
Abstract
Low socioeconomic status (SES) is associated with earlier onset of age-related chronic conditions and reduced life-expectancy, but the underlying biomolecular mechanisms remain unclear. Evidence of DNA-methylation differences by SES suggests a possible association of SES with epigenetic age acceleration (AA). We investigated the association of SES with AA in more than 5,000 individuals belonging to three independent prospective cohorts from Italy, Australia, and Ireland. Low SES was associated with greater AA (β = 0.99 years; 95% CI 0.39,1.59; p = 0.002; comparing extreme categories). The results were consistent across different SES indicators. The associations were only partially modulated by the unhealthy lifestyle habits of individuals with lower SES. Individuals who experienced life-course SES improvement had intermediate AA compared to extreme SES categories, suggesting reversibility of the effect and supporting the relative importance of the early childhood social environment. Socioeconomic adversity is associated with accelerated epigenetic aging, implicating biomolecular mechanisms that may link SES to age-related diseases and longevity.
Similar content being viewed by others

Introduction
Lower socioeconomic status (SES) is associated with lower life expectancy and earlier onset of age-related chronic conditions, based on robust worldwide evidence from studies using a range of SES and health indicators1,2,3,4. A recent study investigating the association of low SES with health in more than 1.7 million individuals, confirmed low SES as an independent predictor of premature mortality, with an associated effect size comparable to, and independent of that of the main non communicable disease (NCD) risk factors, including smoking, physical inactivity, higher alcohol intake, obesity, and hypertension5. Although SES is an overarching health determinant, with NCD risk factors unevenly distributed between SES strata, SES-health associations are only partly explained by the unhealthy lifestyle habits of individuals with lower SES5,6.
Despite extensive research efforts, the biological mechanisms that mediate the impact of SES on age-related conditions are still not fully understood. Systemic inflammation and immunological impairment in response to psychosocial stressors have been proposed as mechanisms through which SES is biologically embedded. Pivotal studies in macaques detected altered levels of expression and methylation in inflammatory and immune function-related genes depending on dominance rank (a proxy for SES), which were reversible following changes in dominance rank7,8. In humans, low SES has been associated with higher inflammatory status, the latter measured using epigenetic9,10,11,12, transcript13, and protein14,15 biomarkers. Interestingly, SES in early life appears particularly important in predicting higher inflammation status later in life14. Recent studies have employed multi-biomarker indicators of physiological function such as allostatic load, which incorporates markers of stress response, inflammation, and cardio-metabolic function, to link social adversity to health status4,16,17.
Accelerated biological aging, which may be measured through a multiple biomarker approach, is likely to be the result of several different physiological and pathological changes during the life-course, and therefore may represent an overarching mechanism linking SES and health. Recently, Horvath developed a multi-tissue predictor, that allows the age of most tissues and cell types to be estimated based on DNA methylation (DNAm) at 353 CpG sites18, while Hannum et al.19 developed a blood-specific DNAm age predictor based on levels of 71 CpG sites. These predictors, known as ‘epigenetic clocks’, allow estimation of whether an individual is experiencing accelerated or decelerated aging by defining age acceleration (AA) as the difference between DNAm age and chronological age. Recently, the ‘intrinsic’ AA defined as the residual from the regression of AA on chronological age and white blood cell (WBC) percentages, was proposed as a more reliable estimate of biological aging as it captures cell-intrinsic properties of the aging process that exhibit some conservation across various cell types and organs20.
The present study examines the association of SES with intrinsic AA in 5,111 adults from three large prospective cohorts: the Italian component of the European Prospective Investigation into Cancer and Nutrition (EPIC, Italy), the Melbourne Collaborative Cohort Study (MCCS, Australia), and The Irish LongituDinal study on Aging (TILDA, Ireland). We used a standardized measure of educational attainment (that is directly comparable across countries) as a proxy for SES. Further, we used a measure that incorporates SES in early life and adulthood (based on occupational position) to explore the association between SES changes across the life-course and AA. Finally, we investigated the role of NCD risk factors in modulating the SES-AA association.
Results
Study Populations
After pre-processing, quality controls, and sample filtering, 5,111 subjects were included in the analyses. There were several differences by study in the distribution of demographic variables and NCD risk factors (Table 1). Table 2 shows demographic and covariate information by SES categorized as ‘low’, ‘medium’ and ‘high’ (Methods). In the linear regression adjusted for study area, obesity (lower BMI in higher SES group), diet (healthier diet in higher SES group), and physical activity (lower percentage of inactive individuals in higher SES group), were associated with SES, whereas no significant differences were observed for smoking habits and alcohol consumption (Table 2).
Age Acceleration (AA) measures
We estimated the epigenetic age of each blood sample using both the approach by Horvath based on 353 CpGs18, and the approach by Hannum et al. based on 71 CpGs19. Out of the 418 age-related CpGs (347 specific for the Horvath measure, 65 specific for the Hannum measure, and 6 CpGs that are in common between the two indicators), we detected 414 CpGs (99%) in EPIC, 416 (99.5%) in MCCS, and 396 (95%) in TILDA. The CpGs identified in EPIC and MCCS but not in TILDA are those that are not present in the new Illumina 850k methylation BeadChip, that has been used for the Irish cohort only (see Supplementary Methods). All the probes had less than 5% missing in the study sample. For the SES-AA associations, we used the epigenetic age measures computed after imputation of missing data, after verifying the concordance with those obtained without the imputation procedure (Pearson correlation coefficients > 0.99; p < 2 × 10−16).
Both DNAm age estimates were highly correlated with chronological age (Pearson correlation coefficients ranging from 0.73 to 0.80). Also, the Horvath and Hannum estimates were highly correlated with each other (Pearson correlation coefficients ranging from 0.80 to 0.92; Figure S1).
Hannum DNAm age was a slightly better predictor of chronological age than Horvath DNAm age. The Pearson correlation coefficients ranged from 0.73 to 0.77 for the Horvath measure, and from 0.74 to 0.80 for the Hannum measure. The average absolute difference (± standard deviation) between DNA methylation predicted age and chronological age was 4.03 ± 3.56 for Horvath measure, and 3.72 ± 3.25 for Hannum measure. Given the above, and since Hannum AA is more specific for DNA methylation in blood cells, we present associations of SES with the Hannum intrinsic AA (referred to hereafter as merely ‘AA’) in the main text. Associations with Horvath intrinsic AA are presented in the supplementary material.
Table 3 shows the results for the association of NCD risk factors with intrinsic AA measures. All the NCD risk factors with the exception of physical activity were significantly associated with epigenetic aging, AA being higher in men, obese people, smokers, habitual alcohol drinkers, and individuals with unhealthy diets.
SES – AA association
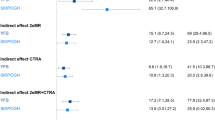
In the meta-analysis of the three study results, SES was associated with AA (Table 4, Fig. 1a) in the basic adjusted model. The effect sizes (interpretable as years of increase in epigenetic age) were β = 0.75 (95% confidence interval (CI): 0.17, 1.34; p = 0.01) and β = 0.99 (95% CI: 0.39, 1.59; p = 0.001) comparing participants of medium SES and low SES with those of high SES, respectively. The estimated linear trend of increased AA per decrease in SES was β = 0.42 (95% CI: 0.15, 0.68; p = 0.002). Similar effect estimates were observed in the single cohorts, with the association being significant in EPIC Italy and MCCS (Table 4). Comparable results were observed using Horvath intrinsic AA as the outcome (Table S1), as well as using alternative SES indicators like the index of relative socio-economic disadvantage (IRSD): β = 0.91 (95% CI: 0.21, 1.62; p = 0.01, Table S6), and household income: β = 0.66 (95% CI: −1.03, 2.35; p = 0.45, Table S7). Further, we show that different SES indicators are strongly associated among themselves (Table S8).

SES–AA association: Bar-plots indicating the estimated effect sizes (in years) and standard errors of the association of SES with Hannum AA (a: model 1 with basic adjustments, b: model 2 adjusted for NCD risk factors), and life-course SES trajectory with Hannum AA (c: model 1 with basic adjustments, d: model 2 adjusted for NCD risk factors).
Basic adjusted model comparison with NCD risk factors adjusted model
We evaluated the change in the magnitude of the SES-AA association due to the inclusion of NCD risk factors in the model, by comparing the estimated effects (βs) of the basic regression model (adjusted for age and sex) with those of the models including different NCD risk factors. NCD risk factors considered were smoking status, BMI, alcohol intake, Mediterranean Diet Score, and physical activity. Additional covariates were first added to the basic adjusted model one-by-one and finally included all together in the ‘fully adjusted’ model (Table 5).
None of the NCD risk factors were associated with a statistically significant reduction in the effect size for the SES-AA association. In the fully adjusted model, the decrease in the effect size was close to being significant. The effect size attenuation for the linear trend was 12% (95% CI: −0.02, 0.26; p = 0.09), suggesting that only part of the SES-AA association could be explained by these risk factors (Table 5, Fig. 1b). The most substantial contribution to the reduction of the effect size for the SES-AA association was observed when including smoking in the regression model, which attenuated the estimate for linear trend by 6% (95% CI: −11, 22). Effect size reductions due to the inclusion of other NCD risk factors were small and non-significant.
Life-course SES trajectory – AA association
The life-course SES trajectory is based on father’s occupational position (a proxy for early life SES), and highest occupational position (a proxy for adulthood SES). It is defined as a categorical variable with four levels, corresponding to four possible SES trajectories: high SES in childhood - high SES in adulthood (stable professional, the reference group), high SES in childhood - low SES in adulthood (any downward mobility), low SES in childhood - high SES in adulthood (any upward mobility), and low SES in childhood - low SES in adulthood (stable unskilled). The measure was not available for the MCCS cohort.
In the meta-analysis of EPIC Italy and TILDA results (Table 6, Fig. 1c), we observed a positive trend of increase in AA with decreasing SES (β = 0.78, 95% CI: −0.33, 1.89; p = 0.17, comparing the extreme categories: stable unskilled and stable professional), with some attenuation after further adjustment for NCD risk factors (Table 6, Fig. 1d, β = 0.77; 95% CI: −0.37, 1.92; p = 0.19), although these associations did not reach statistical significance. Interestingly, individuals who experienced changes in SES over their lifetime (any upward or downward social mobility) had an AA that was intermediate between the stable professional and the stable unskilled groups, and AA was greater for individuals with low SES in childhood than for those with low SES in adulthood (Table 6, Fig. 1c,d).
The estimated effect size was greater and significant in men compared with women, with the association being significant in men when comparing the extreme categories (Table S3), though there was no evidence of SES-gender interaction (Tables S4 and S5).
Sensitivity analysis
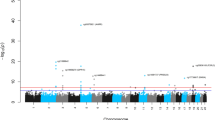
Figure 2 shows repetitions of meta-analyses for SES and life-course SES trajectory stratified by sex and age (younger than, and older than 60 years old). The estimated magnitude of the association was overall consistent across genders and age groups (Fig. 2), with no evidence of SES-gender and SES-age interaction (Table S5).

Sensitivity analysis: Forest-plots indicating the estimated effect sizes (in years; black dots) and 95% confidence intervals (horizontal lines) for the association of SES (a: three studies meta-analysis) and life-course SES trajectory (b: EPIC Italy and TILDA meta-analysis) with Hannum AA, estimated each time in different subsets of the overall sample.
Further, SES-AA associations were assessed in subsets of the overall sample, excluding each time individuals exposed to NCD risk factors: current smokers, habitual drinkers, physically inactive, obese (BMI > 30), and individuals with unhealthy diet (Mediterranean diet score <= 4) (Fig. 2). The estimated effects were comparable to those estimated on the whole sample but with lower statistical significance due to the reduced statistical power. Finally, we examined the SES-AA associations after exclusion of individuals exposed to at least one NCD risk factor and of incident cancer/cardiovascular disease cases. This procedure led to the analysis of 375 Italians, 1,106 Australians and 68 Irish NCD risk factor free individuals (non-obese, non-smokers, physically active, non-drinkers and with a healthy diet). Notably, the association of SES with AA was still significant in this NCD risk factor free subsample (test for linear trend β = 0.62; 95% CI: 0.06, 1.18; p = 0.03; N = 1,549; Fig. 2a).
Other SES indexes – AA association
In MCCS and TILDA cohorts we tested the association of AA with alternative SES indicators. In MCCS, AA was also significantly associated with the index of relative socioeconomic disadvantage (IRSD, Table S6) that is a general index summarizing the socioeconomic conditions of individuals within a given area defined by the Australian Bureau of Statistics21. In TILDA, increasing AA with decreasing income was observed, with estimates comparable to those described for the other SES variables. However, the association was not statistically significant due to the reduced statistical power (Table S7).
Discussion
We explored the association between multiple SES indicators and epigenetic aging in three large cohort studies involving more than 5000 individuals. Lower SES was associated with accelerated epigenetic aging compared to higher SES, with in-between effects for intermediate SES.
In the three cohorts, adult SES was assessed using the highest level of educational attainment, a good proxy for SES that is usually completed before the onset of many chronic diseases, therefore reducing the risk of reverse causation22. Further, we have analyzed alternative SES indicators based on occupational position, household income, and a deprivation index. We investigated the association of these different SES indices with the ‘intrinsic’ AA defined as the residuals from the regression of AA on chronological age and WBC percentages20. According to Horvath and colleagues, this measure is a more reliable indicator of accelerated aging than ‘crude’ AA because it captures cell-intrinsic properties of the aging process that exhibit some preservation across various cell types and organs20. From a statistical point of view, the described procedure allows us to avoid bias due to the correlation of AA with chronological age and the estimated percentages of WBC.
The main finding of this study is the observed graded relationship between SES (variously defined) and AA. The biological (epigenetic) age of low SES individuals was estimated to be on average one year higher than for high SES subjects. We obtained consistent results using two alternative SES indicators: the IRSD and the household income (Tables S6 and S7), due to the high association between different SES indicators (Table S8). According to a recent meta-analysis investigating the association of epigenetic age with mortality in more than 13,000 individuals20, our estimate corresponds to an increased risk of death in the monitored time period (around 11 years of follow up on average) with a hazard ratio (HR) ranging between 1.01 and 1.04, depending on the AA measure.
The increased AA associated with low SES was lower than that estimated for smoking (1.5 years increase in AA for current smokers compared with never smokers), and comparable to that estimated for obesity (one year increase in AA per 10 unit increase in BMI), unhealthy diet (one year increase in AA comparing Mediterranean diet score extreme categories), and alcohol consumption (0.75 years increase in AA for habitual alcohol consumers compared to no/moderate consumers). These results are consistent with our recent meta-analysis of 48 independent cohorts involving more than 1.7 million subjects5, that estimated a two year reduction in life expectancy for low SES compared to high SES individuals, with an effect comparable to those of other risk factors.
SES trajectory and reversibility of the effect
Our results also suggest that the relationship between SES and AA may be responsive to early life social influences. Recent research shows that effects of in-utero and early life exposures (including those associated with SES) may be stored in cells through epigenetic modifications that can be sustained for decades23,24,25,26. It can be speculated that long-term effects of early life exposures may be due to their impact on somatic stem cell populations, which persist as a form of cellular memory, including changes in DNAm patterns. One of the aims of this study was to investigate whether the association of SES with AA is reversible. It was possible to address this question in the Italian and Irish cohorts using the cross-classification of childhood and adulthood social class (early life SES was not available for the MCCS cohort). Our results suggest that AA for individuals who experienced downward mobility (high childhood SES, low adulthood SES) is more comparable to that of the stable professional group, and lower than that for the upwardly mobile (low childhood SES, high adulthood SES), supporting previous observations of the relative importance of the early childhood social environment27,28,29. Moreover, AA was higher for individuals whose childhood SES was low and remained low in adulthood compared with those experiencing upward mobility (low SES in childhood, high SES in adulthood). This pattern is consistent with some degree of reversibility of the unfavorable effect of childhood social adversity.
The role of NCD risk factors in modulating SES-AA association
Due to the relationship of NCD risk factors with both SES and AA (Tables 2 and 3), we evaluated regression models adjusted for potential confounders, and then sequentially adjusted for mediators, to assess the change in the estimated effect of SES on AA. The concepts of ‘confounder’ and ‘mediator’ are often misinterpreted, particularly when referring to SES, which is an overarching determinant of health. Mediators and confounders are similar except for the direction of the relationship between them and the main exposure, in this case, SES30. In our analyses, chronological age and sex were potential confounders because they influence SES (not vice versa) and affect the outcome (AA), but they do not belong to the causal pathway between SES and health. Instead, lifestyle-related behaviors (smoking habits, BMI, alcohol intake, physical activity, and diet) have to be considered as mediating factors as they are influenced by SES and are simultaneously major risk or protective factors for health-related outcomes. They contribute to the SES-AA association by being located on the presumed causal pathway between SES and health5.
In our analyses, we did not observe a significant reduction of the association magnitude due to the inclusion of mediators in the regression model. The associations were robust to adjustment for mediators, although with slightly reduced effect size. The most significant contribution to the partial effect attenuation was observed when including smoking in the statistical model. This pattern of findings suggests that only part of the observed association between SES and AA could be explained by unhealthy lifestyle habits of individuals with lower SES, with smoking being one of the most significant mediators. The residual association is likely attributable to an altered inflammatory status, that is known to be associated at the same time with unhealthy lifestyle (e.g. smoking, poor physical activity)31,32, lower SES14, and accelerated aging33. Further, health inequalities across different SES groups have been explained with disparities in the allostatic load (AL) index17,34. AL is a commonly used metric of health based on the hypothesis that multiple exposures to stressors lead to a progressive dysregulation of different physiological systems34. Disparities in AL have been described as a consequence of childhood social adversities16,35, further supporting our findings on the relative importance of the early life social environment.
We conducted extensive sensitivity analyses confirming the SES-AA association in different subsets of the overall sample and using different SES indicators. Further, to rule out potential ‘collider bias’36, we verified the association in the subgroup of NCD risk factor free individuals (non-obese, non-smokers, non-drinkers, physically active, with a healthy diet, and non-incident cancer/cardiovascular cases; N = 1,549).
Strengths and limitations
Our study has some limitations. The Italian study sample is enriched for incident cases of breast, colon, and lung cancers, lymphoma, and myocardial infarction (though blood samples were collected several years before the disease diagnosis), and the Australian study sample included 11% of controls that were matched to lung cancer cases for smoking status, leading to a slight over-representation of smokers. Conversely, the sample from the Irish TILDA cohort is representative of the national population aged 50 years and over, but the subgroup selected for these analyses was explicitly chosen to examine social mobility associations with AA with the four life-course SES trajectories being equally represented. While most NCD risk factors were measured with the same degree of accuracy as SES, diet is more difficult to measure. No measure of diet comparable to the Mediterranean diet score was available in the TILDA cohort. Therefore we may not have been able to thoroughly assess the contribution of diet as a mediator in the SES-AA association. Also, further investigation is needed to evaluate the role of inflammation in modulating the SES-AA association properly.
Although the association of SES with epigenetic aging was already described in two independent Afro-American cohorts37,38, these studies were based on limited samples of women (N = 100) and teenagers (N = 292). Further, extrinsic epigenetic AA, but not intrinsic epigenetic AA was associated with educational attainment and income in a cross-sectional study involving 4,173 postmenopausal women33. Epidemiology is based on an accumulation of evidence, and the extent to which SES is linked to AA has not been fully investigated in other populations until now. Also, we provide further evidence of the relative importance of the early life social environment, and suggest some degree of reversibility of epigenetic changes, as observed more prominently for smoking-associated methylation25, which has important policy implications.
Policy implications
The implications of long-lasting impacts of life experiences, and particularly SES, on the modulation of epigenetic variations are vast, particularly concerning their public policy significance. While most policies targeted at poverty are focused on adults, such as the unemployed or workers with low incomes (for example, the Earned Income Tax Credit program in the USA), some of these policies also cover infancy. Examples of the latter are Conditional Cash Transfer programs that incentivize schooling and health programs for children39.
Our findings in the Italian and Irish cohorts suggest thatearlier interventions are likely to pay greater dividends than interventions later in life. A related question is whether and to what extent epigenetic changes are reversible, and if they are, which targeted interventions could be most beneficial. Both our previous work12,15 and the current study suggest that the relationship between SES and methylation is more pronounced in individuals whose SES starts low and remains low later in life, compared to those whose status improves during the life-course, while intermediate changes were found in subjects whose SES declined from high to low. This pattern of findings is consistent with some degree of reversibility.
Conclusions
To our knowledge, this is the largest study investigating the relationship between SES and DNAm age (the ‘epigenetic clock’) in peripheral blood in adults. Our results confirm previous observations that SES is a determinant of health that goes beyond the major risk factors for diseases and may involve independent biological mechanisms37,40. Also, our findings support the hypothesis of a life-course accumulation of exposures and suggest some degree of reversibility of the effect, which has significant policy implications. More generally, our approach based on epigenetic measurements may contribute to the identification of SES-specific mechanisms that influence aging and health.
Methods
Details on subject recruitment and relevant demographic and lifestyle variables acquisition are presented in supplementary text.
Socioeconomic status assessment
In all cohorts, the highest level of educational attainment was used as a proxy for SES. To avoid bias due to the different proportions of educational qualifications by gender, birth cohort and study centers, a standardized version education was computed as follows: 1) the highest educational attainment was categorized as primary school or none, vocational or another secondary school, and university or vocational postsecondary school; 2) individuals were grouped for gender, center of recruitment, ethnicity (in MCCS only as Mediterranean or Anglo-Saxon) and 10-year age groups; 3) for each group the proportion of individuals in each educational level was computed; 4) a score was computed by taking into account the distribution of educational level in each group. For example, if within a given group 60% of participants were in the higher educational level, 30% were in the middle educational level and 10% were in the lower educational level, in that group each individual in the higher educational level would be assigned a score of 0.30 (0.60/2), each participant in the intermediate stratum would be assigned a score of 0.75 (0.60 of the first level, plus 0.30/2), and finally, the remaining 10% of subjects in the lower educational stratum would receive a score of 0.95 (0.60 of the first level + 0.30 of the second level + 0.10/2). This calculation was performed for each specific category and provides a continuous score varying from 0 to 1, in which higher values correspond to lower SES2,41. For statistical analysis the defined variable was categorized in tertiles labelling the three categories as ‘high’ (1st tertile), ‘medium’ (2nd tertile) and ‘low’ (3rd tertile) SES.
Life-course SES trajectory
In the EPIC Italy and TILDA cohorts, participants were asked to report their own, their father’s and their partner’s occupational position in a brief questionnaire administered the day of blood collection. Father’s occupational position and highest occupational position (both categorized as ‘low’ and ‘high’) were used as proxies for childhood SES and adulthood SES respectively. The two variables were further combined to create an indicator of the life-course SES trajectory. Further details on the definition of the life-course SES trajectory are given in supplementary material and Stringhini et al.12.
Statistical analyses
To avoid bias due to the different distribution of NCD risk factors by cohort, all the SES-AA associations were tested independently for each study and the results meta-analyzed. We used fixed-effect meta-analysis (inverse variance weights) to obtain pooled estimates for SES-AA associations. The I2 statistic was used to assess the percentage of variance that is attributable to study heterogeneity42. No correction for multiple testing was applied since the SES variables were not mutually independent (Table S8), nor were the two AA measures (Figure S1).
Age Acceleration
DNA methylation age was computed according to the algorithm described by Horvath18, based on a set of 353 age-associated CpG sites, and the one based on 71 blood-specific age-associated CpG sites described by Hannum et al.19. Briefly, the DNA methylation age is computed as a weighted average of the age-related CpGs, with weights defined using a penalized regression model (Elastic-net regularization)18. The few missing values were imputed using the k-nearest neighboring (KNN) imputation algorithm implemented in the R Bioconductor package impute 43. Age acceleration (AA) was defined as the difference between epigenetic and chronological age. Positive values of AA (that is epigenetic age is higher than the chronological age) indicate accelerated aging and vice versa. Since AA could be correlated with chronological age and WBC percentage, we computed the so-called ‘intrinsic’ AA20, defined as the residuals from the linear regression of AA with chronological age and WBC percentages. The latter were estimated using the Houseman algorithm44. Intrinsic AA is not dependent on age and WBC by definition. The two AA measures are referred to as ‘Horvath AA’ and ‘Hannum AA’ respectively.
SES-AA association
The association of SES with AA was investigated by linear regression models using SES as the predictor and AA as the outcome. The ‘basic adjusted’ model (referred to hereafter also as model 1) included age (continuous), sex, recruitment center (EPIC Italy and TILDA only), incident cancer/cardiovascular event (EPIC Italy only), and sample type (MCCS only) as covariates. In all the analyses the higher SES group was used as the reference to assess associations of low SES with epigenetic AA.
Basic adjusted model comparison with NCD risk factors adjusted model
To test for possible reduction in the effect size due to the inclusion of mediators in the model we compared the estimated effect size of model 1 with those of the NCD risk factors adjusted models. The NCD risk factors considered were: smoking status (categorical: never, former, current), BMI (continuous), alcohol intake (categorical: no/moderate, habitual drinkers), Mediterranean Diet Score (ordinal categorical score from 0 to 10, EPIC Italy and MCCS only), and physical activity (ordinal categorical: inactive, moderately inactive, moderately active, active); that were first added to model 1 one-by-one, and finally included all together in the ‘fully adjusted’ model. The reduction in effect size due to the inclusion of mediators in the model was computed as \(1-{{\rm{\beta }}}_{{\rm{m}}}/{{\rm{\beta }}}_{1}\); where β1 is the effect size of model 1 and βm is the effect size of model 1 plus the mediator(s)30,45. Negative value for the effect size reduction occurs when β1 < βm, and should be interpreted as positive confounding45. Confidence intervals and statistical significance of the changes in estimated effect were computed using a block jackknife procedure based on 1,000 resampling46.
Sensitivity and interaction analyses
Sensitivity analyses were performed to confirm significant associations stratifying the analyses by gender and age groups (younger than, and older than 60 years old) and excluding each time current smokers, habitual drinkers, physically inactive individuals, obese individuals (BMI > 30) and those with unhealthy diet (Mediterranean diet score ≤ 4). The difference in the effect of SES on AA by sex and age class (younger than, and older than 60 years old) was tested by adding the interaction term in the regression analysis. Finally, the associations of SES with AA were further verified after exclusion of incident cancer/cardiovascular cases in EPIC Italy, and individuals exposed to at least one NCD risk factor, leading to the analysis of 1,549 NCD risk factor free individuals.
Raw methylation data may be obtained upon request to giovanni.fiorito@iigm.it. All participants gave written informed consent for their samples to be used in genetic and epigenetic studies of health. This study was reviewed and approved by the HuGeF Ethic Committee. This study was conducted following the principles of the Declaration of Helsinki and its subsequent revisions.
References
Marmot, M. G. The status syndrome: how social standing affects our health and longevity. 1st American edn, (Times Books, 2004).
Gallo, V. et al. Social inequalities and mortality in Europe–results from a large multi-national cohort. PLoS One 7, e39013, https://doi.org/10.1371/journal.pone.0039013 (2012).
Stringhini, S. et al. Health behaviours, socioeconomic status, and mortality: further analyses of the British Whitehall II and the French GAZEL prospective cohorts. PLoS medicine 8, e1000419, https://doi.org/10.1371/journal.pmed.1000419 (2011).
Dowd, J. B., Simanek, A. M. & Aiello, A. E. Socio-economic status, cortisol and allostatic load: a review of the literature. Int J Epidemiol 38, 1297–1309, https://doi.org/10.1093/ije/dyp277 (2009).
Stringhini, S. et al. Socioeconomic status and the 25 x 25 risk factors as determinants of premature mortality: a multicohort study and meta-analysis of 1.7 million men and women. Lancet, https://doi.org/10.1016/S0140-6736(16)32380-7 (2017).
Stringhini, S. et al. Association of socioeconomic position with health behaviors and mortality. JAMA 303, 1159–1166, https://doi.org/10.1001/jama.2010.297 (2010).
Tung, J. et al. Social environment is associated with gene regulatory variation in the rhesus macaque immune system. Proc Natl Acad Sci USA 109, 6490–6495, https://doi.org/10.1073/pnas.1202734109 (2012).
Snyder-Mackler, N. et al. Social status drives social relationships in groups of unrelated female rhesus macaques. Anim Behav 111, 307–317, https://doi.org/10.1016/j.anbehav.2015.10.033 (2016).
Borghol, N. et al. Associations with early-life socio-economic position in adult DNA methylation. Int J Epidemiol 41, 62–74, https://doi.org/10.1093/ije/dyr147 (2012).
McGuinness, D. et al. Socio-economic status is associated with epigenetic differences in the pSoBid cohort. Int J Epidemiol 41, 151–160, https://doi.org/10.1093/ije/dyr215 (2012).
Tehranifar, P. et al. Early life socioeconomic factors and genomic DNA methylation in mid-life. Epigenetics 8, 23–27, https://doi.org/10.4161/epi.22989 (2013).
Stringhini, S. et al. Life-course socioeconomic status and DNA methylation of genes regulating inflammation. Int J Epidemiol 44, 1320–1330, https://doi.org/10.1093/ije/dyv060 (2015).
Castagne, R. et al. A life course approach to explore the biological embedding of socioeconomic position and social mobility through circulating inflammatory markers. Sci Rep 6, 25170, https://doi.org/10.1038/srep25170 (2016).
Castagne, R. et al. Biological marks of early-life socioeconomic experience is detected in the adult inflammatory transcriptome. Sci Rep 6, 38705, https://doi.org/10.1038/srep38705 (2016).
Stringhini, S. et al. Association of lifecourse socioeconomic status with chronic inflammation and type 2 diabetes risk: the Whitehall II prospective cohort study. PLoS medicine 10, e1001479, https://doi.org/10.1371/journal.pmed.1001479 (2013).
Barboza Solis, C. et al. Mediating pathways between parental socio-economic position and allostatic load in mid-life: Findings from the 1958 British birth cohort. Soc Sci Med 165, 19–27, https://doi.org/10.1016/j.socscimed.2016.07.031 (2016).
Barboza Solis, C., Fantin, R., Kelly-Irving, M. & Delpierre, C. Physiological wear-and-tear and later subjective health in mid-life: Findings from the 1958 British birth cohort. Psychoneuroendocrinology 74, 24–33, https://doi.org/10.1016/j.psyneuen.2016.08.018 (2016).
Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol 14, R115, https://doi.org/10.1186/gb-2013-14-10-r115 (2013).
Hannum, G. et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Molecular cell 49, 359–367, https://doi.org/10.1016/j.molcel.2012.10.016 (2013).
Chen, B. H. et al. DNA methylation-based measures of biological age: meta-analysis predicting time to death. Aging (Albany NY) 8, 1844–1865, https://doi.org/10.18632/aging.101020 (2016).
Walker, R. & Hiller, J. E. The index of relative socio-economic disadvantage: general population views on indicators used to determine area-based disadvantage. Aust N Z J Public Health 29, 442–447 (2005).
Fox, J. W. Social class, mental illness, and social mobility: the social selection-drift hypothesis for serious mental illness. J Health Soc Behav 31, 344–353 (1990).
Joubert, B. R. et al. Maternal plasma folate impacts differential DNA methylation in an epigenome-wide meta-analysis of newborns. Nat Commun 7, 10577, https://doi.org/10.1038/ncomms10577 (2016).
Joubert, B. R. et al. DNA Methylation in Newborns and Maternal Smoking in Pregnancy: Genome-wide Consortium Meta-analysis. American journal of human genetics 98, 680–696, https://doi.org/10.1016/j.ajhg.2016.02.019 (2016).
Guida, F. et al. Dynamics of smoking-induced genome-wide methylation changes with time since smoking cessation. Hum Mol Genet 24, 2349–2359, https://doi.org/10.1093/hmg/ddu751 (2015).
Demetriou, C. A. et al. Biological embedding of early-life exposures and disease risk in humans: a role for DNA methylation. Eur J Clin Invest 45, 303–332, https://doi.org/10.1111/eci.12406 (2015).
Cohen, S., Janicki-Deverts, D., Chen, E. & Matthews, K. A. Childhood socioeconomic status and adult health. Ann N Y Acad Sci 1186, 37–55, https://doi.org/10.1111/j.1749-6632.2009.05334.x (2010).
Cohen, S. et al. Childhood socioeconomic status, telomere length, and susceptibility to upper respiratory infection. Brain Behav Immun 34, 31–38, https://doi.org/10.1016/j.bbi.2013.06.009 (2013).
Conroy, K., Sandel, M. & Zuckerman, B. Poverty grown up: how childhood socioeconomic status impacts adult health. J Dev Behav Pediatr 31, 154–160, https://doi.org/10.1097/DBP.0b013e3181c21a1b (2010).
MacKinnon, D. P., Krull, J. L. & Lockwood, C. M. Equivalence of the mediation, confounding and suppression effect. Prev Sci 1, 173–181 (2000).
Lee, J., Taneja, V. & Vassallo, R. Cigarette smoking and inflammation: cellular and molecular mechanisms. J Dent Res 91, 142–149, https://doi.org/10.1177/0022034511421200 (2012).
Ertek, S. & Cicero, A. Impact of physical activity on inflammation: effects on cardiovascular disease risk and other inflammatory conditions. Arch Med Sci 8, 794–804, https://doi.org/10.5114/aoms.2012.31614 (2012).
Quach, A. et al. Epigenetic clock analysis of diet, exercise, education, and lifestyle factors. Aging (Albany NY) 9, 419–446, https://doi.org/10.18632/aging.101168 (2017).
McCaffery, J. M., Marsland, A. L., Strohacker, K., Muldoon, M. F. & Manuck, S. B. Factor structure underlying components of allostatic load. PLoS One 7, e47246, https://doi.org/10.1371/journal.pone.0047246 (2012).
Barboza Solis, C. et al. Adverse childhood experiences and physiological wear-and-tear in midlife: Findings from the 1958 British birth cohort. Proc Natl Acad Sci USA 112, E738–746, https://doi.org/10.1073/pnas.1417325112 (2015).
Greenland, S. Quantifying biases in causal models: classical confounding vs collider-stratification bias. Epidemiology 14, 300–306 (2003).
Simons, R. L. et al. Economic hardship and biological weathering: The epigenetics of aging in a U.S. sample of black women. Soc Sci Med 150, 192–200, https://doi.org/10.1016/j.socscimed.2015.12.001 (2016).
Miller, G. E., Yu, T., Chen, E. & Brody, G. H. Self-control forecasts better psychosocial outcomes but faster epigenetic aging in low-SES youth. Proc Natl Acad Sci USA 112, 10325–10330, https://doi.org/10.1073/pnas.1505063112 (2015).
Baird, S., Chirwa, E., McIntosh, C. & Ozler, B. The short-term impacts of a schooling conditional cash transfer program on the sexual behavior of young women. Health Econ 19, Suppl, 55–68, https://doi.org/10.1002/hec.1569 (2010).
Kelly-Irving, M. et al. Adverse childhood experiences and premature all-cause mortality. Eur J Epidemiol 28, 721–734, https://doi.org/10.1007/s10654-013-9832-9 (2013).
Sacerdote, C. et al. Lower educational level is a predictor of incident type 2 diabetes in European countries: the EPIC-InterAct study. Int J Epidemiol 41, 1162–1173, https://doi.org/10.1093/ije/dys091 (2012).
Engels, E. A., Schmid, C. H., Terrin, N., Olkin, I. & Lau, J. Heterogeneity and statistical significance in meta-analysis: an empirical study of 125 meta-analyses. Stat Med 19, 1707–1728 (2000).
Troyanskaya, O. et al. Missing value estimation methods for DNA microarrays. Bioinformatics 17, 520–525 (2001).
Houseman, E. A. et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 13, 86, https://doi.org/10.1186/1471-2105-13-86 (2012).
MacKinnon, D. P., Fairchild, A. J. & Fritz, M. S. Mediation analysis. Annu Rev Psychol 58, 593–614, https://doi.org/10.1146/annurev.psych.58.110405.085542 (2007).
Biswal, B. B., Taylor, P. A. & Ulmer, J. L. Use of jackknife resampling techniques to estimate the confidence intervals of fMRI parameters. J Comput Assist Tomogr 25, 113–120 (2001).
Acknowledgements
This research was supported by the ‘Lifepath’ grant to Paolo Vineis at Imperial College, London, Silvia Polidoro at the HuGeF Foundation, Silvia Stringhini at Lausanne’s University Hospital, Mika Kivimaki at University College London, Richard Layte and Cathal McCrory at the Trinity College Dublin, Dublin, Ireland (European Commission H2020 grant, Grant number: 633666). Giovanni Fiorito, Silvia Polidoro, Giuseppe Matullo and Simonetta Guarrera are supported by the Italian Institute for Genomic Medicine (IIGM), Torino, Italy with support from Compagnia di San Paolo. Silvia Stringhini is supported by the Swiss National Science Foundation (Ambizione Grant n° PZ00P3_147998). Laura Baglietto is supported by a Marie Curie International Incoming Fellowship within the 7th European Community Framework Programme. Panagiotis Georgiadis and Soterios A. Kyrtopoulos were supported by the European Union grant Envirogenomarkers (Grant number 226756). Mika Kivimaki is supported by the UK Medical Research Council (K013351) and NordForsk. The MCCS component of the work was funded by the Australian National Health and Medical Research Council, including project grant numbers 1011618, 1026892, 1026522, 1050198, 623206, and 1043616, and program grant numbers 209057 and 1074383.
Author information
Authors and Affiliations
Contributions
G.F., O.R., S.P. and P.V. designed this study. Sample collection and processing were performed by G.M., P.G., S.A.K., V.K., D.P., S.P., C.S., R.T., G.G.G., and R.L. DNA methylation array experiments were performed by S.P., M.B.A., S.G., A.M.O., and R.A.K. Statistical analyses were performed by G.F., P.A.D., and E.P. M.K., M.C.H., S.S., G.S., A.M.H., R.M., R.K.L., L.B., O.R., C.McC., and R.M. contributed to draft the manuscript and revised it critically. All authors approved the final form of the manuscript for submission.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fiorito, G., Polidoro, S., Dugué, PA. et al. Social adversity and epigenetic aging: a multi-cohort study on socioeconomic differences in peripheral blood DNA methylation. Sci Rep 7, 16266 (2017). https://doi.org/10.1038/s41598-017-16391-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-16391-5
This article is cited by
-
Meta-analysis of epigenetic aging in schizophrenia reveals multifaceted relationships with age, sex, illness duration, and polygenic risk
Clinical Epigenetics (2024)
-
Lifetime employment trajectories and cancer
Scientific Reports (2024)
-
Neighborhood Disadvantage and Parenting in Early Adolescence Predict Epigenetic Aging and Mortality Risk in Adulthood
Journal of Youth and Adolescence (2024)
-
Dietary flavonoids intake contributes to delay biological aging process: analysis from NHANES dataset
Journal of Translational Medicine (2023)
-
The role of genetics and epigenetics in sex differences in human survival
Genus (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
