
Abstract
The Xanthomonadaceae family consists of species of non-pathogenic and pathogenic γ-proteobacteria that infect different hosts, including humans and plants. In this study, we performed a comparative analysis using 69 fully sequenced genomes belonging to this family, with a focus on identifying proteins enriched in phytopathogens that could explain the lifestyle and the ability to infect plants. Using a computational approach, we identified seven phytopathogen-enriched protein families putatively secreted by type II secretory system: PheA (CM-sec), LipA/LesA, VirK, and four families involved in N-glycan degradation, NixE, NixF, NixL, and FucA1. In silico and phylogenetic analyses of these protein families revealed they all have orthologs in other phytopathogenic or symbiotic bacteria, and are involved in the modulation and evasion of the immune system. As a proof of concept, we performed a biochemical characterization of LipA from Xac306 and verified that the mutant strain lost most of its lipase and esterase activities and displayed reduced virulence in citrus. Since this study includes closely related organisms with distinct lifestyles and highlights proteins directly related to adaptation inside plant tissues, novel approaches might use these proteins as biotechnological targets for disease control, and contribute to our understanding of the coevolution of plant-associated bacteria.
Similar content being viewed by others


Introduction
With the advancement of comparative genomics tools and the rapid increase in completely sequenced bacterial genomes, it became feasible to relate their genetic constitution to their lifestyle and interaction with compatible hosts1,2. Comparison among complete genomes of closely related strains with different lifestyles and virulence may elucidate not only mechanisms of infection and adaptation inside the host, but also identify genes responsible for induction of virulence and ability to infect different hosts3,4.
The Xanthomonadaceae family includes endophytes, phytopathogens, human pathogens, as well as soil-associated and marine bacteria. It contains at least 27 genera, including Arenimonas, Kaistibacter, Luteimonas, Lysobacter, Pseudoxanthomonas, Rehaibacterium, Silanimonas, Stenotrophomonas, Thermomonas, Vulcaniibacterium, Xanthomonas, and Xylella 5. As of January 10, 2017, 862 genomes from this family have been completely or partially sequenced6. However, this coverage is not uniform in terms of genera. In particular, Stenotrophomonas, Pseudoxanthomonas, Xylella, and Xanthomonas dominate the sequencing statistics for the Xanthomonadaceae family.
Members of the Xanthomonas genus propagate through injuries on leaves, stems, and fruits, or even through natural openings of their plant hosts, with the wind as the main dispersion agent7. In addition, most Xanthomonas species can occupy both mesophyllic and the vascular environments within their respective hosts, except Xanthomonas albilineans, which is the only xylem-limited species so far described in this genus8. A remarkable feature shared by bacteria of the Xanthomonas genus is the large number and diversity of type III secretion system (T3SS) effectors and transcription activator-like (TAL) type III effector proteins that regulate host gene expression by associating with promoters of plant genes9. Bacteria of the Xylella genus are non-flagellate and xylem-limited, transferred directly inside the host xylem vessels by insect vectors upon feeding10. The known genomes of Xylella species do not encode a type III secretory system (T3SS), but encode type I and type II secretory systems for the export of exoenzymes and other proteins, which allow colonization within the plant xylem11,12,13. Biofilm formation is important for Xylella species survival in unstable environments with high turbulence, pressure oscillation, and limited nutrient availability, such as the xylem vessels and insect foreguts11. Bacteria of the genus Stenotrophomonas are opportunistic human pathogens, plant endophytes, or rhizosphere- and river sediment-associated14,15,16. Members of the Pseudoxanthomonas genus have been found in soil contaminated with hydrocarbons17, bioreactor compost-feedstock enrichment cultures18, and leafy wood soil19. Interestingly while Xanthomonas and Xylella have many members that are phytopathogens, this lifestyle has not been described for any Stenotrophomonas and Pseudoxanthomonas.
As aforementioned, the mechanisms for propagation, colonization, and induction of virulence are distinct among Xanthomonas and Xylella species. Other members of the Xanthomonadaceae family present additional diverse aspects in terms of plant-related lifestyles. Previous comparative genomics work by our group on Xylella and Xanthomonas genomes has shown that the diversity in plant hosts, and in distinct niches within hosts, whose pathologies are phenotypically distinct, can be related to differences in genomic content20,21,22. This work can thus be seen as an expansion of those prior studies, aiming to cover more members of the Xanthomonadaceae with fully sequenced genomes. By expanding the phylogenetic scope we were able to amplify the diversity in plant-associated lifestyles while retaining genomic relatedness, a combination that offers an excellent opportunity to deepen our understanding of bacteria-plant interactions.
Results
Sixty-nine completely sequenced genomes were selected for our comparative study (Table S1 – Supporting Information). Fifty-one belong to the Xanthomonas genus and seven to the Xylella genus, and all of the corresponding organisms are phytopathogens, except Xanthomonas sacchari strain R1, which was isolated from asymptomatic rice seeds (Fig. S1 and Table S1 – Supporting Information). Eight genomes from the Stenotrophomonas genus were included. Five of these are classified as human opportunistic pathogens (strains ISMMS2, ISMMS2R, ISMMS3, D457, K279a), one as endophyte (strain R5513), one as rhizosphere-associated (strain JV3), and one as river sediment-associated (strain ZAC14D2). Three genomes belonging to the genus Pseudoxanthomonas were also included. One is from a species with metabolic potential for xenobiotic degradation (strain BDa59), and the other two (strains 11-1 and J1) were isolated from a bioreactor and leafy wood soil, respectively. A synthesis of current knowledge regarding the species in this study is presented in Fig. S1. A phylogenomic profile based on the core protein families of the 69 species analyzed is shown in Fig. 1.

Comparative genomics of the 69 selected strains. Venn diagrams demonstrating core genome in phytopathogens, non-phytopathogens, and involving all analyzed strains. The phylogenomic tree was based on the core genome. The numbers in nodes represent bootstrap values and the branch lengths are not proportional to evolution time.
The basic question that we tried to answer with this genome set was whether they contain protein-coding gene families that could be used to characterize Xanthomonadaceae plant pathogens. We answer this question in the affirmative, presenting in what follows a set of seven protein families containing 420 members in total that were present in all 58 phytopathogenic strains analyzed and absent in all others (Table S2 – Supporting Information). The only exception was Xanthomonas sacchari R1, which has genes encoding all seven protein families, but has been isolated from a nonsymptomatic host23. A protein family in our study was defined operationally as a group of protein-coding genes clustered by OrthoMCL, with the added manual curation step of including additional genes by inspecting the results of a tBLASTN search (see Methods).
The seven protein families are the following: (1) lipase/esterase (LipA/LesA); (2) secreted chorismate mutase; (3) glycosyl hydrolase (NixF); (4) beta-galactosidase (NixL); two (5 and 6) alpha-L-fucosidase (NixE and FucA1); and (7) VirK protein. Various features of these seven protein families are shown in Table 1, including information regarding presence of a signal peptide, cellular localization, domain analysis (Fig. S3 – Supporting Information), and putative protein-protein interaction results. We now interpret the biological meaning and implications of this general result, focusing on each protein family in turn.
Family 1: Lipase/esterase, virulence inducer and plant immunity modulator
Lipases are a broad group of lipolytic enzymes divided into eight families comprising six subfamilies24,25. They belong to the superfamily of alpha/beta hydrolases together with other enzymes such as esterases, acetylcholinesterases, cutinases, carboxylesterases and epoxide hydrolases. Despite differences in sequence and function, members of this superfamily possess a conserved alpha/beta fold and pentapeptide motif. According to the amino acid composition of the catalytic triad, there are three classes of alpha/beta hydrolases: GGGX-, GX-, and the Y-class26. The GGGX class includes carboxylesterases and short chain length specific lipases that encompass LipA orthologs26. All Xanthomonas strains studied here have one copy of LipA, whereas all Xylella strains have three copies, except for strain M12, which has two copies. Other genes assigned with the lipase or esterase function are found in Xanthomonas genomes; however, these other genes are not orthologous to the family described in this section according to our analysis, since lipA/lesA combine both lipase and esterase and cleave exclusively small chain fatty acids27, besides other functional parameters (Table S3 – Supporting Information)28,29.
We investigated the presence of genes coding for LipA proteins (known as LesA in Xylella spp) in bacteria other than the 69 analyzed here. Interestingly we found that this gene is present in β- and other γ-proteobacteria, but only in plant-associated bacteria (Fig. 2a). This result indicates that this lipase/esterase may be fundamental not only for phytopathogenicity, but also for the association of bacteria with plants. Additional evidence for this hypothesis comes from the fact this lipase/esterase is absent in Xanthomonadaceae bacteria that are not phytopathogens, or indeed in any other members of the γ-proteobacteria family, according to our search results used to construct Fig. 2. The first hit from a non-plant associated bacteria is from Thalassolituus sp. HI0120, with an amino acid identity of only 40%; this protein product does not have the conserved catalytic triad. Although this ortholog from Thalassolittus has not been characterized biochemically, the substitution of the histidine within the catalytic triad suggests that this protein would have distinct biochemical properties30. Moreover, a structural profile of LipA orthologs from X. oryzae indicates that only γ-proteobacteria have genes that code for proteins with the same putative structure topology, with maintenance of the canonical residues directly involved in substrate catalysis (S176, D336 and H377) (Fig. 2b and Table S4 – Supporting Information). In the β-proteobacteria B. graminis, P. violaceinigra, and A. delafieldii the canonical residue H in the respective orthologs is replaced by A, E, and P, respectively. Whether this alters any of the enzymatic properties in this orthologs remains to be evaluated. Additionally, a biochemical characterization of LipA from Xac306 enabled us to verify its importance to virulence. When expressed in E. coli, the enzyme conferred lipase and esterase activity (Fig. 3a–c), and the cell extract was able to cause a hypersensitive response in tobacco leaves (Fig. 3d), while a Xac306 deletion-mutant for lipA lost most of its lipase and esterase activities (Fig. 3e–g) and displayed reduced virulence in citrus (Fig. 3h).

Phylogenetic and structural analysis of secreted lipase. (a) Highlighted in red are γ-proteobacteria (all phytopathogens) whereas blue highlights β-proteobacteria, all associated with soil or plant roots. The numbers associated with nodes represent bootstrap values at the color gradient. The numbers surrounding the phylogeny represent models proposed in “b” taken from the structure of the reference protein (R) obtained from X. oryzae using Protein Data Bank (PDB – c3h2Ia)93. (b) Structural profile of LipA orthologs from X. oryzae (R) highlighting the canonical residues directly involved in substrate catalysis (S176, D336 and H377). The putative protein structures (1 to 11) were obtained using Phyre282. The alignment confidence degree varied from good (red) to bad (blue) in each structure, with the lowest conservation in the region associated with ligand-binding domain (LBD). Check for details at Fig. S9 – Supporting Information.

Characterization of LipA from Xanthomonas axonopodis pv. citri strain 306 pathotype A. Representative results of (a) western-blot of the protein encoded by XAC_RS02605 expressed in E. coli DH5α. (b) Lipase activity around a colony of the transformed E. coli on solid LB medium containing tributyrin, whereas transformant with an empty vector (EV) did not show any activity (n = 3). (c) Esterase activity of secreted extracts measured by fluorescence emitted by degradation of 4-methylumbelliferone (MUB) (n = 4). (d) Hypersensitive response on tobacco leaf infiltrated with cell extracts of E. coli transformed with an empty vector (EV) or containing the XAC_RS02605 cassette (n = 3). (e) Confirmation of disruption of lipA in Xac306 by PCR amplification. (f) Lipase activity around Xac306 colonies (n = 3). (g) Esterase activity of secreted Xac306 extracts (n = 4). (h) Virulence assay on citrus leaf by infiltration of wild type Xac306 and mutant with inactivated lipA. See Methods for more details on assays.
Family 2: Secreted chorismate mutase, a key enzyme in modulation of the plant immune response
All genomes studied here have chorismate mutase (CM); however, non-phytopathogenic species have only one gene with this annotation while phytopathogens have two. The extra gene in phytopathogens is markedly distinct from the other gene, and despite being generally annotated as chorismate mutase, our analysis clearly shows that they belong to two distinct protein families that share a conserved domain. Firstly, the gene present only in phytopathogens averages 576 bp in length, encoding only the CM domain, whereas the other gene present in all taxa is 1,181 bp long, encompassing two other domains besides the CM domain. Secondly, we found that only those genes in the phytopathogens have a signal peptide (Fig. S5a–c – Supporting Information), suggesting that the encoded proteins are secreted. Based on these results we designated the phytopathogen-specific CM gene (and its respective orthologs) as CM-sec and the gene with wide distribution (and its respective orthologs) as CM-nonsec.
The protein coded by CM-nonsec has three domains: an N-terminal chorismate mutase (CM_2 PF01817), an internal prephenate dehydratase (PDT PF00800), and a C-terminal Phe regulatory (ACT PF01842) domain (Fig. S6a – Supporting Information), whereas the protein coded by CM-sec has only a chorismate mutase domain (CM_2 PF01817), which comprises almost the entire length of the protein (Fig. S6b and S7 – Supporting Information).
If CM-sec is secreted, what would be its function? It is known that the secretion of CM by the fungus Ustilago maydis reduces Trp synthesis, which modulates plant metabolism by reducing synthesis of defense compounds31. Based on this functional analogy, we performed a detailed in silico analysis of the CM-sec three-dimensional (3D) structure. The hit in the structural analysis was from Burkholderia thailandensis (PDB 4oj7.1.A) with 98.2% coverage and 39.74% identity, confirming the typical CM domain fold (Fig. S6b – Supporting Information). In addition, quality of conformation analysis between these two sequences indicates that all α-helices have a predicted quality higher than 80%. Furthermore, ten structural parameters were analyzed for this topology and only “relative conservation” shows low values, corroborating the alignment data and the coverage (Fig. S7 – Supporting Information), including conserved catalytic residues and residues involved in pocket conformation (Fig. S6c – Supporting Information).
We reconstructed the phylogenies for the 250 best Blastp hits for both CM-sec and CM-nonsec, using as queries the Xac306 versions. In the CM-sec tree 89% of the orthologs belong to plant-associated bacterial strains (Fig. 4a,b), from different classes (α, β, and γ-proteobacteria). The CM-nonsec tree follows the expected distribution of species within the γ-proteobacteria subdivisions. The branch lengths of the CM-sec tree are relatively small compared to those of the CM-nonsec tree, suggesting more evolutionary conservation for CM-sec genes.

Phylogeny of CM-sec (a) and CM-nonsec (b). Both analysis utilized the first 250 comparison hits from Blastp, standardizing bars to 0.4 residues. It was noticed that for CM-sec the majority of analyzed organisms are plant-associated organisms (check for details at Fig. S10 and S11 – Supporting Information) which are found within three different classes (α, β and γ-proteobacterias), while for CM-nonsec all the analyzed microorganisms are introduced in γ-proteobacteria class maintaining the structural pattern of order belonging to this class. In addition, it is possible to observe that frequency of non-synonymous mutations is smaller in CM-sec indicating that this copy was more conserved during the evolution of the species.
Families 3, 4, 5, and 6: N-glycan degradation enzymes, an intricate mechanism of plant immune system depletion
The next four families that we have found are glycosyl hydrolase (NixF), beta-galactosidase (NixL), and two alpha-L-fucosidases (NixE and FucA1). These four families are associated with N-glycan degradation (Fig. 5)32. According to Boulanger, et al.32 and Dupoiron, et al.33, Xcc has two gene clusters associated with degradation (nix) and internalization of N-glycans and associated monomers (nag). Among the nix genes we find nixE, nixF, and nixL, coding for alpha-L-fucosidase, glycosyl hydrolase, and beta-galactosidase, respectively. Alpha-L-fucosidase catalyzes the α-1,4-glycosidic bond between L-fucose (FUC) and N-acetyl-glycosamine (NAG), glycosyl hydrolase hydrolyzes the β-1,4-glycosidic bound between NAG, and beta-galactosidase hydrolyzes the β-1,4-glycosidic between NAG and galactose (GAL) (Fig. 5b).

Functional and syntenic analysis of genes associated with N-glycans degradation. (a) Composition and structural organization of nix genes in Xcc and Xac306. Each of the individual colors of genes in the cluster corresponds to the respective site of catalysis shown for the N-glycan metabolism in Fig. 5b (adapted from Boulanger, et al.48). Three protein families from our study are encoded by genes included in this cluster and are circled in bold (NixE, NixF, and NixL). For Xac306 a set of genes with correlated functions between nixI and nixJ are highlighted. The region downstream of the cluster has structural variations among the investigated organisms. (b) Composition of an N-glycan with the respective enzymes associated with its degradation. The colors of the proteins identifiers and their catalysis follow the color pattern shown in Fig. 5a and d. The dotted lines refer to the catalysis of the phytopathogen-enriched proteins. (c) Structural organization of nag genes. (d) Composition and structural organization of putative genes associated with N-glycan degradation in Xcc and Xac306. The gene coding the FucA1 family specific to phytopathogens are circled in bold. (e) NixE, NixF, NixL, and FucA1 phylogenies. The red squares highlight positions occupied by Xanthomonas and Xylella. The clade composition of NixE, NixF, and NixL are similar among each other and distinct when compared to FucA1 because these enzymes encoded genes arranged in the same genomic region that compose the nix cluster. Check for details at Figs S12–S15 – Supporting Information.
Comparative analysis of this cluster in Xcc and the one in Xac306 shows the following differences. Xac306 has an additional set of genes between nixI and nixJ: a gene encoding a TonB-dependent receptor (TBDR), a second copy of the beta-galactosidase gene (bga), a cation transporter gene (yhfM), a ribokinase gene (rbk), and a 6-phospho-beta-glucosidase gene (celF). The downstream region in Xcc has transposable elements, genes that encode restriction and modification systems, and the following three T3SS effector genes: xopD, which is involved with suppression of plant defense34 and is found only in Xcc; xopK, which has an unknown function35; and xopL, which is a leucine rich repeat protein36. Other proteins putatively involved in catalysis of glycosidic bonds, such as neuraminidase and asparaginase, are found in the genomic neighborhood of the nix cluster (Fig. 5a).
The fourth protein family related to N-glycan degradation is another copy of alpha-L-fucosidase (FucA1). This family also appears to be associated with a cluster involved in the degradation of this polymer since downstream genes include another copy of beta-galactosidase (bga), an arabinogalactan endo-1,4-beta-galactosidase (galA), and another gene encoding TBDR, all of which are conserved in Xanthomonas (Fig. 5d). The presence of transposable elements was also found in Xcc, not only within the cluster, but also in the upstream flanking region.
According to the CAZy (carbohydrate-active enzymes) database37 there are only two copies of alpha-L-fucosidase (GH4 group) in Xanthomonas and Xylella genomes, and both copies are enriched in phytopathogens. This may be an adaptation to the presence of fucose residues in plant N-glycans, which is not observed in other organisms.
A database search for each of the four proteins involved in N-glycan degradation showed a mixture of bacteria from different phyla (Fig. 5e), which is a distinct distribution pattern when compared to other protein families analyzed in this study. Most of the microorganisms retrieved in the search are associated with plants and soil38.
Family 7: VirK, a new target for study of plant-bacteria interactions
Among the seven protein families highlighted in this study, the most intriguing is VirK. Until now, no study has determined its actual function. To better understand VirK function, several in silico investigations were performed. The encoded protein contains a signal peptide with approximately 22 residues, and secondary structure analysis of the mature protein suggested that VirK consists of 10 α-helices (Fig. S8 – Supporting Information). In silico analysis of the interaction between VirK and other proteins (Fig. 6a) predicts that VirK interacts with several other proteins, as follows: three T3SS-associated proteins, including HrcR, a protein required for formation of the inner membrane ring of the T3SS apparatus39, and the secreted effectors AvrXacE1 (XopE1) and AvrXacE2 (XopE3); XcsH, a T2SS structural protein, and two cellulases; and VirJ, a protein associated with the T4SS apparatus40. Curiously, all of the phytopathogen genomes contained two copies of VirJ, and VirK was associated with both copies. One of the copies is specific to the phytopathogen genomes analyzed here, with the exception of the Xylella strains Ann-1 and MUL0034 (which lack this copy). The computational predictions also suggest that VirK interacts with a copper resistant protein (CutC), with a hypothetical protein whose gene, in some genomes, appears immediately downstream of virK, and with nixI (XAC3076) and asparaginase (XAC3092), both of which are related to N-glycan catalysis.

VirK interaction and phylogenetic analysis. (a) VirK interaction model, adapted from STITCH 4.077. Circles denote nodes corresponding to each protein that interact with VirK (central). Proteins associated with T3SS are shown in pink and proteins associated with T2SS are highlighted in purple. VirJ corresponds to a protein annotated as associated with T4SS92 and phytopathogen genomes have two copies of VirJ. The copies present in all 69 genomes are shown in orange while blue highlights the copy specific to most phytopathogens. VirK associates with a copper resistance protein shown in brown (CutC) and with a hypothetical protein (shown in green) whose gene always appears concatenated downstream from virK. (b) VirK phylogeny. Check for details at Fig. S16 – Supporting Information.
We reconstructed a phylogeny for VirK based on the best 250 Blastp hits (Fig. 6b). The phylogeny suggests pervasive HGT in Proteobacteria, and subsequent selection for maintenance of this gene in specific lineages. The broad distribution of VirK among bacteria associated with plants and its putative interaction partners suggest a possible role for VirK in the modulation of plant immune response during the infection process.
Discussion
In this section, we seek to determine the relevance of the results described in terms of previous work from the literature.
LipA/LesA
In the Xanthomonas genus, lipases/esterases, polygalacturonases, cellulases, pectate lyases, xylanases, and cellobiosidases have been described as fundamental for cell wall degradation41. These enzymes have been analyzed in a study using Xanthomonas oryzae to probe for association with the xylanase XynB. These proteins were found to be required for virulence, since mutations in xynB caused a massive reduction of the virulent phenotype in rice crops42. A high-resolution analysis of LipA structure has shown an all-helical ligand binding module as a distinct functional attachment to the canonical hydrolase catalytic domain28. Point mutations that disrupted the carbohydrate anchor site or blocked the pocket, even at a considerable distance from the enzyme active site, abrogated LipA function in plants, as exemplified by loss of both virulence and the ability to elicit host defense responses. In addition, LipA, cellulase (ClsA) and a putative cellobiosidase (CbsA) were identified in X. oryzae pv. oryzae as type II secretory system (T2SS) effectors that induce the rice defense response, which are then repressed by T3SS effectors to ensure successful infection43.
Lipases/esterases and polygalacturonases have also been studied In the Xylella genus27,44. In Xylella fastidiosa, three new pathogenicity effectors have recently been described, and one of them (LesA) is a lipase/esterase homolog of LipA. The same study demonstrated that paralogs of genes that encode LipA were found in the Xylella genome and that one of these copies is concatenated upstream of lipA. LesA has recently been identified in the secretome of Xylella, and lesA mutants were shown to be not virulent when inoculated into grapevines27. Among cell-wall degrading enzymes identified in the Xylella secretome, three copies of lipase/esterase were among the most abundant (LesA, LesB and LesC). Finally, the same authors demonstrated that the LipA ortholog is secreted into outer membrane vesicles, and the enzyme was also detected in infected host tissue27. The duplicated secretory virulence factors of Xylella fastidiosa (three copies of lipase/esterase) exemplify the positive selection pressures exerted on advantageous genes in such pathogens45. These increased gene copy numbers in Xylella genomes suggest the importance of this enzymatic activity for successful colonization of plants. Literature results show that in Xanthomonas and Xylella the role of lipase/esterase in virulence varies since it is essential in Xylella but dispensable in Xanthomonas, despite both being enzymatically active27.
Although LipA/LesA has a lipase/esterase function and belongs to a large sub-group of α/β-hydrolases, Xanthomonadaceae LipA represents a new catalysis model with a finely adjusted activity28. This finding explains its plant-associated function, characterizing LipA as a notable example of adaptation in phytopathogenic organisms. These authors also suggest that this enzyme is present in a common ancestor of Xanthomonas and Xylella (phytopathogens) and present in Burkholderia species (commensal bacteria associated with plants). Although evolutionarily distant from Xanthomonas and Xylella, our hypothesis is that Burkholderia species would require this molecular speciation because of their association with plants.
Secreted chorismate mutase (CM-sec)
CM is a component of the shikimate pathway (Fig. S4 – Supporting Information) and is required for the conversion of chorismate to prephenate, a precursor for phenylalanine (Phe) and tyrosine (Tyr) synthesis46. In several organisms, chorismate can also be converted to precursors for tryptophan (Trp) synthesis47. In plants, CM acts as a precursor of salicylic acid biosynthesis, an important signaling molecule in the plant immune response48,49.
We identified two CM genes, which we named CM-sec and CM-nonsec. Although both should be able convert chorismate to prephenate, CM-nonsec may also be able to convert prephenate to phenylpyruvate, which would be converted to Phe and Tyr50. Therefore, CM-nonsec likely provides these two amino acids during cell metabolism due to the presence of a Phe-regulatory domain at the C-terminal position that functions to down-regulate this metabolic branch51,52.
In previous studies, X. oryzae pv. oryzae CM-sec knockout mutants were significantly more virulent in rice than the wild type (wt) strain53. In addition to differences in lesion size, disease symptoms were also stronger in CM-sec knockout mutants. When the CM-sec defective strain was complemented with the CM-sec functional gene, the virulence was similar to that of the wt53. In fact, reduction of virulence associated with CM-sec suggests that the protein coded by this gene attenuates the plant’s antibacterial response. Therefore, CM-sec may be an important evolutionary adaptation of phytopathogenic species to help ensure survival of bacteria inside the host. A supporting literature result is that CM-sec also decreases salicylic acid synthesis in plants31. CM-sec is probably secreted via T2SS53. Moreover, differential expression analysis of secreted proteins from an Xac306 wt and an hrpB4 mutant showed that CM-sec is secreted when both strains are grown in XAM154. Since the absence of a functional T3SS in hrpB4 mutants did not affect CM-sec secretion, is was concluded that CM-sec is not secreted through the T3SS. Recently it has been shown that CM is one of the proteins secreted by the T2SS in Xylella fastidiosa 27, but its precise substrates both in Xanthomonas and Xylella pathosystems are yet to be determined.
Proteins involved in N-glycan degradation
Glycans are oligosaccharide chains covalently linked to proteins, and are classified into two types. N-glycans are attached to membrane proteins through a linkage to asparagine in the sequence Asn-Xaa-Ser/Thr, while O-glycans are commonly attached to proteins via Ser/Thr residues55. In plants, N-glycans present a conserved structure composed by monomers of xylose, mannose, fucose, N-acetylglucosamine, and galactose56. Although N-glycans are usually associated with proteins, free N-glycans (F-NG) have been found in different plant tissues57,58. They are either the result of an endoplasmic reticulum (ER) retro-translocation followed by catalysis of endo-β-N-acetylglucosaminidase or N-glycanase, or the result of vesicle trafficking mediated by the Golgi apparatus59. Recently, it has been shown that N-glycans attached to ectodomains of plasma membrane pattern recognition receptors (PRR) likely form initial contact sites between plant cells and invading pathogens60. The N-glycosylation of membrane proteins, both in the ER and in the Golgi apparatus, has been shown to play a crucial role in plant immunity61,62.
X. campestris (Xcc) has two gene clusters required for the degradation (nix) and internalization of N-glycans and associated carbohydrates monomers (nag)32,33. The nix cluster consists of a gene coding for a TonB-dependent receptor (TBDR) and nine genes (nixD-L) with hydrolase function at different glycosidic bonds in the carbohydrate units that compose the N-glycans structure (Fig. 5a,b)33. Xac306 and strains of Xcc have a conserved nag cluster32 (Fig. 5c). Mutations in genes present in both clusters strongly affect the virulence of Xcc in compatible hosts63. Interestingly, L-fucosidase was identified in X. oryzae secretome only when the bacteria were subjected to growth in planta, while glucan 1,4-beta-glucosidase has been detected in planta and in vitro 64. NixE was detected in the culture supernatant of Xcc wt strain, as well as in both T2SS mutant strains, indicating that this protein is secreted in a T2SS-independent manner33. Since T2SS is evolutionarily related to the type IV secretory system (T4SS)65, NixE may be secreted through T4SS. In contrast, NixF and NixL were not detected in the T2SS mutants, suggesting that these proteins are secreted via the T2SS33,66. Based on our results, we hypothesize that Nix proteins could affect the degradation of N-glycans in three different ways: (i) by direct activity on N-glycans associated with plant membrane receptors (EFR and FLS2); (ii) by degradation inside the ER or Golgi apparatus, which prevents formation of mature glycosylated receptors; or (iii) by degradation of free N-glycans. The first two mechanisms may reduce the immune response due to non-recognition of the bacterial EF-Tu and flagellin effectors, and possibly other pathogen-associated molecular patterns (PAMPs).
VirK
Differential expression analyses have indicated that virK/VirK is always induced under infectious conditions or in culture media that mimic survival conditions in planta 67,68. Another study confirmed the differential expression profile of 32 genes from Xac306 that were putatively associated with virulence in medium containing copper69. That work provided additional evidence that the expression profile of virK follows that of genes associated with virulence. It is noteworthy that seven out of 10 proteins differentially expressed in both studies were found to interact with VirK, as determined by STITCH 4.0 (Fig. 6).
VirK was also one of the most highly expressed proteins, in infectious conditions or in simulations of such conditions. Expression of 11 proteins (including VirK) secreted by T2SS from Xac306 was higher in a HrpG* mutant with the ability to induce the expression of T3SS-related genes even under nutrient-rich conditions70. 109 proteins from the secretome of X. oryzae grown in planta and in vitro were identified using 2DE coupled with MALDI-TOF-MS and/or nLC-ESI-MS/MS; VirK and XadA (adhesin, outer membrane protein) were the only proteins associated with virulence detected exclusively in planta 64. In another study, VirK was one of the 64 identified proteins in vascular fluids from infected rice plants, in conjunction with hydrolytic enzymes involved in chemotaxis, membrane proteins, and proteins classically related to PAMPs such as flagellin and EF-Tu68. An analysis of the secretome of Xac306 wt and hrpB4 (associated with T3SS) mutant cultivated in rich vs. virulence induction medium confirmed that VirK together with cellulases and polygalacturonases is one of 14 proteins detected only in the wt secretome when the bacteria were grown in virulence induction medium54. All of these reports support the hypothesis that secretion of VirK is important for pathogen-host interactions, although its precise function remains unknown.
Reports in the literature show that VirK is secreted by the T2SS70 or by the T4SS65. Additional evidence for secretion through the T2SS is given by the finding that HrpG regulates VirK in Ralstonia solanacearum 71, as some proteins secreted by the T2SS are regulated by HrpX/G, a two-component system that plays global roles in coordinating different virulence traits of Xanthomonas axonopodis pv. citri 72.
Co-evolutionary implications
Our analysis suggests that the seven protein families are examples of molecular speciation involved in the survival maintenance of phytopathogens inside plants. The proteins belonging to these seven protein families are putatively secreted by the T2SS and can be classified as effector proteins, directly or indirectly correlated to the induction or evasion of the plant immune system. We hypothesize that the evolution of the corresponding genes is a result of the ‘arms race’ between plants and these phytopathogens73. We further speculate that our results are not restricted to the phytopathogens in the Xanthomonas and Xylella genera, but apply as well to all plant-associated bacteria that have true orthologs of these proteins.
According to the model proposed in Fig. 7, inside plant tissues CM-sec would be related to displacement of plant metabolism for synthesis of aromatic amino acids. As a result, there is a reduction in the production of salicylic acid, a plant hormone that induces the plant defenses against a variety of biotic and abiotic stress by modulating biochemical, morphological, and physiological mechanisms. Likewise, other studies have shown that the lipase/esterase, LipA, is associated with degradation of the plant cell wall. Although the action of this enzyme disrupts the plant’s first defensive barrier so that invading phytopathogens are able to colonize plant tissues, byproducts of this degradation are potential activators of innate immune responses in plants. Moreover, although N-glycans are structures that may be present as free polymers in plant cells, they are essential structures of receptors on the plant cell membrane. Thus, insertion of this polymer into these proteins occurs in the Golgi apparatus, and deficiency in this process culminates in receptors that cannot transduce molecular signals. In fact, loss of the transduction capacity associated with receptors such as EFR and FLS2, which recognize EF-Tu and flagellin proteins, respectively, would reduce the plant immune response. In addition, degradation of N-glycans linked to these receptors on the plant surface by Nix enzymes decreases the immune response while simultaneously facilitating the penetration of bacteria into the plant tissue. Finally, although VirK has no known function to date, our results provide additional evidence that it is a secreted protein, and it is specific to bacteria associated with plants. In addition, VirK is associated with proteins involved in the T2SS, leading us to hypothesize that VirK acts as a new effector associated with reduction of the plant defense response.

Overview of the seven plant-associated specific proteins and its correlation with induction and evasion of the plant immune system. The arrows in black highlight possible activities of these seven enzymes putatively secreted by T2SS. The Nix nomenclature summarizes the activity of enzymes that are part of the whole nix cluster, with particular emphasis on NixE, NixF and NixL. The blue arrows highlight induction of the immune response mediated by DAMP/PTI (mediated by lipase action) and PAMP (mediated by EF-Tu and flagellin), and evasion of immune response induced by ETI (mediated by T3SS effectors). The immune system activation responses (in red) trigger the production of reactive oxygen species (ROS), induction of systemic acquired resistance (SAR) and generation of calloses, necrosis, and programmed cell death (PCD). It is possible that Nix operates in two contexts: in free N-glycans (F-NG) or in N-glycans associated with membrane receptors (EFR and FLS2). The action of Nix proteins could degrade N-glycans that should be linked to these receptors at Golgi and directly degrade N-glycans from receptors on the plant surface. Both byproducts of N-glycans or cell wall degradation may still be used as a carbon source for bacterial metabolism. DAMP: Damage-Associated Molecular Patterns. PTI: PAMP-Triggered Immunity. ETI: Effector-Triggered Immunity.
Conclusion
Our study identified seven protein families whose characteristics make them potential molecular targets in dealing with a variety of plant diseases. These results demonstrate the importance and potential of comparative genomics studies to elucidate intriguing biological processes, and the fact that genomic approaches are transforming our understanding of co-evolutionary interactions between bacteria and plants. Furthermore, given the inferred importance of selection in maintaining, or in some cases, duplicating, the genes mentioned in this study, for survival of the respective phytopathogenic lineages, we speculate that some plant-infecting Xanthomonadaceae are transitioning to a mutualistic equilibrium with their plant hosts. This equilibrium may eventually succeed when bacterial virulence and plant development are both successfully adjusted, a possible outcome in the evolution of parasitic bacteria. Therefore, the present study may broaden our knowledge of plant-parasite interactions within the Xanthomonadaceae family, while also contributing to knowledge of host-parasite evolution in general. A complete understanding of the molecular basis of plant disease resistance will enable the application of these discoveries to generate plants that contain tuned disease-resistance pathways that are durable and recognize a wide spectrum of pathogens.
Methods
Genome selection
We included in our study 69 completely sequenced genomes available at NCBI database as of October 2015 belonging to the genera Pseudoxanthomonas (3), Stenotrophomonas (8), Xanthomonas (51), and Xylella (7) – Fig. S1 and Table S1 (Supporting Information).
Prediction of protein families
Analysis of the presence or absence of each protein family among the 69 genomes allowed us to predict unique, flexible and core protein families that are shared by a sub-set of bacterial strains using Orthologsorter74 available at http://jau.facom.ufms.br/xanthomonadaceae2/orthologsorter/. This tool, whose interface was developed specifically for this purpose enables the user to identify sets of specific protein families from each genome or shared among selected genomes according to a specific interest. Only seven protein families were found from the selection of proteins shared exclusively among the 58 phytopathogens analyzed (see results and discussion). These seven families are comprised by the 420 proteins considered in this work and were found by asking Orthologsorter which families contain at least one protein from the phytopathogen genomes and also do not contain any genes of the other 11 non-pathogenic ones. Protein families have been found by OrthoMCL75, that is a very well-known genome-scale software for grouping orthologous protein sequences, based on Blast reciprocal matches and MCL clustering methods.
In order to confirm whether these seven protein families are specific to the phytopathogens analyzed, avoiding problems related to annotation heterogeneity, an amino acid versus nucleotide tBLASTN alignment was performed using the protein sequences of Xac306 genes as queries and a BLAST database composed of the genome sequences from the 69 fully-sequenced genomes as subject with maximum e-value set to 10−10. This method generated no new hits compared to the information initially gathered from the annotation of the 68 genomes (excluding Xac306) (Table S5 – Supporting Information).
Phylogenomic profile
The phylogenomic profile was predicted based on the core protein families. First OrthoMCL found 10 432 families, of which only 846 had exactly one protein from each genome. These families were aligned and concatenated (295 895 columns) using MUSCLE76. After the removal of non-informative columns (columns that may not be conserved, including columns with too many gaps, or that may be saturated by multiple substitutions) using GBlocks77, only 1 362 columns remained. The tree was then generated from the 1 362 columns using RAxML78 with the PROTCAT model, rapid bootstrapping with 100 replicates, and maximum likelihood search.
Phylogenetic analysis of the seven proteins families
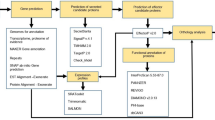
For phylogenetic analysis of each one of the seven protein families specific to the phytopathogens analyzed, we used protein sequences from Xanthomonas axonopodis pv. citri strain 306 pathotype A – Xac306 (NCBI accession NC_003919.1) as the query for a Blastp search79, with the maximum number of hits set to 250. Protein multiple alignment was performed using the G-INS-i algorithm in Mafft 7.2580 with 16 iterative refinement steps. TrimAl v1.4 was used to remove columns with an excess of gaps81. IQTree v1.3.1182 was used to predict the best phylogenetic model out of 468 tested and to estimate the best maximum likelihood (ML) tree with branch support obtained by 1 000 bootstrap pseudo replicates. Tree visualization, rooting, and branch coloring were all performed in FigTree v1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/). Due to unknown relationships among higher-order bacteria clades that appeared in gene trees (e.g., Proteobacteria vs. Actinobacteria), we used midpoint rooting to infer polarity of evolution. Branch coloring among higher-order bacteria lineages was kept the same across the studied genes. Figure S2 (Supporting Information) shows the pipeline of computational analyses performed in this work. Additional computational methods are described in Supplementary Methods.
Strains and growth conditions
The wild type Xanthomonas axonopodis pv. citri strain 306 (Xac306wt) and mutants were grown in Nutrient Broth (NB) medium with aeration (220 rpm) at 28 °C. Plate cultures were prepared in the same medium with the addition of 1.5% agar. NB medium was supplemented with kanamycin (10 μg/mL) for selective growth of the lesA mutants. Xac306wt strain used in this study was kindly provided by Dr. Chuck Farah, University of São Paulo, Brazil. For heterologous expression of LesA from Xac306wt, Escherichia coli strain DH5α cells were grown in Luria-Bertani (LB) medium supplemented with kanamycin (50 μg/mL) at 37 °C and 120 rpm.
Heterologous expression of LipA
Heterologous expression of LipA was performed by cloning XAC0501 in pJexpress 401 vector (DNA2.0, USA). The insertion of the cloned gene was confirmed by PCR using primers flanking lipA, followed by transformation of E. coli strain DH5α competent cells. Cells were grown in LB broth supplemented with kanamycin (50 μg/mL) at 37 °C and 120 rpm until OD600 0.8, when 1 mM IPTG was added and cells cultured for additional 3 h at 30 °C. Total protein was extracted from E. coli cells using a lysis buffer (1 M Tris-HCl pH 7.5; 5 M NaCl; 10 mg/mL lysozyme; 1% glycerol; 100 mM benzamidine; 0.5 M EDTA; 100 mM PMSF). The protein concentration was determined according to Bradford’s method using BSA as the standard.
Western blot analysis
To detect LipA, 10 μg of total protein extracted from E. coli cells was evaluated by SDS-PAGE and transferred by electroblotting to nitrocellulose membrane (BioRad, USA). HRP-conjugated anti-Flag antibody was diluted in PBS-M 1% (PBS plus 1% skim powdered milk 1:1000), followed by incubation of 3 h. Blocking and washing steps were performed with PBS-M 5% (PBS plus 5% skim powdered milk) and PBS-T 0.1% (PBS plus 0.1% Tween 20), respectively. Developments were carried out using ECL Plus western blotting detection reagents (GE Life Sciences, USA).
Lipase and esterase activity assays
Tributyrin (C4) (Sigma-Aldrich, USA) was used as substrate for LipA activity in a plate assay. To test the lipase activity of the total protein extract from E. coli, the triglyceride substrate (1%; v/v) was prepared in a buffer containing 100 mM Tris-HCl (pH 8.0), 25 mM CaCl2, 2% agarose and solidified in Petri dishes. Total protein extracts (1 µg/μL) were added to the wells and assayed for a zone of clearance for 24 to 48 h at room temperature (23 °C). For the E. coli and Xac306 lipA mutant lipase activity assay, tributyrin (1%; v/v) was emulsified in LB and NB agar medium, respectively. The plates were incubated at 37 and 28 °C for about five days, enough time for the emergence of tributyrin degradation halos surrounding the inoculation holes. Esterase activity was also determined using 4-methylumbelliferyl butyrate (4-MUB) substrate as previously described by Vaneechoutte, et al.83.
Hypersensitive response assay in Nicotiana tabacum leaves
Total proteins from empty vector and LipA-expressing E. coli were used for syringe infiltration in greenhouse-grown Nicotiana tabacum leaves (plants were approximately two months of age and 30 cm height). The infiltration occurred with the aid of a needleless syringe in spots previously pierced with a needle. Ninety μg of protein (0.3 μg/μL) was infiltrated into each leaf spot. HR-like lesions in the infiltrated area were photographed 24 h after inoculation.
Isolation of lipA mutants
The mutagenesis cassette was chemically synthesized (GenScript, USA) after insertion of a kanamycin resistance gene in the central portion of the target gene XAC0501 (within Ser-200, the first amino acid of the active triad, maintaining the 5′ and 3′ unchanged to allow homologous recombination). For confirmation of the mutated locus, oligonucleotides Xac0501MutF: TGACCACTCACGCTTCTTCC and Xac0501MutR: CAACCATCGTACCCACTCTATC were used for PCR amplification.
Citrus leaf infection
Mutant and wild type strains of Xac306 were grown in liquid medium NB, centrifuged (5000 x g, 10 minutes, 4 °C) and washed twice with 25 mM CaCl2. The optical density was adjusted to OD600 of 0.5. This suspension was then infiltrated into the lower surface of leaves of orange seedlings (Citrus sinensis, Valencia), approximately 50 cm in height. The infiltration occurred with the aid of a syringe in spots previously pierced with a needle to drench the leaf blade. The treatments of this experiment were done in triplicate. Lesions in the infiltrated area were photographed twenty-five days after inoculation.
Data Availability
All data used in this work is publicly available at NCBI6.
References
Gao, X. Y., Zhi, X. Y., Li, H. W., Klenk, H. P. & Li, W. J. Comparative genomics of the bacterial genus Streptococcus illuminates evolutionary implications of species groups. PloS one 9, e101229, https://doi.org/10.1371/journal.pone.0101229 (2014).
Endo, A. et al. Comparative genome analysis and identification of competitive and cooperative interactions in a polymicrobial disease. The ISME journal 9, 629–642, https://doi.org/10.1038/ismej.2014.155 (2015).
Ji, B. et al. Comparative genomic analysis provides insights into the evolution and niche adaptation of marine Magnetospira sp. QH-2 strain. Environmental microbiology 16, 525–544, https://doi.org/10.1111/1462-2920.12180 (2014).
Carlier, A. L. & Eberl, L. The eroded genome of a Psychotria leaf symbiont: hypotheses about lifestyle and interactions with its plant host. Environmental microbiology 14, 2757–2769, https://doi.org/10.1111/j.1462-2920.2012.02763.x (2012).
Saddler, G. S. & Bradbury, J. F. In Bergey’s Manual of Systematics of Archaea and Bacteria (2015).
Sayers, E. W. et al. Database resources of the National Center for Biotechnology Information. Nucleic acids research 37, D5–15, https://doi.org/10.1093/nar/gkn741 (2009).
Brunings, A. M. & Gabriel, D. W. Xanthomonas citri: breaking the surface. Molecular plant pathology 4, 141–157, https://doi.org/10.1046/j.1364-3703.2003.00163.x (2003).
Pieretti, I. et al. The complete genome sequence of Xanthomonas albilineans provides new insights into the reductive genome evolution of the xylem-limited Xanthomonadaceae. BMC Genomics 10, 616, https://doi.org/10.1186/1471-2164-10-616 (2009).
Ryan, R. P. et al. Pathogenomics of Xanthomonas: understanding bacterium-plant interactions. Nature reviews. Microbiology 9, 344–355, https://doi.org/10.1038/nrmicro2558 (2011).
Redak, R. A. et al. The biology of xylem fluid-feeding insect vectors of Xylella fastidiosa and their relation to disease epidemiology. Annual review of entomology 49, 243–270, https://doi.org/10.1146/annurev.ento.49.061802.123403 (2004).
Mansfield, J. et al. Top 10 plant pathogenic bacteria in molecular plant pathology. Molecular plant pathology 13, 614–629, https://doi.org/10.1111/j.1364-3703.2012.00804.x (2012).
Simpson, A. J. et al. The genome sequence of the plant pathogen Xylella fastidiosa. The Xylella fastidiosa Consortium of the Organization for Nucleotide Sequencing and Analysis. Nature 406, 151–159, https://doi.org/10.1038/35018003 (2000).
Delepelaire, P. T. Type I secretion in gram-negative bacteria. Biochimica et biophysica acta 1694, 149–161, https://doi.org/10.1016/j.bbamcr.2004.05.001 (2004).
Vinuesa, P. & Ochoa-Sanchez, L. E. Complete Genome Sequencing of Stenotrophomonas acidaminiphila ZAC14D2_NAIMI4_2, a Multidrug-Resistant Strain Isolated from Sediments of a Polluted River in Mexico, Uncovers New Antibiotic Resistance Genes and a Novel Class-II Lasso Peptide Biosynthesis Gene Cluster. Genome announcements 3, https://doi.org/10.1128/genomeA.01433-15 (2015).
Youenou, B. et al. Comparative Genomics of Environmental and Clinical Stenotrophomonas maltophilia Strains with Different Antibiotic Resistance Profiles. Genome biology and evolution 7, 2484–2505, https://doi.org/10.1093/gbe/evv161 (2015).
Pak, T. R. et al. Whole-genome sequencing identifies emergence of a quinolone resistance mutation in a case of Stenotrophomonas maltophilia bacteremia. Antimicrobial agents and chemotherapy 59, 7117–7120, https://doi.org/10.1128/AAC.01723-15 (2015).
Dean, B. J. Recent findings on the genetic toxicology of benzene, toluene, xylenes and phenols. Mutation research 154, 153–181 (1985).
Choi, E. J. et al. Comparative genomic analysis and benzene, toluene, ethylbenzene, and o-, m-, and p-xylene (BTEX) degradation pathways of Pseudoxanthomonas spadix BD-a59. Appl Environ Microbiol 79, 663–671, https://doi.org/10.1128/AEM.02809-12 (2013).
Hou, L., Jiang, J., Xu, Z., Zhou, Y. & Leung, F. C. Complete Genome Sequence of Pseudoxanthomonas suwonensis Strain J1, a Cellulose-Degrading Bacterium Isolated from Leaf- and Wood-Enriched Soil. Genome announcements 3, https://doi.org/10.1128/genomeA.00614-15 (2015).
Moreira, L. M. et al. Novel insights into the genomic basis of citrus canker based on the genome sequences of two strains of Xanthomonas fuscans subsp. aurantifolii. BMC Genomics 11, 238, https://doi.org/10.1186/1471-2164-11-238 (2010).
Moreira, L. M., De Souza, R. F., Digiampietri, L. A., Da Silva, A. C. & Setubal, J. C. Comparative analyses of Xanthomonas and Xylella complete genomes. Omics: a journal of integrative biology 9, 43–76, https://doi.org/10.1089/omi.2005.9.43 (2005).
Van Sluys, M. A. et al. Comparative genomic analysis of plant-associated bacteria. Annual review of phytopathology 40, 169–189, https://doi.org/10.1146/annurev.phyto.40.030402.090559 (2002).
Fang, Y. et al. Genome sequence of Xanthomonas sacchari R1, a biocontrol bacterium isolated from the rice seed. Journal of biotechnology 206, 77–78, https://doi.org/10.1016/j.jbiotec.2015.04.014 (2015).
Arpigny, J. L. & Jaeger, K. E. Bacterial lipolytic enzymes: classification and properties. The Biochemical journal 343(Pt 1), 177–183 (1999).
Rosenau, F. & Jaeger, K. Bacterial lipases from Pseudomonas: regulation of gene expression and mechanisms of secretion. Biochimie 82, 1023–1032 (2000).
Pleiss, J., Fischer, M., Peiker, M., Thiele, C. & Schmid, R. D. Lipase engineering database: Understanding and exploiting sequence–structure–function relationships. Journal of Molecular Catalysis B: Enzymatic 10, 491–508, https://doi.org/10.1016/S1381-1177(00)00092-8 (2000).
Nascimento, R. et al. TheType II Secreted Lipase/Esterase LesA is a Key Virulence Factor Required for Xylella fastidiosa Pathogenesis in Grapevines. Scientific reports 6, 18598, https://doi.org/10.1038/srep18598 (2016).
Aparna, G., Chatterjee, A., Sonti, R. V. & Sankaranarayanan, R. A cell wall-degrading esterase of Xanthomonas oryzae requires a unique substrate recognition module for pathogenesis on rice. The Plant cell 21, 1860–1873, https://doi.org/10.1105/tpc.109.066886 (2009).
Tamir-Ariel, D., Rosenberg, T., Navon, N. & Burdman, S. A secreted lipolytic enzyme from Xanthomonas campestris pv. vesicatoria is expressed in planta and contributes to its virulence. Molecular plant pathology 13, 556–567, https://doi.org/10.1111/j.1364-3703.2011.00771.x (2012).
Jaeger, K. E., Dijkstra, B. W. & Reetz, M. T. Bacterial biocatalysts: molecular biology, three-dimensional structures, and biotechnological applications of lipases. Annual review of microbiology 53, 315–351, https://doi.org/10.1146/annurev.micro.53.1.315 (1999).
Djamei, A. et al. Metabolic priming by a secreted fungal effector. Nature 478, 395–398, https://doi.org/10.1038/nature10454 (2011).
Boulanger, A. et al. Identification and regulation of the N-acetylglucosamine utilization pathway of the plant pathogenic bacterium Xanthomonas campestris pv. campestris. J Bacteriol 192, 1487–1497, https://doi.org/10.1128/JB.01418-09 (2010).
Dupoiron, S. et al. The N-Glycan cluster from Xanthomonas campestris pv. campestris: a toolbox for sequential plant N-glycan processing. J Biol Chem 290, 6022–6036, https://doi.org/10.1074/jbc.M114.624593 (2015).
Canonne, J. et al. The Xanthomonas type III effector XopD targets the Arabidopsis transcription factor MYB30 to suppress plant defense. The Plant cell 23, 3498–3511, https://doi.org/10.1105/tpc.111.088815 (2011).
Furutani, A. et al. Identification of novel type III secretion effectors in Xanthomonas oryzae pv. oryzae. Molecular plant-microbe interactions: MPMI 22, 96–106, https://doi.org/10.1094/MPMI-22-1-0096 (2009).
Jiang, W. et al. Identification of six type III effector genes with the PIP box in Xanthomonas campestris pv. campestris and five of them contribute individually to full pathogenicity. Molecular plant-microbe interactions: MPMI 22, 1401–1411, https://doi.org/10.1094/MPMI-22-11-1401 (2009).
Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M. & Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic acids research 42, D490–495, https://doi.org/10.1093/nar/gkt1178 (2014).
Deattie, G. A. In Plant-Associated Bacteria (ed S. S. Gnanamanickam) Ch. 1, 1–56 (Springer, 2007).
Buttner, D. Protein export according to schedule: architecture, assembly, and regulation of type III secretion systems from plant- and animal-pathogenic bacteria. Microbiol Mol Biol Rev 76, 262–310, https://doi.org/10.1128/MMBR.05017-11 (2012).
Del Giudice, M. G. et al. VirJ Is a Brucella Virulence Factor Involved in the Secretion of Type IV Secreted Substrates. The Journal of biological chemistry 291, 12383–12393, https://doi.org/10.1074/jbc.M116.730994 (2016).
Esquerré-Tugayé, M. T., Boudart, G. & Bernard Dumas, B. Cell wall degrading enzymes, inhibitory proteins, and oligosaccharides participate in the molecular dialogue between plants and pathogens. Plant Physiology and Biochemistry 38, 157–163, https://doi.org/10.1016/S0981-9428(00)00161-3 (2000).
Rajeshwari, R., Jha, G. & Sonti, R. V. Role of an in planta-expressed xylanase of Xanthomonas oryzae pv. oryzae in promoting virulence on rice. Molecular plant-microbe interactions: MPMI 18, 830–837, https://doi.org/10.1094/MPMI-18-0830 (2005).
Jha, G., Rajeshwari, R. & Sonti, R. V. Functional interplay between two Xanthomonas oryzae pv,. oryzae secretion systems in modulating virulence on rice. Molecular plant-microbe interactions: MPMI 20, 31–40, https://doi.org/10.1094/MPMI-20-0031 (2007).
Roper, M. C., Greve, L. C., Warren, J. G., Labavitch, J. M. & Kirkpatrick, B. C. Xylella fastidiosa requires polygalacturonase for colonization and pathogenicity in Vitis vinifera grapevines. Molecular plant-microbe interactions: MPMI 20, 411–419, https://doi.org/10.1094/MPMI-20-4-0411 (2007).
Laine, A. L. Role of coevolution in generating biological diversity: spatially divergent selection trajectories. Journal of experimental botany 60, 2957–2970, https://doi.org/10.1093/jxb/erp168 (2009).
Kast, P. et al. A strategically positioned cation is crucial for efficient catalysis by chorismate mutase. J Biol Chem 275, 36832–36838, https://doi.org/10.1074/jbc.M006351200 (2000).
Bekal, S., Niblack, T. L. & Lambert, K. N. A chorismate mutase from the soybean cyst nematode Heterodera glycines shows polymorphisms that correlate with virulence. Molecular plant-microbe interactions: MPMI 16, 439–446, https://doi.org/10.1094/MPMI.2003.16.5.439 (2003).
Vlot, A. C., Dempsey, D. A. & Klessig, D. F. Salicylic Acid, a multifaceted hormone to combat disease. Annual review of phytopathology 47, 177–206, https://doi.org/10.1146/annurev.phyto.050908.135202 (2009).
Maeda, H. & Dudareva, N. The shikimate pathway and aromatic amino Acid biosynthesis in plants. Annual review of plant biology 63, 73–105, https://doi.org/10.1146/annurev-arplant-042811-105439 (2012).
Schmit, J. C. & Zalkin, H. Chorismate mutase-prephenate dehydratase. Phenylalanine-induced dimerization and its relationship to feedback inhibition. J Biol Chem 246, 6002–6010 (1971).
Grant, G. A. The ACT domain: a small molecule binding domain and its role as a common regulatory element. J Biol Chem 281, 33825–33829, https://doi.org/10.1074/jbc.R600024200 (2006).
Zhang, S. et al. Chorismate mutase-prephenate dehydratase from Escherichia coli. Study of catalytic and regulatory domains using genetically engineered proteins. J Biol Chem 273, 6248–6253 (1998).
Degrassi, G., Devescovi, G., Bigirimana, J. & Venturi, V. Xanthomonas oryzae pv. oryzae XKK.12 contains an AroQgamma chorismate mutase that is involved in rice virulence. Phytopathology 100, 262–270, https://doi.org/10.1094/PHYTO-100-3-0262 (2010).
Ferreira, R. M. et al. Unravelling potential virulence factor candidates in Xanthomonas citri. subsp. citri by secretome analysis. PeerJ, https://doi.org/10.7717/peerj.1734 (2016).
Marino, K., Bones, J., Kattla, J. J. & Rudd, P. M. A systematic approach to protein glycosylation analysis: a path through the maze. Nature chemical biology 6, 713–723, https://doi.org/10.1038/nchembio.437 (2010).
Maeda, M. & Kimura, Y. Structural features of free N-glycans occurring in plants and functional features of de-N-glycosylation enzymes, ENGase, and PNGase: the presence of unusual plant complex type N-glycans. Frontiers in plant science 5, 429, https://doi.org/10.3389/fpls.2014.00429 (2014).
Priem, B., Gitti, R., Bush, C. A. & Gross, K. C. Structure of ten free N-glycans in ripening tomato fruit. Arabinose is a constituent of a plant N-glycan. Plant physiology 102, 445–458 (1993).
Suzuki, T. & Funakoshi, Y. Free N-linked oligosaccharide chains: formation and degradation. Glycoconjugate journal 23, 291–302, https://doi.org/10.1007/s10719-006-6975-x (2006).
Priem, B. et al. Isolation and characterization of free glycans of the oligomannoside type from the extracellular medium of a plant cell suspension. Glycoconjugate journal 7, 121–132, https://doi.org/10.1007/BF01050375 (1990).
Haweker, H. et al. Pattern recognition receptors require N-glycosylation to mediate plant immunity. J Biol Chem 285, 4629–4636, https://doi.org/10.1074/jbc.M109.063073 (2010).
Saijo, Y. ER quality control of immune receptors and regulators in plants. Cellular microbiology 12, 716–724, https://doi.org/10.1111/j.1462-5822.2010.01472.x (2010).
Kang, B. S. et al. N-Glycosylation process in both ER and Golgi plays pivotal role in plant immunity. Journal of Plant Biology 58, 374–382, https://doi.org/10.1007/s12374-015-0197-3 (2015).
Boulanger, A. et al. The plant pathogen Xanthomonas campestris pv. campestris exploits N-acetylglucosamine during infection. mBio 5, e01527–01514, https://doi.org/10.1128/mBio.01527-14 (2014).
Wang, Y. et al. Secretome analysis of the rice bacterium Xanthomonas oryzae (Xoo) using in vitro and in planta systems. Proteomics 13, 1901–1912, https://doi.org/10.1002/pmic.201200454 (2013).
Peabody, C. R. et al. Type II protein secretion and its relationship to bacterial type IV pili and archaeal flagella. Microbiology 149, 3051–3072, https://doi.org/10.1099/mic.0.26364-0 (2003).
Szczesny, R. et al. Functional characterization of the Xcs and Xps type II secretion systems from the plant pathogenic bacterium Xanthomonas campestris pv vesicatoria. The New phytologist 187, 983–1002, https://doi.org/10.1111/j.1469-8137.2010.03312.x (2010).
Astua-Monge, G. et al. Expression profiling of virulence and pathogenicity genes of Xanthomonas axonopodis pv. citri. J Bacteriol 187, 1201–1205, https://doi.org/10.1128/JB.187.3.1201-1205.2005 (2005).
Gonzalez, J. F. et al. A proteomic study of Xanthomonas oryzae pv. oryzae in rice xylem sap. Journal of proteomics 75, 5911–5919, https://doi.org/10.1016/j.jprot.2012.07.019 (2012).
Palmieri, A. C., do Amaral, A. M., Homem, R. A. & Machado, M. A. Differential expression of pathogenicity- and virulence-related genes of Xanthomonas axonopodis pv. citri under copper stress. Genetics and molecular biology 33, 348–353, https://doi.org/10.1590/S1415-47572010005000030 (2010).
Yamazaki, A., Hirata, H. & Tsuyumuv, S. HrpG regulates type II secretory proteins in Xanthomonas axonopodis pv. citri. Journal of General Plant Pathology 74, 138–150, https://doi.org/10.1007/s10327-008-0075-7 (2008).
Jalan, N. et al. Comparative genomic analysis of Xanthomonas axonopodis pv. citrumelo F1, which causes citrus bacterial spot disease, and related strains provides insights into virulence and host specificity. J Bacteriol 193, 6342–6357, https://doi.org/10.1128/JB.05777-11 (2011).
Guo, Y., Figueiredo, F., Jones, J. & Wang, N. HrpG and HrpX play global roles in coordinating different virulence traits of Xanthomonas axonopodis pv. citri. Molecular plant-microbe interactions: MPMI 24, 649–661, https://doi.org/10.1094/MPMI-09-10-0209 (2011).
Anderson, J. P. et al. Plants versus pathogens: an evolutionary arms race. Functional plant biology: FPB 37, 499–512, https://doi.org/10.1071/FP09304 (2010).
Farias, N. C. & Almeida, N. F. Orthologsorter: Inferring Genotyping and Functionality from Ortholog Protein Families Master thesis, Federal University of Mato Grosso do Sul, (2013).
Li, L., Stoeckert, C. J. Jr & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome research 13, 2178–2189, https://doi.org/10.1101/gr.1224503 (2003).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic acids research 32, 1792–1797, https://doi.org/10.1093/nar/gkh340 (2004).
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Molecular biology and evolution 17, 540–552 (2000).
Stamatakis, A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690, https://doi.org/10.1093/bioinformatics/btl446 (2006).
Boratyn, G. M. et al. BLAST: a more efficient report with usability improvements. Nucleic acids research 41, W29–33, https://doi.org/10.1093/nar/gkt282 (2013).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular biology and evolution 30, 772–780, https://doi.org/10.1093/molbev/mst010 (2013).
Capella-Gutierrez, S., Silla-Martinez, J. M. & Gabaldon, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973, https://doi.org/10.1093/bioinformatics/btp348 (2009).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Molecular biology and evolution 32, 268–274, https://doi.org/10.1093/molbev/msu300 (2015).
Vaneechoutte, M., Verschraegen, G., Claeys, G. & Flamen, P. Rapid identification of Branhamella catarrhalis with 4-methylumbelliferyl butyrate. Journal of clinical microbiology 26, 1227–1228 (1988).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic acids research 28, 27–30 (2000).
Mitchell, A. et al. The InterPro protein families database: the classification resource after 15 years. Nucleic acids research 43, D213–221, https://doi.org/10.1093/nar/gku1243 (2015).
Schultz, J., Milpetz, F., Bork, P. & Ponting, C. P. SMART, a simple modular architecture research tool: identification of signaling domains. Proceedings of the National Academy of Sciences of the United States of America 95, 5857–5864 (1998).
Sonnhammer, E. L., Eddy, S. R., Birney, E., Bateman, A. & Durbin, R. Pfam: multiple sequence alignments and HMM-profiles of protein domains. Nucleic acids research 26, 320–322 (1998).
Dhillon, B. K. et al. IslandViewer 3: more flexible, interactive genomic island discovery, visualization and analysis. Nucleic acids research 43, W104–108, https://doi.org/10.1093/nar/gkv401 (2015).
Bendtsen, J. D., Nielsen, H., Widdick, D., Palmer, T. & Brunak, S. Prediction of twin-arginine signal peptides. BMC bioinformatics 6, 167, https://doi.org/10.1186/1471-2105-6-167 (2005).
Kall, L., Krogh, A. & Sonnhammer, E. L. A combined transmembrane topology and signal peptide prediction method. Journal of molecular biology 338, 1027–1036, https://doi.org/10.1016/j.jmb.2004.03.016 (2004).
Nielsen, H., Engelbrecht, J., Brunak, S. & von Heijne, G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein engineering 10, 1–6 (1997).
Kuhn, M. et al. STITCH 4: integration of protein-chemical interactions with user data. Nucleic acids research 42, D401–407, https://doi.org/10.1093/nar/gkt1207 (2014).
Berman, H. M. et al. The Protein Data Bank. Nucleic acids research 28, 235–242 (2000).
Acknowledgements
Thanks to all members of the Guttman laboratory (University of Toronto) and members of the Laboratory of Biochemistry and Molecular Biology (LBBM, Federal University of Ouro Preto, UFOP) for their support and suggestions. We are also grateful to Sandeep Chakraborty for assistance in the protein structural interpretations. Funding for this work was provided by the following agencies: National Council of Technological and Scientific Development (CNPq), Fundect (TO007/2015), and Coordination for the Improvement of Higher Education Personnel (CAPES) (the BIGA Project, CFP 51/2013, process 3385/2013). We also thank Science without Borders for the fellowship to R.A.B.A.
Author information
Authors and Affiliations
Contributions
R.A.B.A. and L.M.M. designed the work and selected the genomes. N.F.A., S.T., J.D.C., and D.G. performed genome comparisons and identified the protein families. L.A.D. validated the proteins identified. J.S.L.P. and N.F.A. designed phylogenetic trees. L.C.P., R.N., P.A.Z., L.R.G., A.M.D. constructed de mutants and performed all experimental assays. R.A.B.A., E.B.F., J.C.S., L.M.M., and J.S.L.P. interpreted findings. D.G., L.C.P., S.C., R.N., P.A.Z., and A.M.D. contributed additional interpretations and general manuscript comments. R.A.B.A., L.M.M., J.S.L.P., P.A.Z., and J.C.S. wrote the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Assis, R., Polloni, L.C., Patané, J. et al. Identification and analysis of seven effector protein families with different adaptive and evolutionary histories in plant-associated members of the Xanthomonadaceae. Sci Rep 7, 16133 (2017). https://doi.org/10.1038/s41598-017-16325-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-16325-1
This article is cited by
-
Comparative transcriptomics reveals a highly polymorphic Xanthomonas HrpG virulence regulon
BMC Genomics (2024)
-
An overview of plant resistance to plant-pathogenic bacteria
Tropical Plant Pathology (2023)
-
Gut microbiota is involved in the antidepressant effects of adipose-derived mesenchymal stem cells in chronic social defeat stress mouse model
Psychopharmacology (2022)
-
Comparison and interpretation of characteristics of Rhizosphere microbiomes of three blueberry varieties
BMC Microbiology (2021)
-
Whole genome sequencing and identification of host-interactive genes in the rice endophytic Leifsonia sp. ku-ls
Functional & Integrative Genomics (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
