
Abstract
In marine oxygen (O2) minimum zones (OMZs), the transfer of particulate organic carbon (POC) to depth via the biological carbon pump might be enhanced as a result of slower remineralisation under lower dissolved O2 concentrations (DO). In parallel, nitrogen (N) loss to the atmosphere through microbial processes, such as denitrification and anammox, is directly linked to particulate nitrogen (PN) export. However it is unclear (1) whether DO is the only factor that potentially enhances POC transfer in OMZs, and (2) if particle fluxes are sufficient to support observed N loss rates. We performed a degradation experiment on sinking particles collected from the Baltic Sea, where anoxic zones are observed. Sinking material was harvested using surface-tethered sediment traps and subsequently incubated in darkness at different DO levels, including severe suboxia (<0.5 mg l−1 DO). Our results show that DO plays a role in regulating POC and PN degradation rates. POC(PN) degradation was reduced by approximately 100% from the high to low DO to the lowest DO. The amount of NH4 + produced from the pool of remineralising organic N matched estimations of NH4 + anammox requirements during our experiment. This anammox was likely fueled by DON degradation rather than PON degradation.
Similar content being viewed by others


Introduction
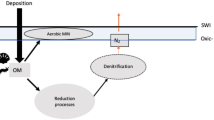
Concentrations of dissolved oxygen (DO) may decline in the future ocean, potentially leading to an expansion of oxygen minimum zones (OMZs)1,2. Although earth system models do not all project increased hypoxia3,4 and although the causes of this expansion are still debated5, over the past 50 years supporting evidence for this occurring has been gathered globally6. In well-oxygenated waters, temperature7,8 and plankton community structure9,10,11,12 are known as the main factors affecting the particulate organic carbon (POC) degradation rate. In oxygen deficient waters however, the importance of temperature and plankton community on remineralisation rate may differ as oxygen itself may play a role13,14,15,16,17. Several studies13,15,16,17 in OMZs have based their estimates of POC degradation rate only on the attenuation of particle flux18. Only refs14,17 used direct estimates of particle degradation by looking at dissolved CO2 variations in enclosed degradation experiments. Also, the quality of the organic matter may play a role in dictating the sensitivity of degradation as the remineralisation rate of N-rich amino acids seems to be less affected by DO deficiency than that of non amino acid POC components15,19.
In OMZs, enhanced POC fluxes could be explained by (1) the fact that microbial respiration using non-O2 electron acceptors may not be as energetically efficient as in oxygenated waters, leading to lower particles remineralisation rates, or by (2) the low abundance in zooplankton may lead to the loss of the packaging function. This would in turn reduce the amount of fast sinking particles20 produced from a pool of slow sinking (or suspended) particles present within the OMZ via grazing and production of fecal pellets.
It is therefore not clear whether previously observed trends in POC degradation rate in the studies cited above were only caused by the low DO concentrations or by differences in ecosystem structure and/or temperature and/or organic matter quality. To mechanistically test the impact of DO concentrations on POC degradation rate, one would therefore need to examine the degradation of POC under experimental conditions where only the DO concentration is changed. This implies the need for a survey of two regions where oxygen varies but all other environmental conditions such as temperature, organic matter quality, and important aspects of community structure such as bacterial, phytoplankton and zooplankton composition are fixed. This is however challenging, given that sediment trap deployments in the ocean are technically difficult to perform and artificial aggregates often differ considerably from natural ones.
Remineralising particulate nitrogen (PN) fuels NH4 + production that subsequently sustains the production of N2 gas (via anammox). Whether anammox or denitrification is the dominant process in OMZ is still debated21,22,23. Sinking OM could thus be degraded through alternative pathways17. More precisely, the heterotrophic microbial community in OMZs preferentially uses N-rich OM, leading a strong decoupling between C and N. This preferential use of N-rich organic matter could be responsible of a large loss of N2 from the ocean. However, PN flux estimates (and subsequent NH4 + production from remineralization) may not be sufficient to fully sustain the observed loss of N2 induced by anammox and denitrification24. This suggests that either (1) another source of NH4 + production occurs in oxygen deficient zones, with microaerobic respiration proposed as a source of ammonium during organic matter degradation24, or (2) estimates of NH4 + production rate from remineralisation of PN presented in ref.21 could be biased low, either via underestimation of surface PN export/remineralisation rate or omission of NH4 + production from the DOM pool which is, on occasion, supplied below the oxycline25. Collectively, this highlights the high current uncertainty in the contribution of PN degradation to the N budget in OMZ.
The discussion above provides motivation for direct measurements of export and OM remineralisation in oxygen deficient areas. Here, we present results from a bottle degradation experiment testing the influence of DO concentration on POC and PN degradation. Sinking material was collected using surface tethered sediment traps in the Gotland Deep (Baltic Sea), where DO concentrations are close to detection limit at 100–150 m. Harvested sinking material was subsequently incubated for 6 days in seawater, replete with associated microbial communities, collected at the same depth as the sediment traps in order to test the effect of different DO concentrations on POM degradation. Particulate OC (POC) and N (PN) were monitored every other day alongside the geochemical quality of the organic matter (using a degradation index26) and the bacterial abundance and biomass. Nutrients concentrations were also measured and used as indicators of OM microbial regeneration pathways.
Results and Discussion
Biogeochemical conditions
surface satellite-derived Chl-a concentrations in the Gotland Deep peaked in mid-June (8–10 mg m−3, Julian day 160) during sampling (Fig. 1A). Chl-a concentrations increased constantly from mid-May (Julian day 135) to the sampling period. Figure 1B shows vertical profiles of temperature (°C), Chl-a (mg m−3) and DO concentration (mg l−1). Surface temperature (10 °C) dropped to 5 °C between depths of 35 and 75 m. Temperature then increased to 8 °C below 75 m and remained stable down to 220 m. Chl-a peaked in the subsurface (2–3 mg m−3) and decreased to background levels below 35 m. DO concentrations decreased significantly below 60 m, reaching a minimum of 0.4 mg l−1 at 110 m, which is typical for the deep Baltic Sea27. However, below 140 m DO concentrations increased up to 3 mg l−1. This was likely due to an inflow of dense North Sea water that had previously entered the Baltic Sea during the strong winter storms observed in late 2014–early 201528,29.

(A) Satellite chlorophyll a concentration time series (MODIS) centered around the sampling site (see methods). Days are in Julian days and the trap deployment period is highlighted in grey. Vertical profiles of (B) temperature, fluorescence-derived chlorophyll-a, and DO, (C) nitrite, nitrate and phosphate, at the sampling site. Note the log10 scale used in panel (C).
Phosphate (PO4 3−) and nitrate (NO3 −) concentrations increased with depth down to 75 m (2.3 and 6.1 µmol l−1 respectively). Below 75 m, phosphate remained constant, but NO3 − decreased strongly down to 0.1 µmol l−1 at the depth of the DO concentration minimum (110 m). Nitrite (NO2 −) concentration peaked at the same depth, i.e. 110 m with 0.1 µmol O2 l−1. This suggests a preferential use of NO3 − relative to PO4 3−. Plotting our estimated Baltic N* values30 (see methods) versus DO concentration (Figure S1), it clearly appears that the excess of PO4 3− is related to DO deficiency in the water column. Along with the nitrite accumulation observed at 110 m (Fig. 1C), this suggests that losses of N, likely through denitrification processes related to low DO concentrations, occurred in the water column.
In the following section, we describe the effect of DO concentration on the degradation of sinking POM. We collected sinking material using surface tethered sediments traps at various DO levels including oxic, suboxic and anoxic concentrations (Fig. 1B) corresponding to the following depths (40, 60, 110, 180 m)31. We then incubated (12 °C, complete darkness, 6 days) sinking material in in situ water (collected with CTD rosette) in gas-tight glass bottles, keeping the DO levels at similar concentrations as in the water column (at 40, 60, 110, 180 m depths, Fig. 1B). Particles were maintained in constant suspension using a modified plankton wheel rotating at 2 rpm (rotation per minute). We estimated how quantitative our sampling procedure was by looking at the variations in total Si (dissolved silicate + biogenic silica concentrations). Our attempt at completing a budget for Si failed due to uncertainties related to the sampling procedure, which ranged from 0 (day 4, 40 m) to 22% (day 4, 60 m). On average, uncertainties due to the sampling procedure was 4% in the 40 m samples, 12% in the 60 m samples, 8% in the 110 m samples, and 3% in samples collected at 180 m (Figure S2). These errors were estimated from the duplicates samples of both DSi and BSi (Figure S2).
Organic matter dynamics during the incubation experiments
Figures 2A and B present the variations of POC and PN concentration (µmol l−1) in the incubation bottles over the time course of the experiment. At the start of the experiment DO concentrations in experimental bottles corresponding to depths of 40, 60 110 and 180 m were 10.0, 11.4, 0.5, and 5.7 mg l−1 respectively. Although these are slightly different from the DO concentrations observed on site, they correspond to the depth pattern measured in the water column within the vicinity of the trap sampling site31 (Fig. 1B). The concentration of DO measured in individual artificial aggregates has previously been found to decrease from the periphery to the core of artificially produced aggregates32. Suboxia has been observed within the core of aggregates when the concentration of DO in the background water fell below 1.9 mg l−1 33. We thus assume anoxia within the sinking particles collected at 110 m in our experiment. However, sinking particles collected in the 180 m treatment may still have had sufficient DO concentrations for microbes to perform aerobic respiration (Fig. 1B) as previous studies34 have showed that microbial activity switches from oxic respiration to N-based respirator pathways at concentrations less 0.1 mg l−1.

Variations in (A) POC and (B) PN concentrations (µmol l−1) over the course of the experiment. Error bars indicate the standard deviation from the duplicate samples. Circles represent 40 m samples, squares 60 m, inverted triangles 110 m and triangles 180 m samples. The DO concentration (mg l−1) in each bottle (when taken down) is indicated by the colour scale in both (A) and (B) panels.
At day 0, POC and PN concentrations varied between the different depth treatments (POC: 148 µmol l−1 for 40 m to 172 µmol l−1 for 110 m, Fig. 2A; PN: 16 µmol l−1 for 180 m to 20 µmol l−1 for 60 m, Fig. 2B). However, POC(PN) concentrations varied widely during the first days of the experiment, including increases in POC(PN) (Fig. 2A). Such increases in enclosed systems (the so-called ‘bottle effect’) is on occasion observed in the initial days of such experiments before stabilization11,12. This may be associated with constant exchanges of the organic carbon (OC) between the dissolved and the particulate phase and/or bacterial growth12.
In order to assess the variability in the concentration of POC(PN) with time during our experiment, we performed repeat linear regressions (POC, PN, DOC concentrations vs time) that included and excluded low DO samples (110 m). The results from the regressions are summarized in Table S3. We found that the concentration of DOC was significantly correlated (p < 0.05) with time (Table S3) at all depths and also when excluding 110 m samples. Their respective slopes (Table S3) were however not significantly different. When considering all time steps, no significant relationships were found between both POC:PN ratios, POC(PN) concentrations and time (Table S3). We also tested our results using data from days 4–6 only, based on the patterns observed in Fig. 2A and B. Using these time steps no correlations were found between POC:PN ratios or DOC concentration and time. Likewise, no correlations were found between POC(PN) concentrations and time. However, when 110 m samples were excluded from the regressions, POC(PN) concentrations showed significant decreases with time (Table S3).
Processes of POM aggregation/dissolution and DOM adsorption/desorption are generally considered as being a function of ecological factors, such as mineral content of the sinking particles, but microorganisms can also participate in DOM/POM exchanges via alteration of the chemical nature of OM35 and solubilizing POM into DOM through cell lysis and release of ectoenzymes. Whilst we can exclude particle disaggregation by zooplankton, which we purposely eliminated here (see methods), disaggregation induced by the degradation of sticky organic matter by microbes cannot be excluded36. Abiotic factors such as sunlight can be ruled out in our experiment. Also the mineral content, such as biogenic silica, in the sinking particles was relatively low during at the study site31 in comparison to fluxes in other ocean provinces37,38, so we assumed little interaction of this component. The content of transparent exopolymere particles (TEP)39 and coomassie stainable particles (CSP)40 (Fig. 3) could be used to qualitatively estimate the transfer OM from the dissolved to particulate phase and could potentially explain the large augmentation in POM vs DOM during the initial part (days 0 to 4) of the experiment (note that the augmentation observed at 60 m day 4 is likely due to a quantitative sampling issue, as shown in Figure S2). We believe that DOM coagulation played an important role during our experiment. POM concentration increases (Fig. 2) were often accompanied by increases in C:N ratios. This supports the idea that DOM (C:N = 16.3 ± 1.6 on average) was adding to the POM pool (C:N = 8.2 ± 0.5 on average).

Content of transparent expolymers (TEP) and Coomassie Stainable Particles (CSP) at day 6 in µm−2 l−1 in the different depth treatments.
Bacterial growth is another factor that likely influenced POM concentrations during the experiment. Using cell counts, we estimated the amount of C resulting from bacterial growth using cell to carbon conversion factors41,42. Figure 4A shows the abundance of bacteria in the various samples during the experiment. The abundance of bacteria increased in all treatments, plateauing at day 2 in 110 m and 180 m treatments and day 4 in 40 m and 60 m treatments. We observed bacterial growth during our experiment, suggesting that particle associated bacteria recovered from exposure to the brine during collection. In the incubation bottles the salinity was close to in situ conditions given that particles were diluted in seawater collected at the same depths as the particles (see methods). The largest increase in bacterial abundance was observed in samples from 40 m, while the smallest increase was observed in 110 m samples. Bacterial abundance in samples from 60 m closely followed the pattern observed in 40 m samples, whereas abundance in samples from 180 m showed similar variations as the 110 m samples (Fig. 4A). We chose a conversion factor of 20 fgC cell−1 42 to estimate the amount of carbon added to what we considered as POC (larger than 0.7 µm, see methods). The upper(lower) ranges using a conversion factor of 50(2) fgC cell−1 41 are presented in Fig. 4B,C,D,E. Increases in organic carbon (OC) generated by bacterial growth from day 2 to day 6 (using 20 fgC cell−1) were 11, 6, −5 and 1 µmol l−1 for sample taken at 40, 60, 110 and 180 m respectively (Fig. 4B,C,D,E). Using the lower cellular carbon conversion factor (2 fgC cell−1) lowers these numbers 10 fold (Fig. 4B,C,D,E). Using the upper conversion factor (50 fgC cell−1), generates increases of 27, 15, -12 and 2 µmol OC l−1 for samples taken at 40, 60, 110 and 180 m respectively. Given our observed increase in POC of about 10 µmol l−1 in the 40 m samples, the conversion factor of 20 fgC cell−1 is appears the most applicable to use. Between day 4 and day 6 bacteria abundance was constant and hence contributed to no OC generation (Fig. 4A). The growing and changing microbial community (Fig. 4A) from day 0 to day 4 could have produced a significant amount of POC, as explained in the paragraph above (Fig. 4B–E). We however show that microbial community growth (and therefore potential creation of POC) was reduced after day 2, since the bacteria abundance was steady during three measurement time steps (day 2, 4, 5 and 6).

(A) Abundance of bacteria (cell ml−1) and associated standard deviation from duplicates. Black line represents 40 m samples; red, 60 m; blue, 110 m and green, 180 m. The estimated equivalent in carbon (µmol l−1) is presented in (B) for 40 m, (C) for 60 m, (D) for 110 m and (E) for 180 m. The bold line represents estimates following cell biomass conversion factor42, the shaded area around the line represents for lower and upper values using lower and values for the cell biomass conversion factor as described in ref.41. See text for more detailed information.
We therefore use degradation calculated between days 2 and 6. The bacterial community appeared to have reached a steady state condition from day 4 onwards, but DOM coagulation seemed to have occurred throughout the entire duration of our experiment. For both these reasons we chose to restrict our examination of POM degradation by looking at data from day 2, day 4, day 5 and day 6, and ignoring data from day 0. We do acknowledge that by adopting this approach we lose characterization of the initial step of degradation; however this is not of great concern for this study where the focus is on testing the mechanistic impact of DO concentrations on POM degradation rather the absolute consumption rates.
POM degradation
We distinguished differences between the treatments by plotting losses of POC (Fig. 5A, Table S1) and PN (Fig. 5B, Table S2) in µmol l−1 as calculated from the difference in POC(PN) concentrations between days 2 and 6 (see justification above). Additionally, we calculated the losses of TOC and TN also as determined from the difference in TOC(N) concentrations during the experiment (Fig. 5).

Losses of OC (A) and ON (B) in µmol l−1 as calculated from the difference in OC(N) concentrations relative to day 2. Circles represent TOC(N) and squares POC(N). Black lines/symbols represent 40 m samples; red, 60 m; blue, 110 m and green, 180 m.
POC loss (Fig. 5A) was the largest in 60 m depth samples (−37 µmol l−1). Surprisingly, ΔPOC2–6 was positive in the 40 and 110 m depth samples (9 and 7 µmol l−1 respectively). Our quantitative Si budget (Figure S2) suggested that the bottles corresponding to day 4 at 60 m had the largest deviation of all, suggesting that POC concentrations in this bottle could have been overestimated. We therefore believe that the large ΔPOC2–6 observed at 60 m should be treated as a potential overestimate. PN loss rates seem to follow the same pattern as POC (Fig. 5); again ΔPN at 110 m were negative. It is unclear why ΔPOC(N)2–6 at 40 and 110 m was negative (Fig. 2C and D). Rates of POC degradation were reduced by 116% from the high DO concentration to the low DO concentration (Table S1). The reduction is higher than 100% because of the small gain of POC observed in the low DO treatment with material from 110 m depth. Similarly, the reduction in PN degradation rates was about 100% (Table S2). Looking at the variation in concentrations of TOC (total organic carbon calculated by adding POC and DOC concentrations) and TON (total organic nitrogen calculated by adding PN and DON concentrations) relative to day 2 (Fig. 5), little OC or ON is lost in the 110 m samples while the larger loss occurs in the 180 m samples. Samples from 60 m however present only a small decrease in TOC, but a larger decrease in TON (Fig. 5). Temporally, changes seem to be greater in OC relative to ON. For instance, the variations of TOC and POC loss at 60 m (Fig. 5A, red symbols) are compared to the variations of ON at the same depth (Fig. 5B, red symbols). The trends we observed in both OC and ON degradation provide two main messages: (1) the material collected at 110 m seems to experience lower degradation rates than the material collected at other depths, and (2) while samples from 40 and 60 m have dissimilar degradation rates, the OM from 180 m have experienced the most intense remineralisation over the time course of our experiment. In the following section we investigate what causes the patterns in OC and ON degradation described above.
Control on POC(PN) degradation rate, DO vs organic matter quality
As stated in introduction, DO concentration can potentially have a large impact on particle remineralisation rate13,14,16,17 through either influencing prokaryotic metabolic pathways of organic matter degradation43 or by reducing the production of fast sinking particles generated through zooplankton fecal re-packaging20. Zooplankton were purposely excluded (see methods section) during our experiment for the sake of focusing on bacterial degradation only.
To fully test the influence of DO concentrations of sinking particle degradation rate (induced by prokaryotic organisms), one would have to produce a homogenous cohort of sinking particles with similar morphological/geochemichal characteristics and then estimate their degradation rate in vitro. This was done previously using sinking particles artifically produced from phytoplankton cultures, however this presents the potential risk of including prokaryotic communities that differ from natural communities found in the field. Specifically, culture-associated prokaryotic communities11,12 differ from natural communities44,45 and are often less active in degrading sinking particles46,47. This is the reason why we here used natural aggregates. However, an implication of this is that we need to consider other parameters, in addition to DO concentration, which could potentially drive patterns in OC/ON degradation rates. We here consider an additional parameter linked to the quality of the organic matter: the degradation index (DI) which is based on the amino acid content of particles26.
Figure 6 presents the losses of TOC(N) and POC(N) at day 6 relative to day 2 (from Fig. 5) as a function of DO concentration with depth (Fig. 6A,C) and the initial organic matter DI26 of trap material (start of incubation) and incubation bottle at day 6 on a color scale. We chose to look at losses at day 6 relative to day 2 as these cover the longest period of time, hence maximizing the signal to noise ratio. Although the reproducibility of such experiments is challenging to assess, we note that our TOC(N) and POC(N) losses provide independent support for these rates of 18(2)μM and 3(1) μM respectively. This is based on regressions between TOC(N) loss and sampling days. POC(N) losses co-vary with DO concentration if we only consider samples taken at 60, 110 and 180 m. If the 40 m samples are included, the relationship collapses. For TOC(N) losses we observed no trend relative to DO concentrations.

Losses of OC and ON in µmol l−1 (calculated from the difference in OC(N) concentrations at day 6 relative to day 2) as a function of DO concentration (mg l−1). Color bars represent (A,B) the sampling depth (m), (C,D) the degradation index26 of the material observed in sediment trap material before incubation and (E,F) the degradation index26 of the material observed in the incubation bottle at the end of the experiment (day 6). Circles represent TOC(N) and squares POC(N).
We find that in contrast to DOM, DO concentration is a potential driver for POM degradation. However, the differences in POC and PN losses observed between the samples taken from 40 and 60 m (with a similar DO concentration) indicate that factors other than DO concentration played a significant role in driving the magnitude of both POC and PN losses. It is unlikely that the reduced remineralisation rates observed in 40 m samples result from a lack/delay of colonization48 by bacteria, given the enhanced bacterial biomass at this depth (Fig. 4).
We hypothesize that the differences in degradation rates between 40 m and 60 m samples may be due to the OM composition13 or rates of chemoautotrophy. OM lability as diagnosed from the DI26 is a useful metric to compare OM composition. Lower DI indicates how much more degraded the OM is and vice versa 26. Figure 6C,D show the DI in the trap material before incubation (starting conditions) and Fig. 6E,F show the DI after the incubation (day 6). The DI is consistently higher in the surface relative to greater depths in both traps and incubation samples (Fig. 6). The DI was also consistently lower in the incubation compared to the traps at all depths. The DI in bottles from 40 m is higher (-0.05) than at 60 m (-0.18), consistent with the “starting point” observed in the trap material (0.82 at 40 m and 0.67 at 60 m). Given the similarity of the DIs (both in trap material and incubation bottles) at 40 and 60 m, but the large difference in POC loss rate between 40 and 60 m, we believe that DI has a moderate control on POC degradation rate at high DO concentrations. The influence of the lability of trap material on the POC degradation rates for samples from 110 (0.63) and 180 m (0.40) is less clear given their very different DO levels (Fig. 2) and cannot be tested. We did not measured depth resolved rates of chemoautotrophy at our experiment station, however chemoautotrophy rates were positively correlated with the concentration of NH4 + in June 2015 in the Baltic Sea49. At the location of the trap deployment the vertical profile of NH4 + concentration shows that concentrations at 110 m and 40/60 m were higher than at 180 m (Fig. S4). Assuming that chemoautotrophy rates follow the depth pattern of NH4 + concentrations49 this suggests that chemoautotrophy rates were similar at 40 and 60 m, and therefore cannot explain why POC and PN losses observed at 40 m are lower than the ones observed at 60 m. We propose that one possibility could be the amount of ballast mineral (biogenic silica, terrigenous material and calcite) present at 40 and 60 m, which is known to slow down POM remineralization11,12,50,51. Unfortunately the total mass flux was not measured, which prevents any conclusions being drawn with regards to the amount of mineral ballast present at each depth.
In any case, our results show that: (1) DO concentration restricts the POC(PN) degradation rate at low DO, and (2) POM degradation rates exhibited high variability at high DO, and may have been controlled by variability in degradation state, protection by ballast mineral, the nutrient environment, amongst other factors.
Microbial remineralisation pathways
In the previous section we looked at the influence of DO concentration and OM composition on the rate at which particulate matter degraded. We now take a closer look at what may happen to the degraded particulate matter, specifically with regards to N. Although we do not have information on N transformation rates such as those provided by isotopic N compound labeling analyses21,52,53, we can investigate potential microbial metabolic pathways of remineralisation by looking at indirect evidence for N transformation rates, such as the variations in nutrients over time. We consider here the concentrations of NO3 −, NO2 − and NH4 + following the N cycle under low DO conditions proposed in ref.43. Due to the complexity of the N cycle in low DO waters (see Fig. 3 in ref.43), the variations in nutrient concentration arises from five N remineralisation pathways: (1) ammonification (remineralisation of ON into NH4 +), (2) anammox (conversion of NH4 + and NO2 − into N2), (3) denitrification (reduction of NO3 − and ON in N2O/N2), (4) nitrate reduction and (5) dissimilatory nitrate reduction to NH4 + (DNRA, reduction of NO3 − and ON into NH4 +) during our experiment.
We observed some loss in dissolved inorganic N (DIN) during our experiment (Table S2) in the bottles corresponding to 110 and 180 m samples. In samples from 40 and 60 m the total concentration of DIN increased over the course of the experiment. To further investigate the origin of this loss we plotted Fig. 7 that shows the gain/loss of NO3 −, NO2 − and NH4 + concentrations between day 4 and day 6 as a percentage of the concentration at day 6 relative to the concentration at day 4. Negative percentages represent a decrease in concentration (consumption/reduction) from day 4 to day 6, while positive percentages represent an increase over the same time scale (accumulation/production). The variation in nutrient concentration (µmol l−1) over the course of the experiment is presented in Figure S5. This approach was used to concisely and qualitatively illustrate nutrient variations during our experiment (Fig. 7). NO3 − increased or remained stable for all depth treatments apart from 180 m. It is unclear why NO3 − concentration decreased with time (Fig. 7) for the 180 m treatments. However, this has been observed in other degradation studies11 and is potentially due to the balance between the actual remineralisation of ON and the reduction of NO3 − through pathways 3, 4 and 5 (see text above). NO2 − concentration increased in all depths treatments; however, the increase observed in samples from 110 m was larger than at other depths while NO2 − concentration did not change in the 40 m samples. Similarly, NH4 + concentration increased (or remain similar) in all the oxygenated depth treatments (40, 60, 180 m) but in 110 m sample, the NH4 + concentration decreased.

Variations of NO3 −, NO2, NH4 + (as expressed as percentages of gain or loss between day 4 and day 6) over the course of our experiment. Negative percentages represent a decrease in concentration (consumption) from day 4 to day 6 while positive percentages represent an increase over the same time scale (accumulation). The variations of nutrients concentration (µmol l−1) are presented in Figure S4.
Nonetheless, in the 110 m samples the lack of accumulation of NH4 + could result from less (or no) ammonification given the small amount of ON degraded between day 4 and day 6 (see previous section). This however does not explain the consumption of NH4 +, which must result from anammox processes only. While our approach only provides indirect qualitative evidence, we speculate that anammox may have occurred in the Gotland basin. Anammox has previously been reported in the Gotland basin during some years53 while others52 found no evidence for it. One possible explanation is the stoichiometry of the OM. Loss of N through denitrification or anammox is primarily balanced by the stoichiometry of the OM. The lower the C:N ratio in sinking POM, the larger the proportion of annamox over total N loss54. It is then possible that late season N loss driven by denitrification observed in52 was due to different POM C:N ratios compared to those observed in spring (our study). In the Baltic Sea, sinking POM C:N ratios are known to show a large inter-annual variability55. This could potentially explain anammox in the Gotland basin in late spring.
It is possible to estimate anammox rates that could have occurred at 110 m in our samples. In the Baltic, anammox bacteria represent no more than 1.5% of the total abundance of bacteria53. In parallel, we know that in OMZs anammox bacteria have NH4 + consumption rates of 2–4 fmol cell−1 d−1 22,53,56. This yields a consumption of 0.2 to 0.4 µmol l−1 d−1 of NH4 + by anammox bacteria (or 0.4–0.7 µmol l−1 of NH4 + consumed between day 4 and day 6). This is comparable with the total drawdown of NH4 + (0.2 µmol l−1) observed in 110 m samples between day 4 and day 6 (Figs 7 and S4). The consumption of ON in our 110 m bottles (Fig. 2B) between day 4 and day 6 was 0.4 µmol l−1 (Figure S3, Table S2). Using the relationship21 between PN export flux and annamox rates (anammox = 0.7 X PN export) would yield an NH4 + consumption of 0.3 µmol l−1. This matches our estimation of anammox rates described above. However, the loss of ON was mainly driven by the loss of DON and not PON (Figure S3, Table S2). We therefore believe that the consumption of NH4 + by anammox observed at 110 m may have been fueled primarily by ammonification from DON and not PN.
Summary
Our results have relevance for two distinct processes. Firstly, for the drivers of the biological carbon pump, as it is currently unclear whether low DO concentration is the direct cause of reduced carbon flux attenuation observed in OMZs relative to oxygenated waters14 (and references therein). Our results suggest that:
-
(1)
DO concentration reduces the POC(PN) bacterial degradation rate at low DO concentrations, but other factors may be as important at higher concentrations. We found that the quality of the POM as determined from the DI index26 has a limited importance.
Additionally, our experiment has relevance for the N cycle. The NH4 + produced from remineralising sinking PN is speculated to fuel anammox processes resulting in loss of N2 gas from the system21. However, it is unclear whether the NH4 + supply from remineralising sinking PN is sufficient to fully sustain the observed loss of N2 induced by anammox24. We show that:
-
(2)
NH4 + supply from remineralising organic nitrogen potentially matches estimates of anammox rates in the Gotland basin.
-
(3)
DON ammonification can also be a source of NH4 + for anammox bacteria.
Methods
Study Area, sampling and analysis of ancillary data
The June 2015 cruise took place onboard the F.S. Alkor (GEOMAR) in the Gotland basin (Fig. 1). Water samples were collected with Niskin bottles via deployment of a SeaBird CTD system. Dissolved oxygen concentration (DO) profiles were taken from the CTD optode and calibrated against DO concentration measurements performed using a semi-automated whole bottle Winkler titration unit57. Oxygen concentration in the incubations bottles was measured using a calibrated OXY-4 mini from Presens. In the bottles corresponding to 110 m samples (low DO concentration see Fig. 1), the DO concentration in the bottles at the start of the experiment was measured using Pyroscience sensor TROXR430 specifically designed for low DO concentration.
Sediment traps deployment and sinking particles collection
Surface tethered sediment traps were deployed in the Gotland Deep on the 11th of June and recovered on the 13th (Fig. 1), Further details are provided in ref.31. The design of the traps and the drifting array follows58, with 12 individual Particle Interceptor Traps (PITs) mounted on a polyvinylchloride (PVC) cross frame59. Traps were deployed at four depths (40, 60, 110 and 180 m) with 12 PITs per depth. Prior to deployment, each PIT was filled with 1.5 l filtered surface seawater (0.2 µm pore size cartridge) collected from the ships underway seawater system, up to 3/4 of the PITs’s height. A brine solution was prepared by dissolving 50 g l−1 of NaCl with filtered surface seawater. This was subsequently filtered through a 0.2 µm cartridge to remove any particulate material. 0.5 l of this brine solution was then slowly pumped into each PIT with a peristaltic pump beneath the 1.5 l (3/4) of filtered seawater. Only the lowest 1/4 (0.5 l) were chosen to be filled with this solution to not lose the aspect ratio. Samples treatment we basically followed the recommendations by ref.60. Samples from each depth were flushed over a 500 µm mesh to remove zooplankton swimmers. Samples were subsequently split in aliquots five aliquots per depth, four were used for flux characterization31 and 1 for the experiment presented here. In the experiment aliquots, particles were left settling for four hours in the dark under controlled temperature (12 °C) in order to maximize the amount in particles transferred in the incubation bottles (see following section).
Dissolved organic carbon, nitrogen and silica
DOC and TDN from CTD and experiment were analyzed from 20 ml samples filtered through 0.7 µm combusted GF/F filter onto pre combusted glass vials. Samples were analyzed using a TOC-VCSH (Shimadzu) analyser following procedures described in refs61,62. Nitrates (NO3 −), nitrites (NO2 −), phosphates (PO4 3-), silicates (DSi) and ammonium (NH4 +) concentrations from CTD and experiment were measured suing similar methods as in refs63,64.
Particulate organic carbon, particulate nitrogen, biogenic silica, transparent exopolymers particle, coomassie stainable particles and amino-acids
Samples for POC and PN from CTD, traps and experiment were filtered onto a pre-combusted GF/F filters and stored at −20 °C until further analysis. Filters were then acidified over-night in an extractor in presence of concentrated fuming HCl. Filters were dried off and analyzed for C and N using a CHN analyser (Euro EA, Hechatech) following procedures described in ref.65. Samples for BSi were filtered at low vacuum (<200 mbar) onto 0.4 µm cellulose acetate filters and stored at −20 °C until further analysis. Filters were digested in NaOH at 85 °C for 135 mins, then the pH was raised to 8 using HCl and silicates were measured following ref.66.
Transparent exopolymere particles (TEP) and Coomassie stainable particles (CSP) from traps and experiment were analyzed by microscopy according to protocol presented in ref.67.
Dissolved free (DFAA), dissolved hydrolysable (DHAA), and total hydrolysable amino acids (THAA) were analyzed. For the dissolved amino acids, 20 mL of seawater were filtered through 0.45 μm syringe filters with low protein binding affinity (GHP membrane, Acrodisk, Pall Corporation). Total amino acids were analyzed directly in the unfiltered seawater sample. Samples were stored at −20 °C until analysis. For the determination of hydrolyzable amino acids, samples were hydrolyzed at 100 °C in 6 N HCl (Suprapur® Hydrochloric acid 30%) and 11 mM ascorbic acid for 20 h. Amino acids were separated by high performance liquid chromatography (HPLC), after ortho-phthaldialdehyde derivatization68,69, using a fluorescence detector (Excitation/Emission 330/445 nm). A pre-mixed standard containing: aspartic acid (Asp), glutamic acid (Glu), serine (Ser), arginine (Arg), glycine (Gly), threonine (Thre), alanine (Ala), tyrosine (Tyr), valine (Val), phenylalanine (Phe), isoleucine (Ileu), leucine (Leu), and γ-Aminobutyric acid (Gaba), was used for identification and quantification.
Bacterial abundance
Bacterial cells from experiment were counted using a flow cytometer (FACSCalibur, Becton, Dickson). 4 ml of sample were collected, fixed with 200 µl of 25% glutaraldehyde and stored at -20 °C for further analysis. Samples were defrosted and sonicated to detach the particles-associated cells, filtered through a 50 µm mesh and analyzed on the flow cytometer following methods presented in ref.70.
Incubation design
Sinking material from all depths (see section above) was then transferred into 1.2 l gas tight incubation bottles. 300 ml of sediment trap slurry split was mixed to 900 ml of CTD water sampled at the trap deployment location and depths. DO concentrations were then measured to control whether the concentration in the bottles actually corresponded to the actual DO concentration in the water column. The DO concentration in bottles corresponding to the DO minimum (110 m, see Fig. 2A) was significantly higher than the DO concentration observed at the same depth on the water column (Fig. 1A). Therefore, only the 110 m bottles were bubbled using a gas mix of N50 N2 and N45 CO2 (99.8675/0.1325%) to lower the DO concentration to the one observed in the water column.
Bottles were then place on a plankton wheel rotating at two rotations per minute (2 rpm) and kept in a temperature controlled room (12 °C) in complete darkness. Bottles were taken down on days 2, 4, 5 and 6 after the experiment. Clear aggregates were present in each bottle when taken down on sampling days. We measured DO, nutrients, ammonium, DOC, TDN concentrations, total bacteria abundance amino acid concentrations and particulate material POC, PN and TEP concentrations at every time steps.
References
Matear, R. J. & Hirst, A. C. Long-term changes in dissolved oxygen concentrations in the ocean caused by protracted global warming. Global Biogeochemical Cycles 17, https://doi.org/10.1029/2002GB001997 (2003).
Doney, S. C., Bopp, L. & Long, M. C. Historical and future trends in ocean climate and biogeochemistry. Oceanography 27, 108–119 (2014).
Bopp, L. et al. Multiple stressors of ocean ecosystems in the 21st century: projections with CMIP5 models. Biogeosciences 10, 6225–6245 (2013).
Cabre, A., Marinov, A., Bernardello, R. & Bianchi, D. Oxygen minimum zones in the tropical Pacific across CMIP5 models: mean state differences and climate change trends. Biogeosciences 12, 5429–5454 (2015).
Deutsch, C. et al. Centennial changes in North Pacific anoxia linked to tropical trade winds. Science 345, https://doi.org/10.1126/science.1252332 (2014).
Stramma, L., Johnson, G. C., Sprintall, J. & Mohrholz, V. Expanding oxygen-minimum zones in the tropical oceans. Science 320, 655–658, https://doi.org/10.1126/science.1153847 (2008).
Marsay, C. M. et al. Attenuation of sinking particulate organic carbon flux through the mesopelagic ocean. Proceedings of the National Academy of Sciences of the United States of America 112, https://doi.org/10.1073/pnas.1415311112 (2015).
Iversen, M. H. & Ploug, H. Temperature effects on carbon-specific respiration rate and sinking velocity of diatom aggregates – potential implications for deep ocean export processes. Biogeosciences 10, 4073–4085 (2013).
Buesseler, K. O. & Boyd, P. W. Shedding light on processes that control particle export and flux attenuation in the twilight zone of the open ocean. Limnology and Oceanography 54, 1210–1232 (2009).
Iversen, M. H. & Ploug, H. Ballast minerals and the sinking carbon flux in the ocean: carbon-specific respiration rates and sinking velocity of marine snow aggregates. Biogeosciences 7, 2613–2624 (2010).
Le Moigne, F. A. C., Gallinari, M. & Laurenceau, E. & De La Rocha, C. L. Enhanced rates of particulate organic matter degradation by microzooplankton are diminished by added ballast minerals. Biogeosciences 10, 5755–5765, https://doi.org/10.5194/bg-10-5755-2013 (2013).
Engel, A. et al. Investigating the effect of ballasting by CaCO3 in Emiliania huxleyi: II. Decomposition of particulate organic matter. Deep-Sea Research Part II-Topical Studies in Oceanography 56, 1408–1419 (2009).
Devol, A. H. & Harnett, H. E. Role of the oxygen minimum zone in transfer of organic carbon to the deep ocean. Limnology and Oceanography 25, 1684–1690 (2001).
Keil, R. G., A., N. J., Biladeau, C., Van der Elst, E. & Devol, A. H. A multiproxy approach to understanding the “enhanced” flux of organic matter through the oxygen-deficient waters of the Arabian Sea. biogeosciences 13, 2077–2092 (2016).
Pantoja, S., Sepulveda, J. & Gonzalez, H. E. Decomposition of sinking proteinaceous material during fall in the oxygen minimum zone off northern Chile. Deep Sea Research I 51, 55–70 (2004).
Roullier, F. et al. Particles size distribution and carbon flux across the Arabian Sea Oxygen Minimum Zone. Biogeosciences 11, 4541–4557, https://doi.org/10.5194/bgd-10-19271-2013 (2014).
Van Mooy, B. A. S., Keil, R. G. & Devol, A. H. Impact of suboxia on sinking particulate organic carbon: Enhanced carbon flux and preferential degradation of amino acids via denitrification. Geochimica Et Cosmochimica Acta 66 (2002).
Martin, J. H., Knauer, G. A., Karl, D. M. & Broenkow, W. W. Vertex - Carbon Cycling in the Northeast Pacific. Deep-Sea Research Part A 34, 267–285 (1987).
Engel, A., Wagner, H., Le Moigne, F. A. C. & Wilson, S. T. Particle export fluxes to the oxygen minimum zone of the Eastern Tropical North Atlantic. Biogeosciences 14, 1825–1838, https://doi.org/10.5194/bg-14-1825-2017 (2017).
Löscher, C. R. et al. Water column biogeochemistry of oxygen minimum zones in the eastern tropical North Atlantic and eastern tropical South Pacific Oceans. Biogeosciences 12, 4495–4556 (2015).
Kalvelage, T. et al. Nitrogen cycling driven by organic matter export in the South Pacific oxygen minimum zone. nature Geoscience 6, 228–234, https://doi.org/10.1038/ngeo1739 (2013).
Kuypers, M. M. M. Massive nitrogen loss from the Benguela upwelling system through anaerobic ammonium oxidation. Proceedings of the National Academy of Sciences of the United States of America 102, 6478–6483 (2005).
Thamdrup, B. Oceanography: Coastal oceanic nitrogen loss. Nature Geoscience 6, 160–161, https://doi.org/10.1038/ngeo1749 (2013).
Kalvelage, T. et al. Aerobic Microbial Respiration In Oceanic Oxygen Minimum Zones. PLOS One 10, doi:0.1371/journal.pone.0133526 (2015).
Franz, J. et al. Dynamics and stoichiometry of nutrients and phytoplankton in waters influenced by the oxygen minimum zone in the eastern tropical Pacific. Deep Sea Research I 62, 20–31 (2012).
Dauwe, B., Middelburg, J. J., Herman, P. M. J. & Heip, C. H. R. Linking diagenetic alteration of amino acids and bulk organic matter reactivity. Limnology and Oceanography 44, 1809–1814 (1999).
Hansson, M. & Andersson, L. Oxygen Survey in the Baltic Sea 2013 -Extent of anoxia and hypoxia, 1960–2013. SMHI Report 49 (2013).
Flinkman, J. Cruise 2/2015, Combine1 (2015).
Mohrholz, V., Naumann, M., Nausch, G., Krüger, S. & Gräwe, U. Fresh oxygen for the Baltic Sea - An exceptional saline inflow after a decade of stagnation. Journal of Marine Systems 148, 152–166 (2015).
Sarmiento, J. L. & Gruber, N. Ocean Biogeochemical Dynamics. Princeton University Press (2006).
Cisternas-Novoa, C., Le Moigne, F. A. C., Roa, J., Wagner, H. & Engel, A. Effect of variable oxygen concentrations on the sinking flux and composition of organic matter in the Baltic Sea. AGU Ocean Science 2016, New-Orleans USA (2016).
Balzano, S., Statham, P. J., Pancost, R. D. & Lloyd, J. R. Role of microbial populations in the release of reduced iron to the water column from marine aggregates. Aquatic Microbial Ecology 54, 291–303 (2009).
Ploug, H. & Bergkvist, J. Oxygen diffusion limitation and ammonium production within sinking diatom aggregates under hypoxic and anoxic conditions. Marine Chemistry 176, 142–149 (2015).
Lam, P. et al. Linking crenarchaeal and bacterial nitrification to anammox in the Black Sea. Proceedings of the National Academy of Sciences of the United States of America 104, 7104–7109 (2007).
He, W., Chen, M., Schlautman, M. A. & Hur, J. Dynamic exchanges between DOM and POM pools in coastal and inland aquatic ecosystems: A review. Science of the Total Environment 551–552, 415–428 (2016).
D. L. Rocha, C. L. & Passow, U. Factors influencing the sinking of POC and the efficiency of the biological carbon pump. Deep-Sea Research Part II-Topical Studies in Oceanography 54, 639–658, https://doi.org/10.1016/j.dsr2.2007.01.004 (2007).
Le Moigne, F. A. C., Pabortsava, K., Marcinko, C. L. J., Martin, P. & Sanders, R. J. Where is mineral ballast important for surface export of particulate organic carbon in the ocean? Geophys. Res. Lett. 41, doi:101002/2014GL061678 (2014).
Le Moigne, F. A. C. et al. On the proportion of ballast versus non-ballast associated carbon export in the surface ocean. Geophys. Res. Lett. 39, L15610, https://doi.org/10.1029/2012GL052980 (2012).
Engel, A., Thoms, S., Riebesell, U., Rochelle-Newall, E. & Zondervan, I. Polysaccharide aggregation as a potential sink of marine dissolved organic carbon. nature 428 (2004).
Cisternas Novoa, C., Lee, C. & Engel, A. Transparent exopolymer particles (TEP) and Coomassie stainable particles (CSP): Differences between their origin and vertical distributions in the ocean. Marine Chemistry 175, 56–71 (2015).
Caron, D. A. et al. The contribution of microorganisms to particulate carbon and nitrogen in surface waters of the Sargasso Sea near Bermuda. Deep Sea Research I 42, 943–972 (1995).
Lee, S. & Fuhrman, J. A. Relationships between Biovolume and Biomass of Naturally Derived Marine Bacterioplankton. Applied and Environmental Microbiology 53, 1298–1303 (1987).
Lam, P. & Kuypers, M. M. Micorbial Nitrogen Cycling Processes in Oxygen Minimum Zones. Annual Review Marine Sciences 3, 317–345, doi:0.1146/annurev-marine-120709-142814 (2011).
Tamburini, C. et al. Effects of hydrostatic pressure on microbial alteration of sinking fecal pellets. Deep-Sea Research II 56, 1533–1546, https://doi.org/10.1016/j.dsr2.2008.12.035 (2009).
Tamburini, C. et al. Pressure effects on surface mediterranean prokaryotes and biogenic silica dissolution during a diatom sinking experiment. Aquatic Microbial Ecology 43, 267–276, https://doi.org/10.3354/ame043267 (2006).
Riou, V. et al. Biodegradation of Emiliania Huxleyi aggregates by natural prokaryotic communities under increasing hydrostatic pressure. Progress in Oceanography (2017).
Sempere, R., Yoro, S. C., Van Wambeke, F. & Charriere, B. Microbial decomposition of large organic particles in the northwestern Mediterranean Sea: an experimental approach. Marine Ecology Progress Series 198, 61–72, https://doi.org/10.3354/meps198061 (2000).
Mislan, K. A. S., Stock, C. A., Dunne, J. P. & Sarmiento, J. Group behavior among model bacteria influences particulate carbon remineralization depths. Journal of Marine Research 72 (2014).
Piontek, J., Endres, S., Le Moigne, F. A. C., Cisternas Novoa, C. & Engel, A. The coupling of pelagic primary production and heterotrophic bacterial production during early summer and its relevance for oxygen consumption in the Baltic Sea. (in prep.).
Hedges, J. I. et al. Evidence for non-selective preservation of organic matter in sinking marine particles. Nature 409, 801–804 (2000).
Keil, R. G. & Cowie, G. L. Organic matter preservation through the oxygen-deficient zone of the NE Arabian Sea as discerned by organic carbon: mineral surface area ratios. Marine Geology 161, 13–22 (1999).
Dalsgaard, T., De Brabandere, L. & Hall, P. O. J. Denitrification in the water column of the central Baltic Sea. geochimica Et Cosmochimica Acta 106, 247–260 (2013).
Hannig, M. et al. Shift from denitrification to anammox after inflow events in the central Baltic Sea. Limnology and Oceanography 52, 1336–1345 (2007).
Babbin, A. R., Keil, R. G., Devol, A. H. & Ward, B. B. Organic Matter Stoichiometry, Flux, and Oxygen ControlNitrogen Loss in the Ocean. Science 344, 406–408 (2014).
Emeis, K. C. et al. Changes in the C, N, P burial rates in some Baltic Sea sediments over the last 150 years—relevance to P regeneration rates and the phosphorus cycle. Marine Geology 167, 43–59 (2000).
Kuypers, M. M. M. Anaerobic ammonium oxidation by anammox bacteria in the Black Sea. nature 422, 608–611 (2003).
Dickson, A. G. Determination of dissolved oxygen in sea water by Winkler titration. In WHP 91-1: WOCE Operations Manual, WOCE Hydrographic Program Office (1994).
Knauer, G. A., Martin, J. H. & Bruland, K. W. Fluxes of particulate carbon, nitrogen, and phosphorus in the upper water column of the northeast Pacific. Deep-Sea Research 26, 97–108 (1979).
Soutar, A., Kliing, P. A., Crill, E., Durffrin, K. W. & Bruland, K. W. Monitoring the marine environment through sedimentation. nature 266, 136–139 (1977).
Buesseler, K. O. et al. An assessment of the use of sediment traps for estimating upper ocean particle fluxes. Journal of Marine Research 65, 345–416 (2007).
Sugimura, Y. & Suzuki, Y. A high-temperature catalytic oxidation method for the determination of non-volatile dissolved organic carbon in seawater by direct injection of a liquid sample. Marine Chemistry 24, 105–131 (1988).
Engel, A. & Galgani, L. The organic sea-surface microlayer in the upwelling region off the coast of Peru and potential implications for air–sea exchange processes. Biogeosciences 13, 989–1007, https://doi.org/10.5194/bg-13-989-2016 (2016).
Le Moigne, F. A. C. et al. Description of the biogeochemical features of the subtropical southeastern Atlantic and the Southern Ocean south of South Africa during the austral summer of the International Polar Year. Biogeosciences 10, 281–295, https://doi.org/10.5194/bg-10-281-2013 (2013).
Koroleff, F. Direct determination of ammonia in natural water as indophenol blue. Information on Techniques and Methods for the Seawater Analysis (1969).
Sharp, J. Improved analysis for particualte organic carbon and nitrogen in seawater. Limnology and Oceanography 68, 984–989 (1974).
Hansen, H. P. & Koroleff, F. In Methods of Seawater Analysis (eds K. Grasshoff, K. Kremling, & M. Ehrhardt) chapter 10 (Wiley-VCH, 1999).
Engel, A. Determination of marine gel particles, in Pratical guidelines for the Analysis In Seawater (O. WurlEd.) ch.7. CRC Press, 125–142 (2009).
Lindroth, P. & Mopper, K. High performance liquid chromatography determination of subpicomole amounts of amino acids by precolumn fluorescence derivatisation with OPA. Analitycal Chemistry 51, 1667–1674 (1979).
Dittmar, T., Cherrier, J. & Ludichowski, K. U. The Analysis of Amino Acids in Seawater In: Practical Guidelines for the Analysis of Seawater CRC-Press, Boca Raton, USA, 125–142, https://doi.org/10.1201/9781420073072.ch7 (2010).
Gasol, J. M. & Del Giorgio, P. A. Using flow cytometry for counting natural planktonic bacteria and understanding the structure of bacterial communities. Scientia Marina 64, 197–224 (2000).
Acknowledgements
Jon Roa, Tania Klüver, Scarlett Sett, Angela Stippkugel, Carola Wagner, Clarissa Karthäuser, Moritz Ehrlich, Sonja Endres, Hannes Wagner, Ruth Flerus, Thomas Browning, Sven Sturm and Christian Begler supported traps preparation and deployments, help with experiment or analysed samples. Thomas J. Browning helped with phrasing of the manuscript. This research was supported by a Fellowship of the Excellence Cluster ‘The Future Ocean’ (CP1403 to F.A.C.L.M.), by the DFG Collaborative Research Center 754 “Climate-Biogeochemistry Interactions in the Tropical Ocean” (to A.E., C.C.N. and F.A.C.L.M) and by a DAAD short term grant (57130097 to C.C.N.). We thank the crew, officers and the captain of the F.S. Alkor.
Author information
Authors and Affiliations
Contributions
F.A.C.L.M designed the experiment, performed deployments/experiment, analysed samples and wrote the manuscript. C.C.N. performed deployments/experiment, analysed samples and contributed to the writing of the manuscript. M.M. performed deployments or helped with the experiment or samples analysis. A.E and J.P designed and conducted the scientific program at sea. All authors commented on the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Le Moigne, F.A.C., Cisternas-Novoa, C., Piontek, J. et al. On the effect of low oxygen concentrations on bacterial degradation of sinking particles. Sci Rep 7, 16722 (2017). https://doi.org/10.1038/s41598-017-16903-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-16903-3
This article is cited by
-
Hypoxia-tolerant zooplankton may reduce biological carbon pump efficiency in the Humboldt current system off Peru
Communications Earth & Environment (2023)
-
The mechanisms and kinetics of phosphate adsorption onto iron-coated waste mussel shell observed from hydrodynamic column
International Journal of Environmental Science and Technology (2022)
-
Microbial Diversity and Metabolic Potential in the Stratified Sansha Yongle Blue Hole in the South China Sea
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
