
Abstract
Enlarged perivascular spaces (EPVS) are reported to be associated with impaired cognitive function and sleep disorders. It is of clinical importance to understand the risk factors for EPVS. Hyperuricemia increases the risk of hypertension and endothelial dysfunction, which are well recognized to be associated with EPVS. Therefore, we postulated that serum uric acid (SUA) might be associated with EPVS. A total of 665 lacunar stroke patients were enrolled in this study. The SUA concentrations of patients with severe EPVS were much higher than those of patients with mild EPVS (for basal ganglia: 5.25 ± 1.40 mg/dl vs. 4.75 ± 1.40 mg/dl, p < 0.001; for white matter: 5.31 ± 1.41 mg/dl vs. 4.88 ± 1.37 mg/dl, p = 0.009). The percentage of subjects with severe EPVS tended to be higher in the highest quartile of SUA (chi-square test: P = 0.002 for basal ganglia and 0.006 for white matter). Spearman correlation analysis indicated that the SUA concentrations were positively correlated with the severity of EPVS (rho > 0, p < 0.05). Multivariate logistic regression analysis showed that high normal SUA was independently associated with a higher severity of EPVS. This finding suggests that high SUA levels might be an independent risk factor for EPVS in lacunar stroke patients.
Similar content being viewed by others

Introduction
Enlarged perivascular spaces (EPVS), or Virchow-Robin spaces, appear as punctate or linear identical signal intensities that are similar to cerebrospinal fluid (CSF) on all MRI sequences in the white matter, basal ganglia and hippocampus1,2. EPVS are perivascular compartments that surround the small penetrating cerebral vessels, serving as an important drainage system for interstitial fluid and solutes in the brain3. It has been identified that EPVS are a marker of cerebral small vessel diseases4,5. Several studies have demonstrated that EPVS were associated with impaired cognitive function1, incident dementia6, depression, and sleep disorders7. Therefore, it is of clinical importance to understand the risk factors for EPVS and to search for future treatment options.
Uric acid (UA) is the end metabolic product of purine nucleotides. It has been reported that increased UA concentrations are associated with endothelial dysfunction8, hypertension9,10,11, diabetes9,12, and elevated serum triglyceride and cholesterol concentrations13, which increase the risk of EPVS. Therefore, we postulated that higher serum uric acid (SUA) concentrations might be associated with EPVS. However, some studies have suggested that UA can exert neuroprotective effects by acting as a free radical scavenger14. Higher level of SUA predicts better outcomes following stroke15,16. Administration of UA reduced ischemic damage and improved outcomes in an experimental model of stroke17. It is apparent that the function of UA remains controversial. An insufficient amount of research has been conducted to investigate the relationship between SUA and EPVS. In the present study, we aimed to explore whether SUA level is associated with EPVS.
Results
Baseline characteristics of the study participants
A total of 665 eligible participants with a mean age of 63 years old were enrolled in this study. None of the participants took uric acid lowering drugs or oestrogen within one week when their SUA concentrations were measured. The clinical and laboratory characteristics of all subjects and subgroups divided by the severity of EPVS are summarized in Table 1. Age, proportion of hypertension and Fazekas scale scores increased and HDL concentrations decreased with the degree of EPVS in basal ganglia increasing.
Association between SUA Concentrations and EPVS
The SUA concentrations of each category divided by the severity of EPVS in different brain regions are presented in Table 1, Figs 1 and 2. As shown in Fig. 1, the SUA concentration increased with the severity of EPVS in basal ganglia (BG-EPVS) increasing. The results of Tukey tests showed that the SUA concentrations of patients with severe BG-EPVS were higher than those of patients with mild BG-EPVS (5.25 ± 1.40 mg/dl vs. 4.75 ± 1.40 mg/dl, p = 0.001). The SUA concentration of the severe BG-EPVS group was higher than that of the moderate group, and the concentration of the moderate group was higher than that of the mild group; however, there were no significant differences (5.25 ± 1.40 mg/dl vs. 4.98 ± 1.38 mg/dl, P = 0.107; 4.98 ± 1.38 mg/dl vs. 4.75 ± 1.40 mg/dl, P = 0.167). As shown in Fig. 2, the SUA concentrations of patients with severe EPVS in white matter (WM-EPVS) were also higher than those of patients with mild EPVS (5.31 ± 1.41 mg/dl vs. 4.88 ± 1.37 mg/dl, p = 0.024) and moderate EPVS (5.31 ± 1.41 mg/dl vs. 4.87 ± 1.39 mg/dl, p = 0.003). However, there was no difference in the SUA concentration when comparing between the mild EPVS group and moderate EPVS group (4.88 ± 1.37 vs. 4.87 ± 1.39, P = 0.998).

Histogram of the mean SUA concentrations in the subgroups divided by the severity of EPVS in basal ganglia. Analysis of variance was used among the three subgroups and Tukey tests were used in multiple comparisons. Statistical significance was accepted at p < 0.05. The histogram error bars indicate the 95% confidence interval (95%CI) of the SUA concentrations.

Histogram of the mean SUA concentrations in the subgroups divided by the severity of EPVS in white matter. Analysis of variance was used among the three subgroups and Tukey tests were used in multiple comparisons. Statistical significance was accepted at p < 0.05. The histogram error bars indicate the 95%CI of the SUA concentrations.
Spearman correlation analysis indicated that the SUA level was positively correlated with the severity of EPVS (for BG-EPVS: Spearman’s rho = 0.149, p < 0.001; for WM-EPVS: Spearman’s rho = 0.104, p = 0.007). However, the correlation was not strong. The results of multivariate logistic regression analysis are presented in Table 2. The odds ratio (OR) of comparing the severe BG-EPVS group to the mild BG-EPVS group was 1.436 (95% CI: 1.223–1.687) after controlling for demographic confounders (model 1) and 1.435 (95% CI: 1.210–1.701) after additionally adjusting for laboratory tests (model 2). The OR value increased to 1.466 (95% CI: 1.217–1.765) after additionally adjusting for Fazekas scale (model 3). Although there were negligible changes in statistical significance after adjusting for the Fazekas scale scores, the risk of the SUA concentration for the severe group did change. Individuals with severe BG-EPVS exhibited a 43.6% higher risk than those with mild BG-EPVS in model 1, and these results increased to 46.6% in model 3. After controlling for demographic confounders, laboratory tests and Fazekas scale scores, the SUA concentration was independently associated with the severity of WM-EPVS (model 1: OR = 1.214, 95% CI: 1.029–1.433; model 2: OR = 1.283, 95% CI: 1.084–1.519; model 3: OR = 1.270, 95% CI 1.074–1.501). However, the risk of the SUA concentration for severe WM-EPVS was lower than that for severe BG-EPVS. The statistical parameters of stepwise logistic regression analysis (forward method) were presented in Table 3, which showed the regression analysis models with statistical significance.
In addition, we further divided all subjects into quartiles by their SUA levels. Quartile 1 (Q1) was SUA ≤ 4.01 mg/dl, quartile 2 (Q2) was 4.01 mg/dl < SUA ≤ 4.91 mg/dl, quartile 3 (Q3) was 4.91 mg/dl < SUA ≤ 5.87 mg/dl, and quartile 4 (Q4) was SUA > 5.87 mg/dl. In the entire group of subjects, the percentages of subjects with severe EPVS tended to be higher in the highest quartile of the SUA level (chi-square test, P = 0.002 and 0.006, respectively) (Figs 3 and 4).

Distribution of the percentage of subjects in each quartile of SUA in different subgroups stratified by the severity of EPVS in basal ganglia. The percentage of subjects with severe EPVS tended to be higher in the highest quartile of SUA level.

Distribution of the percentage of subjects in each quartile of SUA in different subgroups stratified by the severity of EPVS in white matter. The percentage of subjects with severe EPVS tended to be higher in the highest quartile of SUA level.
Discussion
To our knowledge, this is the first study to investigate the relationship between the SUA concentration and EPVS. Our results revealed a positive correlation between SUA and the severity of EPVS in lacunar stroke patients. The SUA concentration increased when the degree of both BG-EPVS and WM-EPVS increased. The percentage of subjects with severe EPVS tended to be higher in the highest quartile of SUA. We found that a higher SUA concentration was an independent risk factor for EPVS by multivariable logistic regression analysis when adjusting for other confounders.
Many studies have explored the etiologies and potential pathophysiological mechanisms of EPVS. An increased permeability of the small vessel walls and blood brain barrier are considered to contribute to the development of EPVS, which has been reported to be associated with damage to microvascular endothelial cells and their tight junctions3,18,19. A large population-based study found that elevated interleukin-6 levels were associated with a higher severity of EPVS in the BG, suggesting that inflammatory responses may participate in the pathogenesis of EPVS20. In addition, leakage of interstitial fluid and structural changes of the microvascular wall may obstruct the drainage space and lead to consequent development of EPVS21,22.
Several mechanisms may be involved in the relationship between EPVS and SUA. One possibility is that high SUA concentration causes urate deposition in the small vessel walls, which directly damage the vascular endothelium and blood brain barrier. Another possible pathway is that UA induces endothelial dysfunction by reducing nitric oxide (NO) synthesis and availability23,24. In addition, high SUA concentration promote low-density lipoprotein oxidation in vitro 25 and stimulate granulocyte adherence to the endothelium23,26. Damage of microvascular endothelial cells and structural changes of the microvascular wall may result in leakage of interstitial fluid, obstruction of the drainage space and consequent development of EPVS27. Taken together, it is reasonable to conclude that high SUA concentration contributes to the development of EPVS through the combined effects of these factors.
The functions of UA are unclear mainly because of its many properties, including inducing oxidative stress, increasing inflammation, reducing NO synthesis and acting as a natural antioxidant26,28. Several studies have shown that low SUA concentration increases the risk of Alzheimer’s29 and Parkinson’s diseases30. In addition, animal experiments have indicated that administering UA reduces ischemic damage in rat models of acute stroke17. However, many studies have reported that elevated SUA concentration increases the risk of white matter hyperintensities (WMH)31,32, silent brain infarction33 and cardiovascular diseases34. Vannorsdall TD et al. found that older adults with high normal SUA concentration was more likely to perform poorly on cognitive measures35. Recent studies have demonstrated that EPVS are associated with impaired cognitive function1,36. In the present study, our results suggested that higher normal SUA concentration was associated with more severe EPVS, which was found to be significant even after controlling for confounding factors. Therefore, we speculated that EPVS might mediate the association between SUA and cognitive impairment, which should be demonstrated in future studies. Our results showed that the SUA concentration was within the normal range even in the severe EPVS group. Schretlen DJ et al. found that mild elevation of SUA (but within the normal range) might increase the risk of cognitive decline and brain ischemia37,38. We hypothesize that even a normal elevation of SUA may be associated with an increased burden of cerebral small vessel diseases, which might have broad public health implications. These findings might hold for the clinical values that respan the normal range of SUA.
There are some limitations in our study. First, our study population was based on lacunar stroke patients in a single centre; therefore, the cohort may not represent the general population. Second, although we found that higher SUA levels were correlated with a higher number of EPVS, the correlation was not strong. In addition, this was a cross-sectional study, and the causal relationship between SUA and EPVS could not be established. Further, our database did not include information on whether females were pre- or postmenopausal nor the dietary habits of the patients, such as the level of purine intake (for example meat and meat products).
In conclusion, our study indicated that elevated SUA concentration was positively correlated with the severity of EPVS. A clinical trial may be useful in determining whether reducing the production of SUA is beneficial in patients with cerebral small vessel diseases.
Methods and Materials
Ethics statement
All subjects provided written informed consent, and the study was approved by the Ethics Committee of Beijing Chaoyang Hospital affiliated to Capital Medical University and was performed in accordance with the Declaration of Helsinki.
Study subjects and data collection
Consecutive small-artery occlusion (SAO) stroke patients defined by the Trial of Org 10172 in Acute Stroke Treatment (TOAST) classification39 were prospectively enrolled in the Neurology Department of Beijing Chaoyang Hospital affiliated to Capital Medical University from Jan 2011 to May 2015. Inclusion criteria were: (1) patients with an NIH Stroke Scale (NIHSS) score < 3; (2) patients undergoing brain MRI scans; (3) patients undergoing SUA measurements at least 7 days after SAO stroke onset; and (4) patients agreeing to participate in our study. The following patients were excluded: (1) patients with other stroke subtypes by the TOAST classification, Parkinson disease, dementia, severe traumatic or toxic or infectious brain injury, and brain tumour; (2) patients with severe heart disease and recent myocardial infarction or angina pectoris disorders, severe infections, severe nephrosis or liver disease, thrombotic diseases and tumour; and (3) patients unable to give written informed consent.
Clinical information including demographic data, past medical history, current smoking habits, alcohol consumption and medications were collected on all patients. Stroke severity was determined by the NIHSS score. All blood samples were collected in the morning after an overnight fasting period and were sent to the clinical laboratory of Beijing Chaoyang Hospital affiliated to Capital Medical University for the measurement of SUA concentrations. The SUA concentrations were measured by same method, using an autoanalyser in the clinical laboratory of our hospital.
Assessments of EPVS and WMH
Neurological image examinations were performed on the same 3.0 T Siemens scanner (Erlangen, Germany) in the Radiology Department. Assessments of EPVS and WMH were performed by two experienced neurologists blinded to clinical information. Disagreements were resolved by consensus. The k statistic of the intra-rater and interrater agreement was 0.85 or above, indicating good reliability.
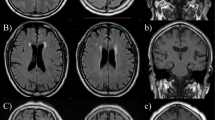
EPVS were defined as CSF-like signal intensity lesions of round, ovoid, or linear shape of <3 mm and located in areas supplied by perforating arteries (Fig. 5). We distinguished lacune from EPVS by their larger size (>3 mm), spheroid shape and surrounding hyperintensities on FLAIR. BG-EPVS and WM-EPVS were separately assessed. The degree of EPVS severity was scored using a scale that was derived from previous studies: 0 = no EPVS, 1 = ≤ 10 EPVS, 2 = 11 to 20 EPVS, 3 = 21 to 40 EPVS, and 4 = > 40 EPVS5,27. The number referred to the highest number of EPVS on one side of the brain. We classified EPVS into three categories: mild = score 0 or 1; moderate = score 2; and severe = score 3 or 4. WMH were scored using the Fazekas scale. A detailed description of these assessments has been previously published40. Periventricular white matter hyperintensities (P-WMH) and deep white matter hyperintensities (D-WMH) were evaluated separately and summed as Fazekas scores.

The severity of enlarged perivascular spaces in basal ganglia and white matter. (A) Mild in basal ganglia; (B) moderate in basal ganglia; (C), severe in basal ganglia; (D), mild in white matter; (E), moderate in white matter; (F), severe in white matter. The arrows were pointing to enlarged perivascular spaces, which appear as punctate or linear identical signal intensities that are similar to cerebrospinal fluid on MRI sequences.
Statistical analysis
Continuous variables were presented as the mean values ± standard deviation and compared with analysis of variance (ANOVA) for factors with both normal distribution and homogeneity of variance. The Tukey test was used for multiple comparisons. Categorical variables were expressed as absolute numbers and percentages. The chi-squared test was used to test the independence of the categorical variables. Spearman correlation analysis was used to calculate the association between SUA levels and the severity of EPVS. Some vascular risk factors, such as age, sex, smoking, alcohol, hypertension, diabetes, serum lipid level, serum glucose level, renal function and the severity of WMH were reported to be associated with cerebral small vessel diseases. The multivariate logistic regression analysis was performed to assess whether SUA concentration was independently associated with EPVS after adjusting for these confounding factors. Given the high number of confounders considered, we adopted the stepwise logistic regression analysis model (forward method). The consequential significance level for entered and removal variables was 0.05 and 0.1, respectively. We also performed the correlation analysis among these variables. The correlation coefficient matrix indicated that the correlation were weak. Analysis was performed with Statistical Package for Social Sciences (SPSS version17.0), and statistical significance was accepted at p < 0.05.
References
Maclullich, A. M. et al. Enlarged perivascular spaces are associated with cognitive function in healthy elderly men. J Neurol Neurosurg Psychiatry 75, 1519–1523 (2004).
Zhu, Y. C. et al. Frequency and location of dilated virchow-robin spaces in elderly people: A population-based 3d mr imaging study. AJNR Am J Neuroradiol 32, 709–713 (2011).
Kwee, R. M. & Kwee, T. C. Virchow-robin spaces at mr imaging. Radiographics: a review publication of the Radiological Society of North America, Inc 27, 1071–1086 (2007).
Wardlaw, J. M. et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 12, 822–838 (2013).
Doubal, F. N., MacLullich, A. M., Ferguson, K. J., Dennis, M. S. & Wardlaw, J. M. Enlarged perivascular spaces on mri are a feature of cerebral small vessel disease. Stroke 41, 450–454 (2010).
Zhu, Y. C. et al. High degree of dilated virchow-robin spaces on mri is associated with increased risk of dementia. Journal of Alzheimer’s disease: JAD 22, 663–672 (2010).
Berezuk, C. et al. Virchow-robin spaces: Correlations with polysomnography-derived sleep parameters. Sleep 38, 853–858 (2015).
Choi, Y. J. et al. Uric acid induces endothelial dysfunction by vascular insulin resistance associated with the impairment of nitric oxide synthesis. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 28, 3197–3204 (2014).
Nakanishi, N. et al. Serum uric acid and risk for development of hypertension and impaired fasting glucose or type ii diabetes in japanese male office workers. European journal of epidemiology 18, 523–530 (2003).
Perlstein, T. S. et al. Uric acid and the development of hypertension: The normative aging study. Hypertension 48, 1031–1036 (2006).
Shankar, A., Klein, R., Klein, B. E. & Nieto, F. J. The association between serum uric acid level and long-term incidence of hypertension: Population-based cohort study. J Hum Hypertens 20, 937–945 (2006).
Dehghan, A., Hoek, M., Sijbrands, E. J., Hofman, A. & Witteman, J. C. High serum uric acid as a novel risk factor for type 2 diabetes. Diabetes Care 31, 361–362 (2008).
Rathmann, W., Funkhouser, E., Dyer, A. R. & Roseman, J. M. Relations of hyperuricemia with the various components of the insulin resistance syndrome in young black and white adults: The cardia study. Coronary artery risk development in young adults. Annals of epidemiology 8, 250–261 (1998).
Li, R., Huang, C., Chen, J., Guo, Y. & Tan, S. The role of uric acid as a potential neuroprotectant in acute ischemic stroke: A review of literature. Neurol Sci 36, 1097–1103 (2015).
Chamorro, A. et al. Prognostic significance of uric acid serum concentration in patients with acute ischemic stroke. Stroke 33, 1048–1052 (2002).
Wang, Z. et al. Serum uric acid levels and outcomes after acute ischemic stroke. Mol Neurobiol 53, 1753–1759 (2016).
Yu, Z. F., Bruce-Keller, A. J., Goodman, Y. & Mattson, M. P. Uric acid protects neurons against excitotoxic and metabolic insults in cell culture, and against focal ischemic brain injury in vivo. J Neurosci Res 53, 613–625 (1998).
Wuerfel, J. et al. Perivascular spaces–mri marker of inflammatory activity in the brain? Brain: a journal of neurology 131, 2332–2340 (2008).
Wardlaw, J. M., Smith, C. & Dichgans, M. Mechanisms of sporadic cerebral small vessel disease: Insights from neuroimaging. Lancet Neurol 12, 483–497 (2013).
Satizabal, C. L., Zhu, Y. C., Dufouil, C. & Tzourio, C. Inflammatory proteins and the severity of dilated virchow-robin spaces in the elderly. Journal of Alzheimer’s disease: JAD 33, 323–328 (2013).
Abbott, N. J. Evidence for bulk flow of brain interstitial fluid: Significance for physiology and pathology. Neurochemistry international 45, 545–552 (2004).
Weller, R. O., Subash, M., Preston, S. D., Mazanti, I. & Carare, R. O. Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and alzheimer’s disease. Brain pathology 18, 253–266 (2008).
Kanellis, J. & Kang, D. H. Uric acid as a mediator of endothelial dysfunction, inflammation, and vascular disease. Seminars in nephrology 25, 39–42 (2005).
Gersch, C. et al. Inactivation of nitric oxide by uric acid. Nucleosides, nucleotides & nucleic acids 27, 967–978 (2008).
Schlotte, V., Sevanian, A., Hochstein, P. & Weithmann, K. U. Effect of uric acid and chemical analogues on oxidation of human low density lipoprotein in vitro. Free radical biology & medicine 25, 839–847 (1998).
Johnson, R. J. et al. Is there a pathogenetic role for uric acid in hypertension and cardiovascular and renal disease? Hypertension 41, 1183–1190 (2003).
Zhang, X. Y. et al. Brain atrophy correlates with severe enlarged perivascular spaces in basal ganglia among lacunar stroke patients. PLoS One 11, e0149593 (2016).
Becker, B. F. Towards the physiological function of uric acid. Free radical biology & medicine 14, 615–631 (1993).
Rinaldi, P. et al. Plasma antioxidants are similarly depleted in mild cognitive impairment and in alzheimer’s disease. Neurobiology of aging 24, 915–919 (2003).
Schlesinger, I. & Schlesinger, N. Uric acid in parkinson’s disease. Movement disorders: official journal of the Movement Disorder Society 23, 1653–1657 (2008).
Shih, C. Y., Chen, C. Y., Wen, C. J., Liu, H. M. & Kuo, H. K. Relationship between serum uric acid and cerebral white matter lesions in the elderly. Nutrition, metabolism, and cardiovascular diseases: NMCD 22, 154–159 (2012).
Sun, M. J. et al. Association between serum uric acid levels and cerebral white matter lesions in chinese individuals. Int J Neurosci 126, 1103–11 (2016).
Heo, S. H. & Lee, S. H. High levels of serum uric acid are associated with silent brain infarction. J Neurol Sci 297, 6–10 (2010).
Fang, J. & Alderman, M. H. Serum uric acid and cardiovascular mortality the nhanes i epidemiologic follow-up study, 1971–1992. National health and nutrition examination survey. Jama 283, 2404–2410 (2000).
Vannorsdall, T. D., Jinnah, H. A., Gordon, B., Kraut, M. & Schretlen, D. J. Cerebral ischemia mediates the effect of serum uric acid on cognitive function. Stroke 39, 3418–3420 (2008).
Chen, W., Song, X. & Zhang, Y. Assessment of the virchow-robin spaces in alzheimer disease, mild cognitive impairment, and normal aging, using high-field mr imaging. AJNR Am J Neuroradiol 32, 1490–1495 (2011).
Schretlen, D. J. et al. Serum uric acid and cognitive function in community-dwelling older adults. Neuropsychology 21, 136–140 (2007).
Schretlen, D. J. et al. Serum uric acid and brain ischemia in normal elderly adults. Neurology 69, 1418–1423 (2007).
Adams, H. P. et al. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. Toast. Trial of org 10172 in acute stroke treatment. Stroke 24, 35–41 (1993).
Fazekas, F., Chawluk, J. B., Alavi, A., Hurtig, H. I. & Zimmerman, R. A. Mr signal abnormalities at 1.5 t in alzheimer’s dementia and normal aging. AJR Am J Roentgenol 149, 351–356 (1987).
Acknowledgements
The authors thank all the study participants. This work is supported by National Natural Science Foundation of China (Grant No. 81271309) and Beijing Municipal Administration of Hospitals’ Youth Programme (QML20150303).
Author information
Authors and Affiliations
Contributions
W.L.H. conceived and designed the experiments. S.N.Y. analyzed the data and drafted the manuscript. Xiaoyu Zhang, Junliang Yuan and Jiangmei Yin collected data. All authors have read and approved the final manuscript to be published.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, S., Zhang, X., Yuan, J. et al. Serum Uric Acid is Independently Associated with Enlarged Perivascular Spaces. Sci Rep 7, 16435 (2017). https://doi.org/10.1038/s41598-017-16715-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-16715-5
This article is cited by
-
Classification of white matter lesions and characteristics of small vessel disease markers
European Radiology (2022)
-
Mechanisms of cognitive dysfunction in CKD
Nature Reviews Nephrology (2020)
-
Cerebral small vessel disease: neuroimaging markers and clinical implication
Journal of Neurology (2019)
-
Association Between the Serum Uric Acid Levels and Lacunar Infarcts in the Elderly
Journal of Molecular Neuroscience (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
