneurol.2016.07.003 (2016).
Roach, E. S., Gomez, M. R. & Northrup, H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol 13, 624–628 (1998).
Northrup, H. & Krueger, D. A. & International Tuberous Sclerosis Complex Consensus, G. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 Iinternational Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol 49, 243–254, https://doi.org/10.1016/j.pediatrneurol.2013.08.001 (2013).
European Chromosome 16 Tuberous Sclerosis, C. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 75, 1305–1315 (1993).
van Slegtenhorst, M. et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 277, 805–808 (1997).
Tee, A. R., Manning, B. D., Roux, P. P., Cantley, L. C. & Blenis, J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol 13, 1259–1268 (2003).
Dibble, C. C. et al. TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol Cell 47, 535–546, https://doi.org/10.1016/j.molcel.2012.06.009 (2012).
Tee, A. R. et al. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci USA 99, 13571–13576, https://doi.org/10.1073/pnas.202476899 (2002).
Tyburczy, M. E. et al. Mosaic and Intronic Mutations in TSC1/TSC2 Explain the Majority of TSC Patients with No Mutation Identified by Conventional Testing. PLoS Genet 11, e1005637, https://doi.org/10.1371/journal.pgen.1005637 (2015).
Nellist, M. et al. Missense mutations to the TSC1 gene cause tuberous sclerosis complex. Eur J Hum Genet 17, 319–328, https://doi.org/10.1038/ejhg.2008.170 (2009).
Sancak, O. et al. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype–phenotype correlations and comparison of diagnostic DNA techniques in Tuberous Sclerosis Complex. Eur J Hum Genet 13, 731–741, doi:5201402 [pii] 10.1038/sj.ejhg.5201402 (2005).
Au, K. S. et al. Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the United States. Genet Med 9, 88–100, doi:10.1097GIM.0b013e31803068c7 00125817-200702000-00006 [pii] (2007).
Niida, Y. et al. Mutational analysis of TSC1 and TSC2 in Japanese patients with tuberous sclerosis complex revealed higher incidence of TSC1 patients than previously reported. J Hum Genet 58, 216–225, https://doi.org/10.1038/jhg.2013.3 jhg20133[pii] (2013).
Yu, T. et al. Novel TSC1 and TSC2 gene mutations in Chinese patients with tuberous sclerosis complex. Clin Neurol Neurosurg 154, 104–108, https://doi.org/10.1016/j.clineuro.2017.01.015 (2017).
Nellist, M. et al. Targeted Next Generation Sequencing reveals previously unidentified TSC1 and TSC2 mutations. BMC Med Genet 16, 10, https://doi.org/10.1186/s12881-015-0155-4 (2015).
Ali, M. et al. Mutation and polymorphism analysis of TSC1 and TSC2 genes in Indian patients with tuberous sclerosis complex. Acta Neurol Scand 111, 54–63, https://doi.org/10.1111/j.1600-0404.2004.00366.x (2005).
Jones, A. C. et al. Comprehensive mutation analysis of TSC1 and TSC2-and phenotypic correlations in 150 families with tuberous sclerosis. Am J Hum Genet 64, 1305–1315, doi:S0002-9297(07)62275-7 [pii]10.1086/302381 (1999).
Dabora, S. L. et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet 68, 64–80, doi:S0002-9297(07)62472-0 [pii]10.1086/316951 (2001).
Martin, K. R. et al. The genomic landscape of tuberous sclerosis complex. Nat Commun 8, 15816, https://doi.org/10.1038/ncomms15816 ncomms15816 [pii] (2017).
Arnold, K., Bordoli, L., Kopp, J. & Schwede, T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201, https://doi.org/10.1093/bioinformatics/bti770 (2006).
Sun, W. et al. Crystal structure of the yeast TSC1 core domain and implications for tuberous sclerosis pathological mutations. Nat Commun 4, 2135, https://doi.org/10.1038/ncomms3135 (2013).
Voukkalis, N. et al. SRPK1 and Akt Protein Kinases Phosphorylate the RS Domain of Lamin B Receptor with Distinct Specificity: A Combined Biochemical and In Silico Approach. PLoS One 11, e0154198, https://doi.org/10.1371/journal.pone.0154198 (2016).
Guex, N. & Peitsch, M. C. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18, 2714–2723, https://doi.org/10.1002/elps.1150181505 (1997).
Kelley, L. A. & Sternberg, M. J. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 4, 363–371, https://doi.org/10.1038/nprot.2009.2 (2009).
Daumke, O., Weyand, M., Chakrabarti, P. P., Vetter, I. R. & Wittinghofer, A. The GTPase-activating protein Rap1GAP uses a catalytic asparagine. Nature 429, 197–201, https://doi.org/10.1038/nature02505 (2004).
Acknowledgements
This paper is dedicated to the memory of Katerina Lambrou. The authors wish to thank the Tuberous Sclerosis Association of Greece for the fruitful collaboration. This work was performed in the framework of “Target Identification for Disease Diagnosis and Treatment (DIAS)” project within GSRT’s KRIPIS action, funded by Greece and the European Regional Development Fund of the European Union under the O.P. Competitiveness and Entrepreneurship, NSRF 2007–2013. Additional financial support to G.E.V. was provided by the Greek Secretariat for Science and Technology (GSRT program 187 gamma), and the American College of Greece (Deree).
Author information
Authors and Affiliations
Contributions
S.A. was in contact with the TSC families, prepared Tables 1 and 2, prepared Figs 1 and 2, and carried out a significant part of the sequencing work. F.F. carried out part of the sequencing work, and part of the MLPA experiments. A.V. carried out part of the MLPA experiments. Y.N. carried out database searches and prepared Table 3. A.D. carried out database searches and prepared Table 4. R.Vo. carried out database searches. R.Vr. carried out database searches. S.Y. provided part of the clinical data included in Table 1. D.J.S. was in contact with the TSC families and contributed to the writing of the manuscript. M.V. carried out prediction analysis for TSC1 R246K and TSC2 I1648F variants and prepared Figs 3 and 4. A.A. wrote part of the manuscript. D.Y. was in contact with the TSC families and the Tuberous Sclerosis Association of Greece, and supervised part of the work. G.E.V. was in contact with the TSC families and the Tuberous Sclerosis Association of Greece, designed the primers, supervised part of the work, coordinated the team and wrote the main manuscript text. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Avgeris, S., Fostira, F., Vagena, A. et al. Mutational analysis of TSC1 and TSC2 genes in Tuberous Sclerosis Complex patients from Greece. Sci Rep 7, 16697 (2017). https://doi.org/10.1038/s41598-017-16988-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-16988-w
This article is cited by
-
Clinical practice recommendations for kidney involvement in tuberous sclerosis complex: a consensus statement by the ERKNet Working Group for Autosomal Dominant Structural Kidney Disorders and the ERA Genes & Kidney Working Group
Nature Reviews Nephrology (2024)
-
Tsc2 mutation rather than Tsc1 mutation dominantly causes a social deficit in a mouse model of tuberous sclerosis complex
Human Genomics (2023)
-
Identification of TSC2 mosaic mutation limited to cortical tuber with TSC targeted sequencing: a case report and literature review
Child's Nervous System (2021)
-
First comprehensive TSC1/TSC2 mutational analysis in Mexican patients with Tuberous Sclerosis Complex reveals numerous novel pathogenic variants
Scientific Reports (2020)
-
Whole exome sequencing identifies a novel intron heterozygous mutation in TSC2 responsible for tuberous sclerosis complex
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
