
Abstract
Enterovirus 71 (EV71) is a key pathogen of hand, foot and mouth disease (HFMD) in children under 6 years of age. The antiviral potency of antioxidant isochlorogenic acid C (ICAC) extracted from foods was evaluated in cellular and animal models. First, the cytotoxicity of ICAC on Vero cells was investigated. The viral plaques, cytopathic effects and yield induced by EV71 infection were obviously reduced by ICAC, which was consistent with the investigation of VP1 transcripts and protein expression. Moreover, the mortality, weight loss and limb paralysis of mice caused by EV71 challenge were remarkably relieved by ICAC injection, which was achieved through decreases in the viral load and cytokine secretion in the mouse brain. Further biochemical assays showed that ICAC modulated several antioxidant enzymes involved in reduced and oxidized glutathione (GSH and GSSG) homeostasis, including glutathione reductase (GR), glutathione peroxidase (GPX), and glucose-6-phosphate dehydrogenase (G6PD), resulting in restoration of the GSH/GSSG ratio and reactive oxygen species (ROS) level. Finally, the antiviral effects of ICAC were dose-dependently disrupted by BSO, a biosynthesis inhibitor of GSH. This study indicated that ICAC acted as an antioxidant and prevented EV71 infection by modulating the redox homeostasis of glutathione.
Similar content being viewed by others

Introduction
Human enterovirus 71 (EV71), which belongs to the genus Enterovirus, has been confirmed as a critical pathogen of hand, foot and mouth disease1. EV71 infection can cause severe symptoms, such as pulmonary oedema and brainstem and cerebellar encephalitis, which can lead to respiratory failure and death2,3,4,5. Cytokines in tissues, such as tumour necrosis factor-α (TNF-α), interleukin-6 (IL-6), and monocyte chemotactic protein-1 (MCP-1), can be induced by an excess viral load, resulting in tissue damage and chronic inflammation6, which play key roles in the pathogenicity and severity of EV71 infection7,8,9,10,11. In 2015, a vaccine against EV71 was approved as a new tool to control hand, foot and mouth disease (HFMD) outbreaks12. However, no specific chemical drug targeting EV71 has been approved. Thus, antiviral compounds must be screened for new drug development, and the mechanisms should be carefully discussed.
Viral infection can lead to oxidative stress13. A previous study showed that the redox status modulated host cell susceptibility to viruses14. For example, a shift in the intracellular redox milieu towards the oxidizing end enhanced viral replication and CPE15. Interestingly, the viral infection could be reversed by supplementing with glutathione (GSH), which indicated roles for GSH in antiviral defence16.
GSH plays a central role in reactive oxygen species (ROS) detoxification17. Briefly, glutathione peroxidase (GPX) catalyses the degradation of ROS coupled with the conversion of GSH to its oxidized form (GSSG). Glutathione reductase (GR) is an essential factor responsible for reducing GSSG back to GSH in the presence of NADPH, which is provided by glucose-6-phsophate dehydrogenase (G6PD)17. Upon viral infection, an imbalance in GSH redox homeostasis in host cells is observed15 due to the accumulation of ROS18, which can be reversed by exogenous supplementation of antioxidants15,16. L-buthionine sulfoximine (BSO) is a specific and selective inhibitor of γ- glutamylcysteine synthase and consequently of GSH synthesis19. Previous report20 showed BSO significantly reduced GSH level. Thus, BSO is usually used to deplete GSH in cells.
Isochlorogenic acid C (ICAC), which is a di-O-caffeoyl derivative of chlorogenic acid (CHA), is a well-known antioxidant21,22,23 from herbal plants21 that has revealed more potent effects than other isomers24,25. Previous reports showed that ICAC and its isomers exhibited a broad-spectrum antiviral potency against respiratory syncytial virus (RSV)26, human immunodeficiency virus (HIV)27,28,29, and coxsackievirus30. However, the potential effects of ICAC against EV71 are unknown and should be investigated. Only CHA (the parent nucleus of ICAC) has been reported to inhibit EV71 replication in vitro 31. However, ICAC extracted from Flos Lonicerae was indicated to reverse acetaminophen-induced liver injury via modulating the GSH content32. Thus, the potential regulation of GSH metabolism should be discussed with a focus on ICAC-derived inhibition of EV71 infection.
In this study, the antiviral efficacy of ICAC against EV71 was confirmed in both cellular and animal models. The compound was shown to reduce the mortality of mice upon EV71 challenge by decreasing the viral load and cytokine secretion. Further biochemical assays suggested that ICAC restored the GSH/GSSG ratio by regulating the enzymes responsible for GSH metabolism, resulting in a decreased ROS level. Taken together, the data in this study indicated that ICAC acted as an antioxidant and prevented EV71 infection via modulating GSH redox homeostasis.
Results
Cytotoxicity of ICAC
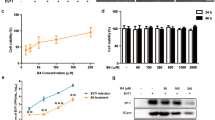
The molecular structure of ICAC, which is also called 4,5-O-dicaffeoylquinic acid, is displayed in Fig. 1A. To investigate the toxicity of ICAC on Vero cells, the cell viability was determined after ICAC supplementation for 48 h. The compound exhibited minor cytotoxicity at concentrations up to 250 µM (Fig. 1B, P > 0.05). However, the cell viability decreased gradually and clearly when the concentration was increased to 375 µM or more in comparison with the viability of the blank control (Fig. 1B, P < 0.05). According to the results, the median toxic concentration (TC50) of ICAC for the cells was approximately 429 µM.

The molecular structure of ICAC and its effects on Vero cell viability. (A) The molecular structure of ICAC. (B) The compound was diluted to various concentrations as indicated in triplicate. The cytotoxicity of ICAC was determined by MTS assay after incubation with the drug for 48 h. The cell viability in DMEM without ICAC (control) was set as 100% (n = 6). All results were expressed as the means ± SEs. Asterisks indicate that the data significantly differ from the control group at the P < 0.05 level according to one-way analysis of variance.
Antiviral effects of ICAC against EV71 in vitro
The antiviral potency of ICAC was estimated in a cellular model. As expected, the plaques and CPE caused by EV71 infection were significantly reduced by ICAC in a dose-dependent manner (Fig. 2A,B). In particular, 100 µM ICAC, which had an inhibitory rate of 63.1%, clearly relieved the CPE caused by viral infection (Fig. 2B, P < 0.05). This concentration was appropriate for the inhibition of EV71 infection at a dose of 100 TCID50 and was utilized in the subsequent experiments. According to the results, the concentration required to obtain the 50% of maximal effect (EC50) and the selection index (SI = TC50/EC50) were approximately 72 µM and 5.92, respectively.

The antiviral effects of IACA against EV71 in Vero cells. Vero cells were infected with 100 TCID50 of EV71 with or without different ICAC concentrations as indicated. Uninfected cells were used as the control group. (A) ICAC blocked the CPE of EV71 infection. (B) Cell viability was detected using the MTS cell proliferation assay kit at 48 hpi. The viability of the control group was set as 100% (n = 6). (C) The virions were collected by freeze-thawing at the indicated time points. The supernatant was harvested for the viral titre assay. (D) The RNA load was determined using a real-time PCR kit specific for the VP1 gene. (E) Protein samples normalized to 40 µg were subjected to 12.5% SDS-PAGE and then transferred to PVDF membrane to detect the EV71 VP1 protein expression levels. The amount of β-Tubulin was used as the internal standard. The VP1 band intensity was analysed and normalized to the corresponding band intensity of β-Tubulin. The band intensity of the EV71 group was set as 100% (n = 3). All results were expressed as the means ± SEs. Asterisks indicate that the data significantly differ from the EV71 group at the P < 0.05 level according to one-way analysis of variance.
To investigate inhibition of the viral yield by ICAC, a time-course experiment was performed. As shown in Fig. 2C, EV71 replicated rapidly from 0–12 hours post-infection (hpi), followed by a gradual titre increase from 12–24 hpi that was delayed by ICAC addition. For instance, ICAC reduced the viral titres by approximately 39-fold at 12 hpi (Fig. 2C, P < 0.05) compared with the titres obtained by EV71 infection alone. Furthermore, the VP1 mRNA transcript and protein levels were analysed. The VP1 transcripts induced by infection were significantly restrained by ICAC administration at 12 hpi (Fig. 2D, P < 0.05). Western blotting analysis indicated that ICAC decreased the VP1 protein expression enhanced by EV71 infection at 12 hpi (Fig. 2E, P < 0.05). Taken together, our data demonstrated the antiviral effects of ICAC against EV71 in the Vero cell model.
ICAC reduced the mortality of mice upon EV71 challenge
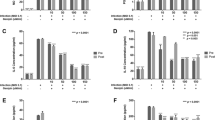
To further confirm the inhibitory effects of ICAC against EV71, a suckling mouse model was utilized. First, the acute toxicity of ICAC was studied. ICAC at doses of less than 25 mg/kg daily for 14 days failed to cause any death or abnormal signs in the mice (Fig. 3A). Thus, the doses used in the subsequent animal experiments were nontoxic to the suckling mice. In the subsequent antiviral study, the model group mice revealed weight loss, paralysis, and mortality from 4–8 days post-infection (dpi), and all the mice died within 11 dpi (Table 1 and Fig. 3B–D). In contrast, ribavirin (10 mg/kg), which was used as a positive control drug, increased the survival rate to 50% and the survival time to 10.1 ± 1.8 days (Table 1 and Fig. 3B, P < 0.05). The weight loss caused by EV71 was also clearly alleviated by ribavirin (Fig. 3C). Moreover, administration of ICAC at a dose of 6.4 mg/kg obviously prevented the EV71-induced death by 60% and raised the survival time to 12.6 ± 0.6 days (Table 1 and Fig. 3B, P < 0.05). The symptoms, including growth inhibition (Fig. 3C), paralysis, and emaciation (Fig. 3D), caused by the viral infection were relieved by treatment with 6.4 mg/kg of ICAC. Additionally, 3.2 mg/kg of ICAC showed protective effects against mortality and growth inhibition in the infected mice (Table 1 and Fig. 3B,C), whereas 1.6 mg/kg of ICAC resulted in only weak effects on EV71 infection.

ICAC reduced the mortality of the EV71-infected mice by regulating the viral load and cytokine secretion. Two-day-old mice were utilized as the animal model. (A) The acute toxicity of ICAC (12.5–200 mg/kg) on mice was evaluated. In the antiviral study, the mice were challenged intraperitoneally with saline (control) or 1×107 TCID50 of EV71. Then, the infected mice were treated with various doses (1.6–6.4 mg/kg) of ICAC or 10 mg/kg of ribavirin for 14 days. The mice in the model group were injected with saline. The survival rates (B) and body weight ratios (C) were recorded continuously until 14 dpi. The body weight ratio was presented relative to the corresponding treatments on the first day (n = 10). (D) Representative photos of limb paralysis caused by EV71 infection (left) and healthy phenotypes of mice treated with 6.4 mg/kg of ICAC (right) at 6 dpi. The brain tissues were sampled at 3, 6, and 9 dpi. (E) The viral loads in the brains were determined by real-time PCR (n = 6). The IL-6 (F), MCP-1 (G), and TNF-α (H) levels in the brains were determined by ELISA (n = 6). All results were expressed as the means ± SEs. Asterisks indicate that the data significantly differ from the EV71 group at the P < 0.05 level according to one-way analysis of variance.
ICAC regulated the viral load and cytokine secretion in EV71-infected mice
The effects of ICAC on the viral load were investigated in the subsequent experiment. As shown in Fig. 3E, the viral load in the mouse brain tissues gradually increased to a peak at 6 dpi, followed by a decline at 9 dpi, which was obviously inhibited by ICAC at the indicated time points (Fig. 3E, P < 0.05). For example, the viral copies in the ICAC-treated mice decreased by approximately 17-fold at 6 dpi (Fig. 3E, P < 0.05), which was consistent with the results observed in the cellular model (Fig. 2D).
To further explore the regulation of cytokine secretion by ICAC, the IL-6, MCP-1, and TNF-α levels in the mouse brains were assessed at 3, 6, and 9 dpi. EV71 challenge resulted in significant IL-6, MCP-1, and TNF-α accumulation in the brain tissues at 3 and 6 dpi (Fig. 3F–H, P < 0.05) compared with the cytokine accumulation in the control group. ICAC relieved the accumulation of IL-6, MCP-1, and TNF-α clearly at 3 and 6 dpi (Fig. 3F–H, P < 0.05). Additionally, MCP-1 secretion in the brain tissues declined to near normal levels in the presence or absence of ICAC administration at 9 dpi (Fig. 3G), but ICAC failed to decrease IL-6 and TNF-α secretion in response to EV71 infection at 9 dpi (Fig. 3F,H). Therefore, ICAC regulated the viral load and the secretion of cytokines, including IL-6, MCP-1, and TNF-α, in EV71-infected mouse brains, which confirmed the antiviral effects of ICAC.
ICAC mitigated the oxidative damage caused by EV71 infection via glutathione metabolism
To investigate the antiviral mechanism, the regulation of glutathione metabolism by ICAC was evaluated. Infection with 100 TCID50 of EV71 for 12 h induced a decrease in the GSH content that was coupled with an increase in GSSG in Vero cells (Fig. 4, P < 0.05). However, the ICAC treatment obviously reduced or eliminated the effects of EV71 infection on GSH and GSSG (Fig. 4, P < 0.05). Moreover, a quite high GSH/GSSG ratio, which is a key parameter for the intracellular redox status, was restored by ICAC addition compared with the ratio in the model group (Fig. 4B). Further analysis of GSH metabolic enzymes provided more insights into the exact mode of the antiviral action of ICAC against EV71. As shown, the activities of GR, GPX and G6PD in Vero cells were decreased by approximately 46.8%, 51.0% and 42.1%, respectively, following exposure to EV71 infection and were recovered by ICAC to varying degrees (Fig. 5). These results were consistent with the regulation of GSH redox homeostasis and the ROS level by ICAC (Fig. 4).

ICAC maintained GSH redox homeostasis in EV71-infected Vero cells. Infected (100 TCID50 of EV71) Vero cells were treated with medium or 100 µM ICAC for 12 h. Uninfected cells were used as the control group. The reduced and oxidized glutathione (GSH and GSSG) contents were analysed by HPLC (n = 3). All results were expressed as the means ± SEs. Asterisks indicate that the data significantly differ from the EV71 group at the P < 0.05 level according to one-way analysis of variance. The GSH/GSSG ratio was also calculated.

ICAC decreased the ROS level induced by EV71 infection via modulating the antioxidant enzymes involved in GSH metabolism. Infected (100 TCID50 EV71) Vero cells were treated with medium or 100 µM ICAC for 12 h. Uninfected cells were used as the control group. The antioxidant enzymes activities (A–C) and ROS levels (D) of the Vero cells were detected (n = 3). All results were expressed as the means ± SEs. Asterisks indicate that the data significantly differ from the EV71 group at the P < 0.05 level according to one-way analysis of variance.
BSO disrupted the antiviral effects of ICAC against EV71 in Vero cells
To validate the potential effects of GSH induced by ICAC, BSO, a previously described inhibitor of GSH biosynthesis was utilized. As shown in Fig. 6, BSO disrupted the cellular defence effects of ICAC in a dose-dependent fusion. For example, when the concentration of BSO was increased to 500 µM, the protection of ICAC on Vero cells upon EV71 was abolished totally (Fig. 6). Meanwhile, results from preliminary experiment showed that BSO only revealed minor cytotoxicity on cellular viability at concentrations up to 2 mM (data not shown), which indicated the concentration used in our study was atoxic to the cells. On the other hand, it was also observed that exogenous GSH failed to inhibit the CPE caused by EV71 infection (data not shown). Thus, the data here exhibited GSH played a key role in ICAC-induced inhibition on EV71 infection, which confirmed the antiviral mechanism of ICAC.

The antiviral effects of ICAC against EV71 were disrupted by BSO administration. Infected (100 TCID50 of EV71) Vero cells, treated with 100 µM ICAC, exposed to various concentrations of BSO (0~500 µM). Uninfected cells were used as the control group. Vero cells infected with 100 TCID50 of EV71 alone were used as the model group. Cell viability was detected using the MTS cell proliferation assay kit at 48 hpi. The viability of the control group was set as 100% (n = 6). Asterisks indicate that the data significantly differ from the ICAC group at the P < 0.05 level according to one-way analysis of variance.
Discussion
ICAC is a natural product from Lonicera japonica, which is a well-known traditional Chinese herb that is widely used in HFMD treatments33. In addition to the capacity of ICAC to inhibit RSV26, HIV27,28,29, and coxsackievirus30 replication, we confirmed that the compound inhibited EV71 infection in cellular and animal models. In this study, ICAC revealed inhibitory potential against EV71 replication in Vero cells. In EV71-challenged mice, ICAC administration remarkably improved the survival rates and symptoms, including emaciation and paralysis. The compound also reduced the viral load and cytokine secretion in the brain. Finally, biochemical analysis showed that ICAC regulated the GSH/GSSG ratio and ROS level via the antioxidative enzymes involved in GSH metabolism. Taken together, the results demonstrated that ICAC inhibited EV71 infection via modulating GSH redox homeostasis in Vero cells.
In this study, the cytotoxicity of ICAC was investigated in the Vero cell model, which demonstrated an atoxic dose range. Vero cells, which are sensitive to EV71 infection34,35,36,37, were chosen as the cellular model to assess the antiviral effects of ICAC against the virus. Herein, the results indicated that ICAC reduced the plaques and CPE caused by EV71 infection in a dose-dependent manner. The time-dependent assay showed suppression of the EV71 yield by ICAC, which explained the protective effect of ICAC on cell viability. Moreover, VP1 transcripts and protein synthesis, which are normally utilized for EV71 identification38,39, were decreased by ICAC treatment, which confirmed the inhibitory effect of ICAC on the EV71 yield. Taken together, these results demonstrate the antiviral capacity of ICAC against EV71. Previous reports have indicated that ICAC exhibits a broad antiviral spectrum26,27,28,29,30,40,41,42, and this study enriches the antiviral spectrum of ICAC. Our results indicate that ICAC may be the active ingredient that inhibits enteroviruses in the aqueous extract of Lonicera japonica 30 and the Reduning injection43. Moreover, ICAC exhibits potential utility in the control of HFMD via the necessary structural modification44.
The suckling mice challenged with EV71 gradually developed HFDM symptoms, including weight loss, paralysis, and death45,46,47. In this study, the mortality, weight loss, and limb paralysis caused by EV71 infection were alleviated significantly by ICAC administration. ICAC showed better effects than ribavirin injection, which is a clinical drug used for HFMD treatment48. Furthermore, ICAC decreased the viral load in the mouse brain49, which was confirmed by the results obtained in the cellular model. Previous research reported that ICAC inhibited HIV replication by inhibiting integrase activity28,29. However, the target through which ICAC reduced the EV71 yield is still unclear. Macrophages are an important target cell and may even be effectors of the EV71 attack6,50. Excessive cytokines and chemokines, including IL-6, MCP-1 and TNF-α, are secreted by the cells in response to the accumulation of viral copies6. In the central nervous system, these factors were proven to be responsible for the pathogenesis of the severe brainstem encephalitis and pulmonary oedema caused by EV71 infection9,10,11,51. In the present study, the steep accumulation of IL-6, MCP-1 and TNF-α in the infected mouse brains52,53 was reduced by ICAC, which correlated with the protection against mortality. Thus, the results revealed that ICAC inhibited EV71-induced mouse deaths by relieving the excessive viral load and cytokine secretion. However, the potential regulatory mechanisms underlying the effects of ICAC on the viral load and cytokine secretion should be discussed further.
To examine the antiviral mechanism of ICAC, we investigated modulation of GSH and its metabolism enzymes by the compound. GSH, which scavenges ROS in response to viral infection54, is an ideal biochemical to protect cells against oxidative stress. Maintenance of GSH redox homeostasis was reported to ameliorate cellular susceptibility to EV71 infection15. In this study, the depletion of the GSH pool induced by EV71 infection18 was reversed by ICAC supplementation, which correlated with the protection of ICAC on the cells upon to EV71 infection. Further biochemical analysis showed that ICAC up-regulated the enzymatic activities of GR, GPX and G6PD, which were responsible for GSH redox homeostasis. Additionally, these enzymes were indicated to play critical roles in the antiviral process. For example, GR and GPX exhibited protective effects against the myocarditis caused by coxsackievirus B355,56. The increase in enzymatic activities by antioxidant administration correlated with the improved tolerance against viral infection57,58,59. Moreover, the increased susceptibility to EV71 infection was reversed by both G6PD expression and exogenous NAC treatment (a well-known antioxidant) in G6PD-deficient cells, which ensured the anti-EV71 role of G6PD15. The up-regulation of the enzymes, responsible for GSH metabolism, not only alleviated the oxidative damage caused by viral infection, but also maintained the redox homeostasis of GSH in Vero cells infected by EV71. Finally, the inhibition effects of GSH induced by ICAC on EV71 infection was proven by BSO addition, which usually used for GSH-depletion. Normally, renal intracellular GSH is degraded by γ-glutamyl transpeptidase into dipeptides and amino acids, which are translocated into cells60,61. Those findings could explain the observation that exogenous supplementation of GSH failed to prevent cells from CPE caused by EV71 infection. The present results indicated that regulation of GSH redox homeostasis could be the potential antiviral mechanism of ICAC against EV71. However, GSH was reported to be a novel factor essential for coxsackievirus virion morphogenesis, which indicated that the function of GSH in viral infection should be limited to the maintenance of redox homeostasis62,63. By contrary, EV71 morphogenesis was not observed to rely on GSH63. Thus, the function and regulation of GSH in EV71 infection and defence may be complex.
In summary, in this study, we showed the antiviral effects of ICAC against EV71 by inhibiting the viral yield. The compound improved the mortality and symptoms caused by viral infection by decreasing the EV71 load and cytokine secretion in the brain. ICAC maintained the GSH/GSSG ratio and its metabolic enzymatic activities. The antiviral effects of ICAC was disrupted by BSO, a biosynthesis inhibitor of GSH. Taken together, these results suggested that ICAC prevented EV71 infection via modulating GSH redox homeostasis.
Methods
Chemicals
Isochlorogenic acid C (ICAC, C25H24O12; CAS No. 32451-88-0; MW 516.45) was purchased from the National Institutes for Food and Drug Control of China (Beijing, China). The MTS cell proliferation assay kit was purchased from Promega Biotech Co., Ltd. (Beijing, China). Glutathione, oxidized glutathione, and L-buthionine sulfoximine (BSO) were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Virus and cells
EV71-infected Vero cells were used as the infection model36,37. The virus strain was isolated and identified clinically (GenBank accession no. HQ882182)40. EV71 was propagated in Vero cells, and the titres were determined as previously described64. The Vero cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% foetal bovine serum (FBS, Gibco) at 37 °C in a humidified incubator with 5% CO2.
Animals
The 2-day-old BALB/c mice (1.6–2.0 g, SPF class) used as the animal model43,44,45 were purchased from Shanghai JSJ Experimental Animal Co. Ltd. (Shanghai, China). The mice were housed in an IVC system (temperature: 23 ± 2 °C, humidity: 40–70%, mechanical air supply, light-dark cycle: 12 h/12 h, illumination: 300 lux) with plenty of food and water. All animal experiment protocols were approved by Jiangsu Laboratory Animal Association (Licence number: SYXK(Jiangsu)2010-0010), which were conducted in accordance with the “Guiding Opinions on PETA’s” promulgated by Ministry of Science and Technology of China in 2006.
Cellular toxicity assay
Vero cells (1×104 cells/well) were seeded into 96-well plates and supplemented with various concentrations of ICAC as indicated in triplicate. The cell viability was detected using the MTS cell proliferation assay kit at 48 hours after addition.
Antiviral study
Vero cells (1×104 cells/well) seeded into 96-well plates were infected with EV71 (100 TCID50) at 37 °C for 2 h. After removing the virus, the cells were treated with various concentrations of ICAC as indicated in triplicate for 48 h. The plaque reduction assay was performed as previously described40,65. The cytopathic effects (CPEs) caused by the viral infection were measured quantitatively using the MTS cell proliferation assay kit according to the user manual. The concentration required for the 50% of maximal effect (EC50) and selection index (SI, SI = TC50/EC50) were calculated as previously described38. For the biochemical and molecular biology investigations, infected Vero cells (1 × 106 cells/well in a 6-well plate) were treated with 100 µM ICAC for 12 h and then lysed for extraction.
Time-course analysis of the EV71 yield
This analysis was performed as previously described9,66,67. Briefly, Vero cells (1 × 106 cells/well) seeded into 6-well plates were infected with EV71 (100 TCID50) at 37 °C for 2 h. After removing the virus, the infected cells were treated with 100 µM ICAC. The viral particles were collected by freeze-thawing at −80 °C at the indicated time points.
Viral load detection
Total RNA was isolated from the samples using the TRIzol reagent (Invitrogen, USA), and cDNA was synthesized using random hexamers with a reverse transcript kit (TaKaRa, China) according to the user manuals. The cDNA was subjected to the EV71 RNA Detection Kit (Shanghai ZJ Bio-Tech Co., Ltd) specific for the VP1 gene. Positive fragments adjusted to a series of concentrations were used as a standard curve.
Western blotting analysis
The samples were treated with RIPA lysis buffer (Beyotime Institute of Biotechnology, China) and centrifuged at 12,000 × g for 15 min at 4 °C. The protein concentrations were determined with a BCA protein assay kit (Beyotime Institute of Biotechnology, China). Western blotting analysis was performed as previously reported67.
Mouse protection assay
Suckling mice were intraperitoneally challenged with EV71 (1×107 TCID50). Infected mice were intraperitoneally injected with ICAC (1.6, 3.2, and 6.4 g/kg, qd) for 14 days. Ribavirin injection (10 mg/kg, qd) was used as a positive control. The model group was injected with the same volume (0.1 mL) of saline. The survival rates and body weights of the mice were monitored daily. For RNA and cytokine extraction, the mice were treated as described above, and the brains were sampled at 3, 6, and 9 dpi.
Cytokine quantification
The brain tissues (~150 mg) were homogenized in 1 mL of 50 mM Tris buffer (pH 8.0) containing 5 M guanidine-HCl. The homogenates were mixed on an orbital shaker at room temperature for 4 h. The samples were diluted with cold PBS with 1× protease inhibitor cocktail (Thermo Scientific, USA) and then centrifuged at 16,000 × g for 20 min at 4 °C. The supernatants were used for the enzyme-linked immunosorbent assay (ELISA). The IL-6, MCP-1 and TNF-α levels were measured using anti-mouse ELISA kits (eBioscience, San Diego, CA, USA) according to the manufacturer’s guidelines.
GSH and GSSG analyses
The GSH and GSSG levels were analysed by high-performance liquid chromatography (HPLC) as previously reported16.
Enzymatic activity assays
The cells were lysed on ice using the Membrane and Cytosol Protein Extraction Kit (Beyotime Institute of Biotechnology, China) for the biochemical assays. The GR activity was determined using the Glutathione Reductase Assay Kit (Abcam, UK) according to the user manual. One unit was defined as the amount of enzyme that catalysed the conversion of 1.0 μmol of GSSG into GSH and generated 1.0 μmol of 5-merapto-2-nitrobenzoic acid under the assay kit conditions per minute at 25 °C. The GPX activity was detected using the Glutathione Peroxidase Assay Kit (Abcam, UK) according to the user manual. One unit was defined as the amount of enzyme that caused the oxidation of 1.0 µmol of NADPH to NADP+ under the assay kit conditions per minute at 25 °C. The G6PD activity was estimated using the Glucose 6-Phosphate Dehydrogenase Kit (Abcam, UK) according to the user manual. One unit was defined as the amount of enzyme that catalysed the conversion of 1.0 μmol of glucose-6-phosphate into 6- phosphoglucono-δ-lactone and generated 1.0 μmol of NADH under the assay kit conditions per minute at 37 °C.
Determination of ROS
Vero cells (1 × 106 cells/well) seeded in 6-well plate were infected and treated as described above. The ROS level was detected using a Cellular Reactive Oxygen Species Detection Kit (Abcam, UK) according to user manual. The relative fluorescence units (RFU) was monitored at Ex/Em = 540/570 nm.
Statistics analysis
All results were expressed as the means ± SEs. The statistical significances of differences in the mean values were assessed with one-way analysis of variance (asterisk) or Chi-square test (cross) at the P < 0.05 level, respectively.
References
Schmidt, N. J., Lennette, E. H. & Ho, H. H. An apparently new enterovirus isolated from patients with disease of the central nervous system. J. Infect. Dis. 129, 304–309 (1974).
Huang, X. et al. Epidemiological and etiological characteristics of hand, foot, and mouth disease in Henan, China, 2008–2013. Sci. Rep. 5, 8904 (2015).
Mizuta, K. et al. Cross-antigenicity among EV71 strains from different genogroups isolated in Yamagata, Japan, between 1990 and 2007. Vaccine 27, 3153–3158 (2009).
Wang, Y. et al. Enterovirus 71 infection in children with hand, foot, and mouth disease in Shanghai, China: epidemiology, clinical feature and diagnosis. Virol. J. 12, 83 (2015).
Yang, F. et al. Enterovirus 71 outbreak in the People’s Republic of China in 2008. J. Clin. Microbiol. 47, 2351–2352 (2009).
Gong, X. et al. Excessive proinflammatory cytokine and chemokine responses of human monocyte-derived macrophages to enterovirus 71 infection. BMC Infect. Dis. 12, 224 (2012).
Han, J. et al. Serum cytokine profiles of children with human enterovirus 71-associated hand, foot, and mouth disease. J. Med. Virol. 86, 1377–1385 (2014).
Lin, T. Y. et al. Different proinflammatory reactions in fatal and non-fatal enterovirus 71 infections: implications for early recognition and therapy. Acta Paediatr. 91, 632–635 (2002).
Wang, S. M. et al. Pathogenesis of enterovirus 71 brainstem encephalitis in pediatric patients: roles of cytokines and cellular immune activation in patients with pulmonary edema. J. Infect. Dis. 188, 564–570 (2003).
Wang, S. M. et al. Cerebrospinal fluid cytokines in enterovirus 71 brain stem encephalitis and echovirus meningitis infections of varying severity. Clin. Microbiol. Infect. 13, 677–682 (2007).
Wang, S. M. et al. Acute chemokine response in the blood and cerebrospinal fluid of children with enterovirus 71-associated brainstem encephalitis. J. Infect. Dis. 198, 1002–1006 (2008).
Mao, Q. Y., Wang, Y., Bian, L., Xu, M. & Liang, Z. EV71 vaccine, a new tool to control outbreaks of hand, foot and mouth disease (HFMD). Expert Rev. Vaccines 15, 599–606 (2016).
Schwarz, K. B. Oxidative stress during viral infection: a review. Free Radic. Biol. Med. 21, 641–649 (1996).
Beck, M. A., Handy, J. & Levander, O. A. The role of oxidative stress in viral infections. Ann. N. Y Acad. Sci. 917, 906–912 (2000).
Ho, H. Y. et al. Glucose-6-phosphate dehydrogenase deficiency enhances enterovirus 71 infection. J. Gen. Virol. 89, 2080–2089 (2008).
Cai, J. et al. Inhibition of influenza infection by glutathione. Free Radic. Biol. Med. 34, 928–936 (2003).
Meister, A. & Anderson, M. E. Glutathione. Annu. Rev. Biochem. 52, 711–760 (1983).
Cheng, M. L., Weng, S. F., Kuo, C. H. & Ho, H. Y. Enterovirus 71 induces mitochondrial reactive oxygen species generation that is required for efficient replication. PLOS One 9, e113234 (2014).
Griffith, O. W. & Meister, A. Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (S-n-butyl homocysteine sulfoximine). J. Biol. Chem. 254, 7558–7560 (1979).
Smith, A. D. & Dawson, H. Glutathione is required for efficient production of infectious picornavirus virions. Virology 353, 258–267 (2006).
Aniya, Y. et al. Free radical scavenging and hepatoprotective actions of the medicinal herb, Crassocephalum crepidioides from the Okinawa Islands. Biol. Pharm. Bull. 28, 19–23 (2005).
Hwang, S. H., Paek, J. H. & Lim, S. S. Simultaneous ultra performance liquid chromatography determination and antioxidant activity of linarin, luteolin, chlorogenic acid and apigenin in different parts of Compositae species. Molecules 21, 1609 (2016).
Guo, W. et al. Isolation of isochlorogenic acid isomers in flower buds of Lonicera japonica by high-speed counter-current chromatography and preparative high performance liquid chromatography. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 981–982, 27–32 (2015).
dos Santos, M. D. et al. Effects of caffeoylquinic acid derivatives and C-flavonoid from Lychnophora ericoides on in vitro inflammatory mediator production. Nat. Prod. Commun. 5, 733–740 (2010).
Park, K. H. et al. The anti-oxidative and anti-inflammatory effects of caffeoyl derivatives from the roots of Aconitum koreanum R. Raymond. Biol. Pharm. Bull. 32, 2029–2033 (2009).
Ooi, L. S., Wang, H., He, Z. & Ooi, V. E. Antiviral activities of purified compounds from Youngia japonica (L.) DC (Asteraceae, Compositae) (Ooi, L. S., Wang, H., He, Z. & Ooi, V. E. Antiviral activities of purified compounds from Youngia japonica (L.) DC (Asteraceae, Compositae). J. Ethnopharmacol. 106, 187–191 (2006).
Heyman, H. M. et al. Identification of anti-HIV active dicaffeoylquinic- and tricaffeoylquinic acids in Helichrysum populifolium by NMR-based metabolomic guided fractionation. Fitoterapia 103, 155–164 (2015).
Robinson, W. E. Jr., Reinecke, M. G., Abdel-Malek, S., Jia, Q. & Chow, S. A. Inhibitors of HIV-1 replication [corrected; erratum to be published] that inhibit HIV integrase. Proc. Natl Acad. Sci. USA 93, 6326–6331 (1996).
Robinson, W. E. Jr. et al. Dicaffeoylquinic acid inhibitors of human immunodeficiency virus integrase: inhibition of the core catalytic domain of human immunodeficiency virus integrase. Mol. Pharmacol. 50, 846–855 (1996).
Yu, Y. et al. Homosecoiridoid alkaloids with amino acid units from the flower buds of Lonicera japonica. J. Nat. Prod. 76, 2226–2233 (2013).
Li, X. et al. Chlorogenic acid inhibits the replication and viability of enterovirus 71 in vitro. PLOS One 8, e76007 (2013).
Jiang, P., Sheng, Y. C., Chen, Y. H., Ji, L. L. & Wang, Z. T. Protection of Flos Lonicerae against acetaminophen-induced liver injury and its mechanism. Environ. Toxicol. Pharmacol. 38, 991–999 (2014).
Chen, X. et al. A laboratory evaluation of medicinal herbs used in china for the treatment of hand, foot, and mouth disease. Evid. Based Complement. Alternat. Med. 2013, 504563 (2013).
Chang, S. C., Lin, J. Y., Lo, L. Y., Li, M. L. & Shih, S. R. Diverse apoptotic pathways in enterovirus 71-infected cells. J. Neurovirol. 10, 338–349 (2004).
Xi, X. et al. The interplays between autophagy and apoptosis induced by enterovirus 71. PLOS One 8, e56966 (2013).
Wang, J. et al. Glycyrrhizic acid as the antiviral component of glycyrrhiza uralensis Fisch. Against coxsackievirus A16 and enterovirus 71 of hand foot and mouth disease. J. Ethnopharmacol. 147, 114–121 (2013).
Weng, T. Y. et al. Lactoferrin inhibits enterovirus 71 infection by binding to VP1 protein and host cells. Antiviral Res. 67, 31–37 (2005).
Liu, J. et al. Lycorine reduces mortality of human enterovirus 71-infected mice by inhibiting virus replication. Virol. J. 8, 483 (2011).
Oberste, M. S., Nix, W. A., Maher, K. & Pallansch, M. A. Improved molecular identification of enteroviruses by RT-PCR and amplicon sequencing. J. Clin. Virol. 26, 375–377 (2003).
Wang, C. Y. et al. Eupafolin and ethyl acetate fraction of Kalanchoe gracilis stem extract show potent antiviral activities against enterovirus 71 and coxsackievirus A16. Evid. Based Complement. Alternat. Med. 2013, 591354 (2013).
Wang, G. F. et al. Anti-hepatitis B virus activity of chlorogenic acid, quinic acid and caffeic acid in vivo and in vitro. Antiviral Res. 83, 186–190 (2009).
Xie, Y. et al. Caffeic acid derivatives: a new type of influenza neuraminidase inhibitors. Bioorg. Med. Chem. Lett. 23, 3556–3560 (2013).
Cao, Z. Y., Chang, X. J., Zhao, Z. P., Cao, L. & Xiao, W. Antiviral effects of Reduning injection against enterovirus 71 and possible mechanisms of action. Chin. J. Nat. Med. 13, 881–888 (2015).
Huang, L. H. et al. Pharmacokinetics of isochlorgenic acid C in rats by HPLC-MS: absolute bioavailability and dose proportionality. J. Ethnopharmacol. 185, 105–109 (2016).
Li, P. et al. Genome analysis of enterovirus 71 strains differing in mouse pathogenicity. Virus Genes 52, 161–171 (2016).
Li, Z. et al. In vivo time-related evaluation of a therapeutic neutralization monoclonal antibody against lethal enterovirus 71 infection in a mouse model. PLOS One 9, e109391 (2014).
Lin, P., Gao, L., Huang, Y., Chen, Q. & Shen, H. An enterovirus 71 strain causes skeletal muscle damage in infected mice. Int. J. Clin. Exp. Pathol. 8, 3460–3468 (2015).
Li, Z. H. et al. Ribavirin reduces mortality in enterovirus 71-infected mice by decreasing viral replication. J. Infect. Dis. 197, 854–857 (2008).
Lee, Y. R. W., Wang, P. S., Wang, J. R. & Liu, H. S. Enterovirus 71-induced autophagy increases viral replication and pathogenesis in a suckling mouse model. J. Biomed. Sci. 21, 80 (2014).
Wang, S. M. et al. Enterovirus 71 infection of monocytes with antibody-dependent enhancement. Clin. Vaccine Immunol. 17, 1517–1523 (2010).
Husseini, R. H., Sweet, C., Collie, M. H. & Smith, H. Elevation of nasal viral levels by suppression of fever in ferrets infected with influenza viruses of differing virulence. J. Infect. Dis. 145, 520–524 (1982).
Hsiao, H. B. et al. Toll-like receptor 9-mediated protection of enterovirus 71 infection in mice is due to the release of danger-associated molecular patterns. J. Virol. 88, 11658–11670 (2014).
Lee, Y. P., Wang, Y. F., Wang, J. R., Huang, S. W. & Yu, C. K. Enterovirus 71 blocks selectively type I interferon production through the 3C viral protein in mice. J. Med. Virol. 84, 1779–1789 (2012).
Giblin, F. J. Glutathione: a vital lens antioxidant. J. Ocul. Pharmacol. Ther. 16, 121–135 (2000).
Beck, M. A., Esworthy, R. S., Ho, Y. S. & Chu, F. F. Glutathione peroxidase protects mice from viral-induced myocarditis. FASEB J. 12, 1143–1149 (1998).
Chen, L. Y., Tian, X. L. & Yang, B. A study on the inhibition of rat myocardium glutathione peroxidase and glutathione reductase by moniliformin. Mycopathologia 110, 119–124 (1990).
Beck, M. A., Williams-Toone, D. & Levander, O. A. Coxsackievirus B3-resistant mice become susceptible in Se/vitamin E deficiency. Free Radic. Biol. Med. 34, 1263–1270 (2003).
Sartori, G. et al. Antiviral action of diphenyl diselenide on herpes simplex virus 2 infection in female BALB/c mice. J. Cell. Biochem. 117, 1638–1648 (2016).
Smith, A. D., South, P. K. & Levander, O. A. Effect of gold(I) compounds on the virulence of an amyocarditic strain of coxsackievirus B3. Biol. Trace Elem. Res. 84, 67–80 (2001).
Griffith, O. W. & Meister, A. Glutathione: interorgan translocation, turnover, and metabolism. Proc. Natl. Acad. Sci. USA 76, 5606–5610 (1979).
Griffith, O. W. & Meister, A. Translocation of intracellular glutathione to membrane-bound γ-glutamyl transpeptidase as a discrete step in the γ-glutamyl cycle: glutathionuria after inhibition of transpeptidase. Proc. Natl. Acad. Sci. USA 76, 268–272 (1979).
Ma, H. C. et al. An interaction between glutathione and the capsid is required for the morphogenesis of C-cluster enteroviruses. PLOS Pathog. 10, e1004052 (2014).
Thibaut, H. J. et al. Binding of glutathione to enterovirus capsids is essential for virion morphogenesis. PLOS Pathog. 10, e1004039 (2014).
Reed, L. J. M. & Muench, H. A simple method of estimating fifty percent endpoints. Am. J. Hyg. 27, 493–497 (1938).
Lin, Y. J. et al. Inhibition of enterovirus 71 infections and viral IRES activity by Fructus Gardeniae and geniposide. Eur. J. Med. Chem. 62, 206–213 (2013).
Cao, Z. et al. Luteoloside acts as 3C protease inhibitor of enterovirus 71 in vitro. PLOS One 11, e0148693 (2016).
Lu, J. et al. Viral kinetics of enterovirus 71 in human abdomyosarcoma cells. World J. Gastroenterol. 17, 4135–4142 (2011).
Acknowledgements
This work was financially supported by National Major New Drugs Innovation and Development Project by Ministry of Science and Technology (2013ZX09402203). The EV71 stain was kindly supplied by Dr. Xilang Wang from Institution of Microbiology and Epidemiology, Academy of Military Medical Sciences, People’s Liberation Army of China (Beijing, China).
Author information
Authors and Affiliations
Contributions
Z.C. planned and performed the cellular experiments, and wrote the manuscript. Y.D. performed the animal experiments. L.C. analyzed the data. G.D. and Z.W. reviewed the data and manuscript. W.X. designed the research. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
We have the following interests. All authors are employed by Jiangsu Kanion Pharmaceutical Co., Ltd. There are no patents, products in development or marketed products to declare. This does not alter our adherence to all the Scientific Reports on sharing data and materials, as detailed online in the guide for authors.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cao, Z., Ding, Y., Cao, L. et al. Isochlorogenic acid C prevents enterovirus 71 infection via modulating redox homeostasis of glutathione. Sci Rep 7, 16278 (2017). https://doi.org/10.1038/s41598-017-16446-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-16446-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
