
Abstract
The new oncologic paradigm of precision medicine is focused on identifying metabolic, proteomic, transcriptomic and genomic variabilities in tumors that can be exploited to tailor treatments and improve patient outcomes. Metabolic changes are a hallmark of cancer, and inhibition of metabolic pathways is now a major strategy in medicinal chemistry for targeting cancers. However, non-invasive biomarkers to categorize metabolic subtypes are in short supply. The purpose of this study was to characterize the intracellular and extracellular metabolic profiles of four prostate cancer cell lines with varying degrees of aggressiveness. We observed metabolic differences between the aggressive prostate cancer cell line PC3 and the even more aggressive, metastatic subline PC3M assessed by hyperpolarized in vivo pyruvate studies, nuclear magnetic resonance spectroscopy, and carbon-13 feeding studies. On further examination of the differences between these two cell lines, we found increased glutamine utilization in the metastatic PC3M subline that led directly to sensitivity to glutaminase inhibitor CB-839. Our study supports the theory that metastatic progression increases glutamine utilization and the inhibition of glutaminolysis could have clinical implications.
Similar content being viewed by others
Introduction
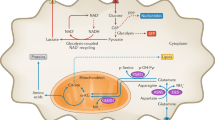
Prostate cancer (PCa) is the second leading cause of cancer death in men in the United States. Despite the approval of multiple new therapies for PCa1,2,3,4,5, metastatic disease remains incurable. The metabolism of normal prostate cells versus cancer cells has been revealed in several biochemistry assays to be significantly different6. For example, the concentration of citrate is higher in normal prostate than in PCa, reaching concentrations as high as 180 mM in prostatic fluid7. The significant reduction in citrate level in PCa8,9,10 results from citrate utilization in other metabolic pathways6,11. Several recent publications have found differences between PCa and normal prostate in expression of metabolic enzymes such as glutaminase (GLS)12,13, acetyl-CoA synthetase14, and the monocarboxylic acid transporters MCT1/MCT415,16,17 (See Fig. 1a). Our long-term goal is to identify the roles of these metabolites in prostate cancer growth and progression.

Metabolic differences between PCa cell lines. In all experiments, metabolite values were determined by MRS and values were normalized as stated per experiment. Statistical significance indicated by an asterisk (*). (a) In this schematic, partial pathways of glycolysis, glutaminolysis, and the citric acid cycle are shown: red type is specific enzymes, red arrows depict the conversion of cofactors such as NAD+, blue type is transporters, and black type is specific metabolites. The dashed box highlights the production of lactate through lactate dehydrogenase (LDH). There are several steps between fructose-6-phosphate and the generation of two equivalences of pyruvate not shown. We were specifically focused on determining the differences in aerobic and anaerobic glycolytic metabolites in PCa cell lines. Abbreviations: HK (hexokinase), PGI (phosphoglucose isomerase), GLDH (glutamate dehydrogenase), GLS (glutaminase), MCT4 and MCT1 (monocarboxylic acid transporters), GLUT1 (glucose transporter), SLC1A5 (glutamine transporter), MPC (mitochondrial pyruvate carrier), MAE (malic enzyme). (b) To compare extracellular metabolite changes among cell lines, concentrations of metabolites within a 24-hour period were determined and changes over that period were graphed as shown. Positive bars are metabolites excreted into the media and negative bars are metabolites consumed by the cells over the 24-hour period (n = 5 samples per cell type). Glutamine uptake by PC3M cells was over three fold higher than that by PC3 cells (P < 0.0001). Lactate production by PC3 was 1.3 fold higher than that by PC3M (P < 0.0001). Statistical significance between metabolite levels was determined using two-way ANOVA with Tukey’s multiple comparisons. (c) To compare intracellular metabolite levels in cultured cells, metabolite concentrations were determined and the fold change compared to the indolent cell line (RWPE1) was plotted. The levels of metabolites succinate and phosphocholine (PCho) were significantly lower in PC3M cell lysates than in PC3 lysates (P < 0.0004). Statistical significance was determined by two-way ANOVA with Tukey’s multiple comparisons. (d) To compare levels of metabolites in ex vivo tumor tissues, PC3 and PC3M tumors were excised from mice and samples prepared for analysis (100 mg per sample, PC3 n = 4, PC3M n = 5). Levels of lactate and taurine were significantly higher in PC3 tumors than in PC3M tumors, while levels of aspartate, glutamate, glutamine and succinate were significantly higher in PC3M tumors than in PC3 tumors. Unpaired t-test using Holm-Sidak method was used to determine statistical significance (**P < 0.02, *P < 0.04).
The major changes that occur in PCa are similar to what is seen in other cancers including increased glycolysis18. The increases in glycolysis in PCa progression has been observed both in cell culture19 and in vivo using a new metabolic imaging technique called hyperpolarized magnetic resonance20. Hyperpolarization can enable a >10,000 fold sensitivity enhancement of the magnetic resonance (MR) signal of contrast agents over Boltzmann polarization20,21,22,23. More importantly, this signal enhancement is retained on the metabolites of the hyperpolarized molecule allowing direct observation and measurement of metabolic flux and real-time metabolic imaging. The most widely used method for providing hyperpolarized contrast agents for metabolic MR imaging is dynamic nuclear polarization (DNP)24,25 and the most studied hyperpolarized 13C tracer with DNP is pyruvate. Recently, the first Phase I study with hyperpolarized 1-13C pyruvate was completed26. Hyperpolarized 1-13C pyruvate is taken up by glycolytic cancer cells quickly and is then converted to 1-13C lactate through lactate dehydrogenase (LDHA)20,26,27,28,29,30. This conversion rate can be measured in real time and in vivo by measuring the integration of the lactate signal over the overall hyperpolarized signal (nLac). Multiple laboratories have used hyperpolarized 1-13C pyruvate to image the transgenic adenocarcinoma of mouse prostate (TRAMP) model31. In the TRAMP model, increases in glycolytic flux were observed in higher histological grade PCa20 and was reduced after hormone therapy32.
A key nutrient for most cancers is glutamine, and one of the critical steps in its utilization is its conversion to glutamate through the glutaminase enzyme33,34,35,36. The GLS gene encodes two splice variants of glutaminase, kidney-type glutaminase (KGA) and glutaminase C (GAC), collectively called GLS137. The GLS2 gene encodes two glutaminase isoforms, LGA (liver-type) and GAB, collectively called GLS237. GLS2 isozymes have been shown to be downregulated in several cancers38, including acute myeloid leukemia39 and breast40,41 and lung cancers42. In contrast, GLS1 is typically upregulated in cancers39. Glutamine can be used not only as a nitrogen source but also as a carbon source and for energy production33,34,35. A recent publication reported that GLS1 was expressed in 68 of 107 (64%) PCa specimens but in only 9 of 37 (24%) benign hyperplastic prostate specimens12.
Androgen receptor (AR) signaling is one of the most critical pathways for maintaining prostate growth and normal function; however, AR activation is also important in prostate cancer pathogenesis and progression43,44. Androgen ablation is usually a first-line drug with and without resection. However, a subset of patients does not benefit from androgen ablation or in time their cancer evolves and begins to be non-responsive. Androgen insensitive PCa, termed castration resistant, can occur by multiple mechanisms including point mutations in AR gene, generation of splicing variants (e.g. AR-V7), amplification of AR gene, or complete ablation of the AR receptor43,44.
We are specifically interested in determining the metabolic differences in castration resistant and metastatic PCa. Our purpose was to further our understanding in the metabolic differences in the extracellular and intracellular metabolic profiles of four PCa cell lines with varying degrees of aggressiveness specifically focusing on glutamine and glucose utilization. Our in vitro metabolic results led us to determine the hyperpolarized metabolic flux of 1-13C pyruvate in xenograft PC3 and PC3M animal models and the testing of CB-839. CB-839 (Calithera Biosciences) is a nanomolar binder to both major isoforms of glutaminase 1 (GLS1), has good oral bioavailability, and is currently in a Phase I study for solid tumors and leukemia (NCT02071862, NCT02071927)39,41.
Results
Production of lactate and uptake of glucose and glutamine differ in prostate cancer cells
Changes in the concentrations of extracellular metabolites (consumed and excreted) over 24 hours of growth were measured in four human PCa cell lines: RWPE-1, RWPE-2, PC3, and PC3M45. Figure 1 summarizes all of the metabolic profiling data both in cell culture and tumor tissue. Changes observed in selected extracellular metabolites among the four cell lines in 24 hours are shown in Fig. 1b. Positive values represent secreted metabolites, while negative values represent consumed metabolites. Glutamine uptake was over 3 times higher in PC3M cells than in PC3 (P < 0.0001), while lactate production was 1.3 times higher in PC3 cells than in PC3M (P < 0.0001). However, glucose uptake and glutamate utilization is similar in RWPE2, PC3, and PC3M cell lines. Significant differences in intracellular metabolite levels were found between PC3 and PC3M both in cell culture and excised tumor tissues. In cell culture, levels of phosphocholine (PCho) and succinate were significantly different (P < 0.0001) in PC3 and PC3M (Fig. 1c). In addition, intracellular glutamine levels were observed to be lower in the aggressive cell lines (PC3 and PC3M) compared to unaggressive lines but were not statistically significant by two-way ANOVA analysis. The metabolic profile of tumor tissue samples (Fig. 1d), which includes both the cells (intracellular) and interstitial spaces (extracellular), revealed the same differences found in PC3 and PC3M in vitro assays: lactate was higher in PC3 tumors than in PC3M tumors, and succinate, aspartate, glutamate and glutamine were higher in PC3M tumors.
Carbon-13 tracing experiments reveal differences in glutamine utilization
To further explore the differences in glutamine and glucose utilization in PC3 and PC3M cells lines, carbon-13 feeding/tracing experiments were performed. Similar lactate values were seen in both cell types and in both tracing experiments; however with the carbon-13 glutamine feeding experiments we selectively found the label in intracellular succinate and proline in the PC3M cell line (Fig. 2). This result, along with our proton spectroscopy results, emphasizes the differences in the utilization of glutamine between PC3 and PC3M.

Carbon-13 feeding studies reveal differences in glutamine utilization. 13C-labeled feeding studies were done to elucidate glucose and glutamine utilization by PC3 and PC3M cells. Four plates of PC3 and PC3M cells were incubated for 24 hours with 1,6-13C glucose or carbon-13 fully labeled glutamine. Metabolites were determined by 13C high-resolution spectroscopy. The results show increased carbon-13 label from glutamine in succinate and proline in PC3M cells. Unpaired t-test using Holm-Sidak method was used to determine statistical significance.
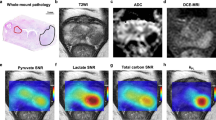
Hyperpolarized pyruvate studies reveal increased glycolytic flux in PC3 tumors
We utilized hyperpolarized 1-13C pyruvate to determine the differences in glycolysis of PC3 and PC3M subcutaneous tumors in nude mice. This conversion rate can be measured in real time and in vivo by measuring the integration of the lactate signal over the overall hyperpolarized signal. A series of slice-selective 13C spectra were collected immediately after injection of tumor-bearing mice with hyperpolarized 1-13C pyruvate to observe the arrival of the compound to the tumor and its conversion to lactate. Pyruvate to lactate conversion was greater in PC3 tumors than in PC3M tumors (0.44 ± 0.09 [n = 5] versus 0.29 ± 0.02 [n = 3], P < 0.03) (Fig. 3). This finding corresponds with the greater extracellular lactate production over 24 hours by PC3 cells than by PC3M cells in culture.

Increased glycolysis observed in PC3 tumors with hyperpolarized pyruvate. (a) The schematic illustrates the different pathways through which hyperpolarized 1-13C pyruvate can be metabolized in the cell. Because of the quick loss of polarization, resonances for lactate and alanine are the main metabolites detected in vivo. (b) Spectra from representative PC3M and PC3 tumor-bearing mice taken 21 s after tail vein injection of hyperpolarized pyruvate. The faster conversion rate in PC3 is readily seen. (c) 1H image of a representative PC3M tumor-bearing mouse. Slice-selective 13C spectroscopy was utilized to capture the 13C signal in the tumor tissue after injection of hyperpolarized pyruvate. (d) Hyperpolarized pyruvate data were processed to generate dynamic curves characterizing the arrival of hyperpolarized pyruvate and its chemical conversion into lactate. Normalized lactate (nLac), defined as the ratio of total cumulative lactate signal to the total carbon-13 signal, was calculated for each 13C scan. Normalized lactate conversion was greater in PC3 tumor-bearing animals (0.44 ± 0.09) than in PC3M tumor-bearing animals (0.29 ± 0.02). The statistical significance of the difference between tumor groups was determined by unpaired two-tailed t-test (P < 0.03).
Drug inhibition assays reveal glutamine utilization variability
PC3 and PC3M cells were grown with and without added glutamine (Fig. 4a) or in the presence of 0.1% DMSO (vehicle control) or 1 μM of GLS1 inhibitor CB-839 for 72 hours (Fig. 4b). Cells were counted after the 72-hour period. Both treatments reduced proliferation of the PC3M cells. Interestingly, both cell lines proliferated without glutamine. Cell viability measurements were performed in both cell lines after incubation for 72 hours with vehicle control or 1 μM CB-839. Levels of ATP were lower in the PC3M cells incubated with CB-839 (Fig. 4c). The ATP level could be rescued by adding 4 mM dimethyl 2-oxoglutarate to the media (Fig. 4d). Dimethyl 2-oxoglutarate is a hydrophobic analog of α-ketoglutarate and has been shown to be metabolized in the citric acid cycle39,41. In cells treated with mTOR inhibitor rapamycin (3 nM or 25 nM) for 12 hours, the ratios of ATP levels in treated and control cells showed reduced ATP in PC3M cells but not in PC3 cells (Fig. 4e). These results confirm that glutamine dependence is higher in the metastatic subline PC3M than in PC3 cells and that glutamine is being utilized to fuel the citric acid cycle. These results also suggest that mTOR activation is more prevalent in the PC3M cells than in the PC3 cells.

Therapeutic vulnerabilities of PC3M cell line observed in drug assays. PC3 and PC3M cells were treated with various metabolites or inhibitors and the effects on cell viability and proliferation were assessed. Unless otherwise noted, the statistical significance of differences between groups was determined by unpaired two-tailed t-test. (a) Comparison of PC3 and PC3M cell counts after 72 hours of growth in the presence or absence of 2 mM glutamine (Gln). (b) Comparison of PC3 and PC3M cell counts after 72 hours of treatment with GLS1 inhibitor CB-839 or vehicle. Five to six replicates were used for each condition. In both experiments (a and b), PC3M proliferation was inhibited to a greater extent than PC3 proliferation (P < 0.03 [+/− Gln], P < 0.00001 [+/− CB-839]). PC3 proliferation was not inhibited by CB-839 but was inhibited by complete exclusion of glutamine in the media. Both cell lines proliferated in the absence of glutamine and in the presence of CB-839. (c) ATP levels in cells treated for 72 hours with vehicle or with 1 μM CB-839 over untreated control. Five to six replicates were used for each condition. Levels of ATP were reduced only in the treated PC3M samples (P < 0.0001). (d). ATP levels were equivalent to those in cells treated with vehicle when cells treated with 1 μM CB-839 were also incubated with 4 mM dimethyl 2-oxoglutarate, showing that DMKG rescued the citric acid cycle. (e) Treatment with mTOR inhibitor rapamycin (3 nM or 25 nM) significantly reduced ATP levels in PC3M cells compared to vehicle control but did not reduce ATP levels in PC3 cells. The statistical significance of the differences was determined by two-way ANOVA (P < 0.00001, seven replicate samples per condition).
Protein levels indicate differences in signaling and glutamine metabolism pathways
Reverse-phase protein array (RPPA) technology46 was used to analyze the cellular protein activity in the lysates from PC3 and PC3M cells (n = 3). Figure 5 highlights a few of the proteins whose levels varied by >0.1 between the cell lines and specifically in the signaling pathway of AKT and AMPK. Levels of phosphorylated S473-AKT (Akt-pS473, P < 0.01, unpaired t-test using Holm-Sidak method for multiple comparisons) was higher in the PC3M cell lysates than in the PC3 cell lysates, while total AKT levels were equal. Levels of p-AMPK were higher in the PC3 lysates than in the PC3M lysates. Both AMPK and AKT are master regulators of cell growth and cellular metabolism. The deviation between the levels of these two proteins could be the underlying cause of the metabolic variability between the two lines.

Signaling pathway variation between PC3 and PC3M cells. Reverse-phase protein array (RPPA) analysis was used to identify differences in expression of various proteins in PC3 and PC3M cells (n = 3 samples per cell type). The x-axis labels represent selected proteins targeted by the antibodies used in the analysis; V is validated and C is course. The validated antibodies have been shown in multiple assays to target only the protein named and not to have off-target binding; course antibodies have off-target binding. PC3M cell lysates expressed higher levels of phosphorylated AKT (Akt-pS473, P < 0.01, unpaired t-test using Holm-Sidak method for multiple comparisons), 4E-BP1, and mTOR than PC3 cell lysates. Expression of AMPK and p-AMPK was higher in PC3 cell lysates.
The levels of GLS1 (GAC and KCA) and GLS2 were determined by Western blot in PC3 and PC3M cell lysates treated or not treated with 1 μM CB-839 (Fig. 6). Expression of GLS2 was higher in PC3 lysates than in PC3M lysates in both treated and untreated samples. Levels of GLS1 were approximately equivalent in both cell lines. GLS2 overexpression is observed in both PC3 and PC3M cells with drug incubation indicating that GLS2 could be compensating for GLS1 inhibition. GLS2 overexpression after drug treatment is the highest in PC3 cells. Reduced GLS2 levels with and without drug present may lead to the greater susceptibility of PC3M to CB-839 treatment.

Glutaminase protein expression in PC3 and PC3M cell lines before and after CB-839 treatment. PC3 and PC3M cells were treated (+) or not treated (−) with GLS1 inhibitor CB-839 (1 μM) and their expression of GLS1 and GLS2 proteins were determined by Western blot. Antibodies specific for GAC and KCA were utilized on two replicate samples, while GLS2 expression was determined on three replicates. PC3 cell lysates had higher levels of expression of GLS2 than PC3M cell lysates regardless of CB-839 treatment. The average density of bands in the GLS2 blots were determined by using ImageJ software. The statistical significance was determined by unpaired two-tailed t-test. The difference in GLS2 expression was significant in the untreated cells (P < 0.03) but because of high variability the difference was not significant in the drug-treated cells. Levels of GLS1 (GAC and KGA) were similar in all cell lysates. The full gels can be seen in the supplemental data.
Discussion
Our results illustrate the metabolic reprogramming that can occur between similar cell lines. Our metabolic results are intriguing because the PC3M line was derived by isolating and growing a lung metastasis of PC3 in a nude mouse47. The PC3M line is more metastatic and aggressive than the initial parent cell line47. Both lines are androgen receptor–null and therefore castration resistant. Glycolysis was greater in PC3 cells than in PC3M cells both in vivo and in vitro as evaluated by metabolic imaging and metabolic profiling. Furthermore, variations in 13C-labeled glutamine uptake and metabolic products in cultured PC3M cells compared to cultured PC3 cells indicate that glutamine is being utilized to a greater extent to feed the citric acid cycle (succinate labeling) in PC3M cells. In addition, higher levels of succinate and aspartate (both metabolites formed from the citric acid cycle) were observed in PC3M tumor tissue. The level of GLS2 expression was lower in PC3M cells than in PC3. Together, these differences lead to the greater vulnerability of PC3M cells to GLS1 inhibitor CB-839. To our knowledge, this is the first study showing that CB-839 reduces cell proliferation and viability in an isogenic PCa line.
This increase in glutamine utilization in more aggressively metastatic cell lines has been observed also in ovarian cancer48, suggesting that glutamine utilization might be directly related to metastatic progression48. Jiang et al. reported that cells in culture require glutamine reductive decarboxylation to form spheroid structures from anchorage-dependent growth49. CB-839 could have a larger effect on metastatic progression than on cell proliferation. Our results reveal that both PC3 and PC3M proliferate even in the complete absence of glutamine, illustrating the dynamic nature of metabolism and the capacity for multiple compounds to be utilized as carbon and nitrogen sources to provide the building blocks for replication.
Our RPPA data revealed differences in levels of phosphorylated AKT (pS473P) and mTOR in PC3M versus PC3 lysates. Incubation with 3 nM rapamycin (an mTOR inhibitor) or 1 μM CB-839 reduced ATP production only in PC3M cells. Phosphorylated AKT is known to activate mTOR, which can stimulate glutamine utilization50, and increases in glutamine concentration can directly modulate the mTOR pathway51. In future studies, we will determine whether regulation of this pathway can further explain the differences in glutamine utilization between these two cell lines.
In conclusion, we observed greater glycolytic flux in PC3 tumors than in PC3M tumors in mice and see increased glutamine utilization in the PC3M cell line. Once we understand the mechanisms underlying this metabolic variation between PCa subtypes, we may be able to use hyperpolarized metabolic imaging to provide a rationale for individualized combinations of metabolic pathway inhibitors and chemotherapy and potentially improve the outcome of prostate cancer patients.
Materials and Methods
Cell lines and culture conditions
Four human PCa lines were used: RWPE-1 (non-tumorigenic, considered benign), RWPE-2 (non-metastatic), PC3 (aggressive, castration resistant), and the aggressive subline PC3M (castration resistant)45. RWPE1, RWPE2, and PC3 cells were purchased from ATCC. PC3M cells were purchased from the MD Anderson cell line core facility, which validates all of the cell lines it provides. Cell lines were tested for mycoplasma every couple of months by MD Anderson cell line core facility. For intracellular metabolite analyses, RWPE1 and RWPE2 cells were cultured in keratinocyte serum-free media supplemented with bovine pituitary extract and recombinant human epidermal growth factor. PC3 cells were cultured in F12-K media supplemented with 10% fetal bovine serum (FBS), and PC3M cells were cultured in RPMI1640 media supplemented with 0.1 mM modified essential media (MEM) and 10% FBS. All media were supplemented with 10 U/mL penicillin and 10 µg/mL streptomycin. For extracellular metabolite determination, PC3 and PC3M cells were seeded in optimized media and, after 24 hours, exchanged into RWPE1/2 media. All media were purchased from Gibco and Corning.
For CB-839 studies and glutamine studies, PC3 and PC3M cells were grown in RPMI1640 media (with or without glucose/glutamine) supplemented with 10% FBS, 10 U/mL penicillin, and 10 µg/mL streptomycin. The media was supplemented with frozen aliquots of glutamine and glucose prior to experiments.
Mouse models
All experimental methods involving mice were performed in accordance with the guidelines and regulations of the MD Anderson Institutional Animal Care and Use Committee (IACUC), and the animal protocols used were approved by that committee. Male nude mice >5weeks old were each injected with ~5 × 106 cells subcutaneously on the rear flank. When the resulting tumors had grown to approximately 1 to 1.3 cm in size, the animals underwent imaging as described in a later subsection. The animals were sacrificed with isoflurane overdose and cervical dislocation 6–48 hours after imaging, and tumor tissues were removed and flash-frozen for metabolic studies.
High-resolution magnetic resonance spectroscopy
Magnetic resonance spectroscopy (MRS) was used for all metabolic analyses. MRS allows chemical resonances of metabolites to be determined in one experiment. For all high-resolution MRS, one-dimensional 1H proton spectroscopy was performed with water suppression on a 500 MHz Bruker Biospin Avance III high-definition nuclear magnetic resonance (NMR) instrument equipped with a Prodigy BBO cryoprobe. The cryoprobe increases the sensitivity of the measurement 3- to 4-fold. All supplies (deuterium oxide [D2O], 3-(trimethylsilyl)-1-propanesulfonic acid-d6 sodium salt [DSS-d6], and potassium phosphate buffer [K2HPO4, pH 7.4]) were purchased from Sigma-Aldrich and used without further purification.
To determine intracellular metabolite levels, cells (0.5–3.0 × 107 per sample) were trypsinized and pelleted at approximately 80% confluence, and metabolites were extracted using an ice-cooled 2:1 methanol to water solution (3 mL) and MP Biomedicals lysing matrix D beads (~500 µL per 107 cells). The homogenates were then subjected to centrifugation for 10 min at 4000 g, the supernatant removed and lyophilized overnight, and the remaining metabolites dissolved in D2O with 0.5 mM DSS-d6 and 50 mM K2HPO4. The one-dimensional 1H-NMR spectra used 256 scans and a spectral width (SW) of 10245 Hz and was referenced to DSS at 0.00 ppm. Water suppression was performed with presaturation. For the metabolites assigned and quantified by using 1H-NMR spectroscopy, four or five replicates of each cell type were analyzed. Data were processed/analyzed with Chenomx (Chenomx, Inc.), MestreNova (Mestrelab Research), and/or Topspin (Bruker Biospin) software. Integrated values of intracellular metabolites were determined by taking the ratio of the resonance for each metabolite over the DSS-d6 peak to the total integration of all resonances in the spectra. This allowed normalization of the probe performance within each sample. Metabolite resonances were identified through reference to either of two online metabolomics databases, Human Metabolome Database (http://www.hmdb.ca)52 or Biological Magnetic Resonance Bank (http://www.bmrb.wisc.edu/metabolomics) and, when necessary, confirmed by spiking the sample with a known amount of the metabolite in question. To easily discern the metabolic profile of the more aggressive PCa lines, the fold difference of each metabolite to RWPE1 (indolent PCa) was graphed (Fig. 1c).
To determine the differential uptake and excretion of some of the dominant media metabolites, five plates (145 × 20 mm) of each cell line were cultured so that there were five replicates at each time point. Because of the large difference in cell size, samples were not normalized to cell count; instead, cell cultures were carefully monitored to allow initiation of the media time course experiment when cells had reached 80% confluence. When cell cultures became approximately 80% confluent, uptake and excretion of metabolites were monitored in the four cell lines over a period of 24 hours in keratinocyte serum-free media. Aliquots of media were taken at the beginning (t = 0) and end of the 24-hour period (t = 24) from five separate tissue culture plates for each cell line. Figure 1b plots the fold change in the integrated value of each metabolite over a 24 hour period.
To determine intracellular metabolite values from subcutaneous tumors, flash frozen tissues (100 mg per sample) were homogenized using a liquid nitrogen–cooled mortar and pestle; metabolites were extracted by a method similar to that used for cell pellets. Integrated values for each metabolite were determined by taking the ratio of the resonance for each metabolite over the DSS-d6 peak to the total integration of all resonances in the spectra. To visualize these differences easier, each integration value was multiplied by a scaling factor to account for the number of protons each chemical resonance corresponded to (1000 (1H), 500 (2H), 250 (4H), Fig. 1d).
Carbon-13 tracing experiments
PC3 and PC3M cells were cultured for 24 hours in glucose/glutamine–free Dulbecco MEM to which either fully labeled 13C-glutamine (final concentration 3 mM) or 1,6-13C glucose (final concentration 10 mM) was added (Fig. 2). After the 24-hour period, the media was removed, and the cells were washed twice with phosphate-buffered saline solution (PBS), trypsinized, pelleted, and then homogenized by the same method as for non-tracing experiments. Labeled compounds (Sigma-Aldrich) were used without any further purification. Lyophilized metabolites were redissolved in 50 mM phosphate buffer (pH 7.4) with 5 mM DSS-d6. Proton decoupled one-dimensional 13C spectroscopy was run (4096 averages, relaxation delay 6s, SW 29760 Hz, 30° flip angle) on each sample. Two-dimensional heteronuclear single-quantum correlation experiments were run on each sample to confirm the assignment of each metabolite (16 averages, relaxation delay 3s, 256 points in 2nd dimension, SW 6500 Hz for 1H, SW 20800 Hz for 13C).
Protein array and Western blotting analyses
For reverse-phase protein array (RPPA) analysis, flash-frozen cell pellets comprising 5 × 106 cells were transferred to the MD Anderson RPPA core facility for processing and analysis. The pellets were subjected to lysis using RPPA lysis buffer, and lysates were serially diluted manually with five 2-fold dilutions of lysis buffer and then printed on nitrocellulose-coated slides53. The NormLinear algorithm, which corrects for protein loading and antibody variations, was used to determine the difference in protein expression between groups of lysates54,55.
For detection of specific proteins via Western blotting, PC3 and PC3M cells were grown in complete RPMI1640 media supplemented with 10% FBS, 10 U/mL penicillin, and 10 µg/mL streptomycin in 6-well plates. When cells became 60% confluent, fresh media was added with or without GLS1 inhibitor CB-839 (1 μM). After 72 hours, the media was removed and the cells were detached using trypsin, counted, and pelleted. Whole-cell lysates were prepared by using RIPA buffer (Thermo Scientific), and total protein concentration was determined by the Pierce BCA protein assay (Thermo Scientific). Protein lysates were denatured by boiling in sodium dodecyl sulfate (SDS) sample buffer for 5 min at 95 °C and then loaded onto a 4–12% mini-precast polyacrylamide gel (Nupage). Membranes were blocked with Odyssey blocking buffer for 60 minutes at room temperature, and then were incubated with antibodies recognizing KCA (20170-AB, ProteinTech)39, GAC (199581-AP, ProteinTech)39, or GLS2 (NBP1-76544, Novus Biologicals) overnight at 4 °C and with β-actin (SC477778, Santa Cruz Biotechnology) for 1 hour at room temperature. Molecular weights were determined using SeeBlue Plus2 Pre-stained protein standard (LC5925, Invitrogen). Secondary antibodies conjugated to infrared (IR) fluorescent dyes were utilized to image bands using the LI-COR Odyssey IR fluorescent system. Densitometry analysis was performed using ImageJ software (Fig. 6, Supplemental Figure).
Imaging procedures and hyperpolarized pyruvate
Ox063 trityl radical (Oxford Instruments) was mixed with neat 1-13C pyruvic acid (Sigma-Aldrich) to a concentration of 15 mM. Aliquots of this solution (20 μL) along with 0.4 μL of 50 mM Gd3+ relaxation agent (Magnevist, Bayer Healthcare) was loaded into a commercial HyperSense dynamic nuclear polarization (DNP) polarizer (Oxford Instruments) and irradiated at a microwave frequency of 94.100 GHz for 30–40 minutes (until the polarization plateau was reached) and then dissolved in 4 mL buffer solution containing 40 mM Tris (7.6 pH preset), 80 mM NaOH, 0.1 g/L EDTA, and 50 mM NaCl.
For imaging, the tumor-bearing mice were anesthetized with 3% isoflurane mixed with oxygen and then maintained with 0.5–1% isoflurane. Animals were placed on a heated pad and their respiration and heart rate monitored during imaging procedures. The neutral (pH 7-8) 80 mM hyperpolarized 1-13C pyruvate solution was injected into each mouse via tail vein catheter.
All imaging and spectroscopy were performed with a dual tuned 1H/13C volume coil (Doty Scientific) or a 1H/13C volume coil (Bruker BioSpin) in a 7T Bruker Biospec horizontal bore MR scanner equipped with a single channel for carbon excitation/reception. Proton anatomic images were taken using a multi-slice T2-weighted RARE sequence. A small 8 M 13C-urea phantom doped with gadolinium-DPTA was next to the tumor for chemical shift referencing. A series of slice-selective 13C spectra (field-of-view 40 × 40 mm, slice-thickness 8–12 mm) were collected immediately after injection of hyperpolarized 1-13C pyruvate. The single slice was placed over the tumor using the multi-slice proton imaging sequence for placement. A total of 90 transients were acquired with a time delay of 2 seconds between each transient (total time 3 minutes). Each transient utilized a 15–20° degree flip angle excitation pulse (gauss pulse) and 2048 data points. Metabolic flux ratios of pyruvate to lactate were determined with a unidirectional model (Fig. 3). Data were processed both in MATLAB (MathWorks Inc) or MestReNova. The dynamic spectra were manually phased and line-broadening was applied (10 to 15 Hz). The area under the spectral peaks for pyruvate and lactate were integrated over the whole array. Normalized lactate (nLac) ratio was calculated as lactate over the sum of pyruvate and lactate signals56.
CB-839, glutamine, and rapamycin cell proliferation and viability assays
For cell viability assays, PC3 or PC3M cells (2,000) in RPMI media were added to wells of a 96-well plate. GLS1 inhibitor CB-839 was dissolved in dimethyl sulfoxide (DMSO) at a concentration of 10 mM. We prepared 100 μl aliquots of the solution which were kept frozen at −20 °C. For each experiment, a separate frozen aliquot of the 10 mM CB-839 solution was utilized to generate our experimental 1 μM CB-839 conditions and any remaining material was disposed of. The plates were incubated in the CO2 incubator for 72 hours. Vehicle control wells were treated with 0.1% DMSO. The cells were then subjected to the Promega CellTiter-Glo Luminescent Cell Viability Assay (G7571). A similar assay was utilized to determine the effect of mTOR inhibitor rapamycin, except cells were seeded at 5000 cells per well at the beginning of the 12-hour incubation.
For the cell proliferation assay, PC3 or PC3M in RPMI media were added to each well of a 24-well plate (5000 cells per well). Cells were treated with CB-839 (1 μM) or 0.1% DMSO. Plates were incubated in a CO2 incubator for 72 hours. Cells were then trypsinized and counted using a Bio-Rad TC20 Cell Counter. In another set of experiments using the same parameters, cells were grown in the presence or absence of glutamine and counted after 72 hours (Fig. 4).
Statistical analyses
All statistical analyses were performed with GraphPad Prism version 6.00 for Windows, GraphPad Software, La Jolla California USA, www.graphpad.com. P values are given for all analyses and statistical significance determined to be any P value below 0.05. All experimental data are shown as mean ±SEM.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Cookson, M. S. et al. Castration-resistant prostate cancer: AUA Guideline. The Journal of urology 190, 429–438 (2013).
de Bono, J. S. et al. Abiraterone and increased survival in metastatic prostate cancer. The New England journal of medicine 364, 1995–2005 (2011).
Ryan, C. J. et al. Phase II study of abiraterone acetate in chemotherapy-naive metastatic castration-resistant prostate cancer displaying bone flare discordant with serologic response. Clinical cancer research: an official journal of the American Association for Cancer Research 17, 4854–4861 (2011).
Efstathiou, E. et al. Effects of abiraterone acetate on androgen signaling in castrate-resistant prostate cancer in bone. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 30, 637–643, https://doi.org/10.1200/JCO.2010.33.7675 (2012).
Chen, E. J. et al. Abiraterone treatment in castration-resistant prostate cancer selects for progesterone responsive mutant androgen receptors. Clinical cancer research: an official journal of the American Association for Cancer Research 21, 1273–1280, https://doi.org/10.1158/1078-0432.CCR-14-1220 (2015).
Mycielska, M. E. et al. Citrate transport and metabolism in mammalian cells: prostate epithelial cells and prostate cancer. Bioessays 31, 10–20, https://doi.org/10.1002/bies.080137 (2009).
Kavanagh, J. P. Isocitric and citric acid in human prostatic and seminal fluid: implications for prostatic metabolism and secretion. Prostate 24, 139–142 (1994).
Costello, L. C., Franklin, R. B. & Narayan, P. Citrate in the diagnosis of prostate cancer. Prostate 38, 237–245 (1999).
Jadvar, H. & Alavi, A. Role of Imaging in Prostate Cancer. PET Clin 4, 135–138, https://doi.org/10.1016/j.cpet.2009.05.003 (2009).
Jadvar, H. Molecular imaging of prostate cancer: a concise synopsis. Mol Imaging 8, 56–64 (2009).
Mycielska, M. E. et al. Citrate enhances in vitro metastatic behaviours of PC-3M human prostate cancer cells: status of endogenous citrate and dependence on aconitase and fatty acid synthase. Int J Biochem Cell Biol 38, 1766–1777 (2006).
Pan, T. et al. Elevated expression of glutaminase confers glucose utilization via glutaminolysis in prostate cancer. Biochem Biophys Res Commun 456, 452–458, https://doi.org/10.1016/j.bbrc.2014.11.105 (2015).
Canape, C. et al. Probing treatment response of glutaminolytic prostate cancer cells to natural drugs with hyperpolarized [5-13C]glutamine. Magnetic resonance in medicine 73, 2296–2305, https://doi.org/10.1002/mrm.25360 (2015).
Schug, Z. T. et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 27, 57–71, https://doi.org/10.1016/j.ccell.2014.12.002 (2015).
Sanita, P. et al. Tumor-stroma metabolic relationship based on lactate shuttle can sustain prostate cancer progression. BMC Cancer 14, 154, https://doi.org/10.1186/1471-2407-14-154 (2014).
Vaz, C. V. et al. Androgen-responsive and nonresponsive prostate cancer cells present a distinct glycolytic metabolism profile. The international journal of biochemistry & cell biology 44, 2077–2084, https://doi.org/10.1016/j.biocel.2012.08.013 (2012).
Vaz, C. V. et al. Androgens enhance the glycolytic metabolism and lactate export in prostate cancer cells by modulating the expression of GLUT1, GLUT3, PFK, LDH and MCT4 genes. Journal of cancer research and clinical oncology 142, 5–16, https://doi.org/10.1007/s00432-015-1992-4 (2016).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674, https://doi.org/10.1016/j.cell.2011.02.013 (2011).
Lodi, A., Woods, S. M. & Ronen, S. M. Treatment with the MEK inhibitor U0126 induces decreased hyperpolarized pyruvate to lactate conversion in breast, but not prostate, cancer cells. NMR in biomedicine 26, 299–306, https://doi.org/10.1002/nbm.2848 (2013).
Albers, M. J. et al. Hyperpolarized 13C lactate, pyruvate, and alanine: noninvasive biomarkers for prostate cancer detection and grading. Cancer research 68, 8607–8615, https://doi.org/10.1158/0008-5472.CAN-08-0749 (2008).
Golman, K., Ardenkjaer-Larsen, J. H., Petersson, J. S., Mansson, S. & Leunbach, I. Molecular imaging with endogenous substances. Proc Natl Acad Sci USA 100, 10435–10439, https://doi.org/10.1073/pnas.1733836100 1733836100 [pii] (2003).
Brindle, K. M., Bohndiek, S. E., Gallagher, F. A. & Kettunen, M. I. Tumor imaging using hyperpolarized 13C magnetic resonance spectroscopy. Magnetic resonance in medicine 66, 505–519, https://doi.org/10.1002/mrm.22999 (2011).
Dutta, P., Martinez, G. V. & Gillies, R. J. A New Horizon of DNP technology: Application to In-vivo 13C Magnetic Resonance Spectroscopy and Imaging. Biophysical reviews 5, 271–281, https://doi.org/10.1007/s12551-012-0099-2 (2013).
Brindle, K. New approaches for imaging tumour responses to treatment. Nature reviews. Cancer 8, 94–107, https://doi.org/10.1038/nrc2289 (2008).
Kurhanewicz, J., Vigneron, D. B. & Nelson, S. J. Three-dimensional magnetic resonance spectroscopic imaging of brain and prostate cancer. Neoplasia 2, 166–189 (2000).
Nelson, S. J. et al. Metabolic imaging of patients with prostate cancer using hyperpolarized [1-13C]pyruvate. Science translational medicine 5, 198ra108, https://doi.org/10.1126/scitranslmed.3006070 (2013).
Chen, A. P. et al. High-speed 3T MR spectroscopic imaging of prostate with flyback echo-planar encoding. J Magn Reson Imaging 25, 1288–1292, https://doi.org/10.1002/jmri.20916 (2007).
Hurd, R. E., Yen, Y. F., Chen, A. & Ardenkjaer-Larsen, J. H. Hyperpolarized 13C metabolic imaging using dissolution dynamic nuclear polarization. Journal of magnetic resonance imaging: JMRI 36, 1314–1328, https://doi.org/10.1002/jmri.23753 (2012).
Golman, K. In, ‘t Zandt, R. & Thaning, M. Real-time metabolic imaging. Proceedings of the National Academy of Sciences of the United States of America 103, 11270–11275, https://doi.org/10.1073/pnas.0601319103 (2006).
Chen, H. Y. et al. Assessing Prostate Cancer Aggressiveness with Hyperpolarized Dual-Agent 3D Dynamic Imaging of Metabolism and Perfusion. Cancer research 77, 3207–3216, https://doi.org/10.1158/0008-5472.CAN-16-2083 (2017).
Chen, A. P. et al. Hyperpolarized C-13 spectroscopic imaging of the TRAMP mouse at 3T-initial experience. Magnetic resonance in medicine 58, 1099–1106, https://doi.org/10.1002/mrm.21256 (2007).
Zierhut, M. L. et al. Kinetic modeling of hyperpolarized 13C1-pyruvate metabolism in normal rats and TRAMP mice. Journal of magnetic resonance 202, 85–92, https://doi.org/10.1016/j.jmr.2009.10.003 (2010).
DeBerardinis, R. J. et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA 104, 19345–19350, https://doi.org/10.1073/pnas.0709747104 (2007).
Cheong, H., Lu, C., Lindsten, T. & Thompson, C. B. Therapeutic targets in cancer cell metabolism and autophagy. Nature Biotechnology 30, 671–678, https://doi.org/10.1038/nbt.2285 (2012).
Wise, D. R. & Thompson, C. B. Glutamine addiction: a new therapeutic target in cancer. Trends in biochemical sciences 35, 427–433, https://doi.org/10.1016/j.tibs.2010.05.003 (2010).
Le, A. et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell metabolism 15, 110–121, https://doi.org/10.1016/j.cmet.2011.12.009 (2012).
de la Rosa, V. et al. A novel glutaminase isoform in mammalian tissues. Neurochemistry international 55, 76–84, https://doi.org/10.1016/j.neuint.2009.02.021 (2009).
Hu, W. et al. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proceedings of the National Academy of Sciences of the United States of America 107, 7455–7460, https://doi.org/10.1073/pnas.1001006107 (2010).
Jacque, N. et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 126, 1346–1356, https://doi.org/10.1182/blood-2015-01-621870 (2015).
Cassago, A. et al. Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proceedings of the National Academy of Sciences of the United States of America 109, 1092–1097, https://doi.org/10.1073/pnas.1112495109 (2012).
Gross, M. I. et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Molecular cancer therapeutics 13, 890–901, https://doi.org/10.1158/1535-7163.MCT-13-0870 (2014).
Mohamed, A., Deng, X., Khuri, F. R. & Owonikoko, T. K. Altered glutamine metabolism and therapeutic opportunities for lung cancer. Clinical lung cancer 15, 7–15, https://doi.org/10.1016/j.cllc.2013.09.001 (2014).
Massie, C. E. et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. The EMBO journal 30, 2719–2733, https://doi.org/10.1038/emboj.2011.158 (2011).
Shafi, A. A. et al. Differential regulation of metabolic pathways by androgen receptor (AR) and its constitutively active splice variant, AR-V7, in prostate cancer cells. Oncotarget 6, 31997–32012, https://doi.org/10.18632/oncotarget.5585 (2015).
Russell, P. J. & Kingsley, E. A. Human prostate cancer cell lines. Methods Mol Med 81, 21–39, https://doi.org/10.1385/1-59259-372-0:21 (2003).
Kozlowski, J. M. et al. Metastatic behavior of human tumor cell lines grown in the nude mouse. Cancer Res 44, 3522–3529 (1984).
Yang, L. et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol Syst Biol 10, 728, https://doi.org/10.1002/msb.20134892 (2014).
Jiang, L. et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 532, 255–258, https://doi.org/10.1038/nature17393 (2016).
Csibi, A. et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 153, 840–854, https://doi.org/10.1016/j.cell.2013.04.023 (2013).
Yuan, L. et al. Glutamine promotes ovarian cancer cell proliferation through the mTOR/S6 pathway. Endocr Relat Cancer 22, 577–591, https://doi.org/10.1530/ERC-15-0192 (2015).
Wishart, D. S. et al. HMDB 3.0–The Human Metabolome Database in 2013. Nucleic Acids Res 41, D801–807, https://doi.org/10.1093/nar/gks1065 (2013).
Tibes, R. et al. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther 5, 2512–2521, https://doi.org/10.1158/1535-7163.MCT-06-0334 (2006).
Gonzalez-Angulo, A. M. et al. Functional proteomics can define prognosis and predict pathologic complete response in patients with breast cancer. Clinical proteomics 8, 11, https://doi.org/10.1186/1559-0275-8-11 (2011).
Hu, J. et al. Non-parametric quantification of protein lysate arrays. Bioinformatics 23, 1986–1994, https://doi.org/10.1093/bioinformatics/btm283 (2007).
Lee, J. et al. High-throughput hyperpolarized (13) C metabolic investigations using a multi-channel acquisition system. Journal of magnetic resonance 260, 20–27, https://doi.org/10.1016/j.jmr.2015.08.025 (2015).
Acknowledgements
We want to thank Dr. Mark Titus at MD Anderson Cancer Center and the Department of Scientific Publications at MD Anderson Cancer Center for reading and editing the manuscript. In addition, we thank Calithera Biosciences for the CB-839. The research was funded in part by a grant from the U.S. Department of Defense (CDMRP PC110065, NMZ, SS, YL); by Institutional Research Grants (NMZ, PB) and a Startup grant (PB) from MD Anderson Cancer Center; by grants from the U.S. National Cancer Institute (5 P50 CA 094056-14, PB; U54 CA151668, PB; R21CA185536), and by a grant from the Gulf Coast Consortium (JM, PB). YL was funded partially by the Odyssey Postdoctoral Fellowship. This work also was supported by the National Institutes of Health/NCI Cancer Center Support Grant under award number P30CA016672 and used the characterized cell line core facility, the small animal imaging facility, and the NMR core facility at MD Anderson.
Author information
Authors and Affiliations
Contributions
N.Z. led the research effort and wrote manuscript. N.Z., S.S., C.M., J.L., P.D., Y.L., J.M., L.N. ran experiments, helped design study and analyzed data. W.N. helped with animal models. N.Z., L.J. and P.B. helped design study. All authors read and edited manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zacharias, N.M., McCullough, C., Shanmugavelandy, S. et al. Metabolic Differences in Glutamine Utilization Lead to Metabolic Vulnerabilities in Prostate Cancer. Sci Rep 7, 16159 (2017). https://doi.org/10.1038/s41598-017-16327-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-16327-z
This article is cited by
-
Targeting the glutamine metabolism to suppress cell proliferation in mesenchymal docetaxel-resistant prostate cancer
Oncogene (2024)
-
METTL14 downregulation drives S100A4+ monocyte-derived macrophages via MyD88/NF-κB pathway to promote MAFLD progression
Signal Transduction and Targeted Therapy (2024)
-
Molecular mechanism of glutaminase activation through filamentation and the role of filaments in mitophagy protection
Nature Structural & Molecular Biology (2023)
-
Dual contribution of the mTOR pathway and of the metabolism of amino acids in prostate cancer
Cellular Oncology (2022)
-
Glutamine deficiency promotes stemness and chemoresistance in tumor cells through DRP1-induced mitochondrial fragmentation
Cellular and Molecular Life Sciences (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
