
Abstract
Long non-coding RNAs (lncRNAs) were shown to be the crucial regulators of the many diseases. In this study, the expressions of lncRNAs were investigated in resected 158 hepatocellular carcinomas (HCCs) to evaluate the effects of their expression levels on prognosis. The expression levels of HULC and MALAT1 were shown to be significantly higher in the normal background tissue of HCC than those in the normal liver tissue of metastatic liver tumor without hepatitis (HULC: fold change 14.9, P = 1.7e-06; MALAT1: fold change 17.5, P = 1.2e-06. The formation of capsule was shown to be correlated with the increased expression of HULC (P = 0.041), while the size of HCC under 2 cm was correlated with a decrease in MALAT1 expression (P = 0.019). The levels of serum alpha-fetoprotein above 20 ng/mL indicated a decreased expression of both HULC and MALAT1 (HULC: P = 0.017; MALAT1: P = 0.0036). The increase in the expression levels of MALAT1 in HCC tissues was significantly correlated with better overall survival (HULC: P = 0.099, MALAT1: P = 0.028). Thus, the expression of these lncRNAs in HCC potentially correlates with the HCC malignancy and they represent potential prognostic biomarkers of the resected HCC.
Similar content being viewed by others


Introduction
Hepatocellular carcinoma (HCC) represents the fifth most common malignancy and the third most common cause of cancer-related death worldwide1. Hepatic resection is one of the most effective treatments for non-metastatic HCC2,3,4. However, after the curative resection, ~80% of patients develop intrahepatic recurrence, and 50% die within 5 years5. Therefore, although the surgical resection of early HCC can be curative, the pronounced tendency toward recurrence represents a major concern6.
Intrahepatic HCC recurrence is categorized as intrahepatic metastasis (IM) or multicentric occurrence (MO). IM refers to the HCC foci developing from tumor cells that spread into the remnant liver via the portal vein before or during hepatic resection. MO refers to the development of postsurgical HCC foci due to chronic active hepatitis or cirrhosis, caused by viruses, alcohol, toxins, or other HCC-relevant risk factors7,8,9,10. The results of previous studies indicated that the clinical progression and outcome of IM and MO differ significantly11. Considering these specific characteristics of recurrence, focusing on the analysis of tumor tissue alone may be insufficient. In all HCC cases, the consideration of any correlations between the HCC tissue and the surrounding non-tumor tissue is important12. We demonstrated previously that the alterations in the non-malignant liver tissue gene profiles can be used for the prognostic purposes in HCC13.
Long non-coding RNAs (lncRNAs) were identified as the key players in tumorigenesis and tumor progression. Many reports showed that lncRNA dysregulation is linked to the development of many diseases, including cancer14,15,16. Therefore, we hypothesized that the expression of lncRNAs in the background liver tissue of HCC may be important for HCC prognosis as well. We attempted to identify novel lncRNAs related to HCC prognosis by using the microarray analysis of their expression in the background liver tissue of HCC patients and normal liver tissue without HCC and/or hepatitis. We evaluated the differences in lncRNA expression levels in HCC and corresponding non-tumor tissues, and identified the unique prognostic markers for the HCC.
Methods
Patients and Samples
For microarray analysis, the representative non-tumor liver tissue of a HCC patient (CN, for the corresponding normal) was obtained from a typical HCC patient, a 58-year-old man during hepatectomy. This HCC was caused by the chronic hepatitis C infection, and it recurred three years after the resection. Pathology confirmed the absence of cancerous regions from the CN sample. As controls, non-cancerous liver tissue without hepatitis (SN, for super normal) was obtained from 11 patients with liver metastases who underwent hepatectomy at Nagoya University Hospital, Japan. Their primary diseases were colorectal cancer (n = 5), gastrointestinal stromal tumor (n = 2), or gastric cancer, esophageal cancer, cervical cancer, or tongue cancer (n = 1, each).
For real-time quantitative reverse transcription PCR (RT-qPCR), HCC and CN tissue samples were collected from 158 consecutive patients who underwent curative resection at Nagoya University Hospital between January 1998 and December 2011. Resection was defined as curative when all gross tumors were removed completely; incidentally found small lesions suspected to be HCC that were treated intraoperatively by radiofrequency therapy or microwave coagulation therapy were also regarded as curative cases. Patient characteristics are summarized in Table 1. After surgery, all patients were monitored by performing blood examinations, ultrasonography, and computed tomography. In the cases of possible recurrence, angiography was performed in order to confirm it. The median follow-up duration of all cases was 48.5 months (range, 0.3 to 193.8 months). All tumor tissue samples were histologically confirmed as HCC samples by pathologists. All surgically obtained tissue samples were immediately frozen in the liquid nitrogen and stored at −80 °C until further analyses. This study and all procedures were approved by the Institutional Review Board at Nagoya University and all patients provided written informed consent. All clinical investigations were conducted in accordance with the principles of the Declaration of Helsinki.
Gene Expression Analysis
Microarray
Total RNA was extracted from the fresh-frozen CN and SN tissue samples using a Qiagen miRNeasy mini-kit (Qiagen, Hilden, Germany). Eleven SN samples were pooled so that the individual differences are eliminated. RNA quality was confirmed based on the RNA integrity number ≥8 as measured using an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA). RNA was labeled with cyanine-3 dye using a Quick Amp labeling kit (Agilent, Santa Clara, CA, USA) and hybridized to Agilent whole human genome (4 × 44 K) microarrays for 17 h in a rotating SciGene model 700 oven (SciGene, Sunnyvale, CA, USA). The arrays were scanned (Agilent DNA microarray scanner), and the data were feature-extracted using Feature Extraction software 10.5.1.1 and statistically analyzed using the default settings for GeneSpring GX 11.0.1 software (Agilent, Santa Clara, CA, USA)17.
RT-qPCR
Total cDNA was developed from the RNA extracted from each fresh-frozen tissue with M-MLV Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA). This total cDNA was used as a template for the next step of quantitative PCR. PCR was performed using SYBR Premix Ex Taq II (Takara Clontech, Kyoto, Japan) under the following conditions: denaturing at 95 °C for 10 s, 40 cycles of denaturing at 95 °C for 5 s, and annealing/extension at 60 °C for 30 s. The SYBR Green signal was detected in real-time using StepOne Plus Real-Time PCR System (Life Technologies, Carlsbad, CA, USA). The relative quantification method was used and each gene expression level was determined to be the expression level of the target gene, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), for each sample.
The PCR primers used in current study were for the 84 base pairs fragment of Hepatocellular Carcinoma Up-Regulated Long Non-Coding RNA (HULC) (sense, 5′-ACTCTGAAGTAAAGGCCGGAA-3′; antisense 5′-GCCAGGAAACTTCTTGCTTGA-3′) and for the 14 base pairs fragment of Metastasis Associated Lung Adenocarcinoma Transcript 1 (MALAT1) (sense, 5′-CCAGTTGAATTCACCAGTGGAC-3′; antisense 5′ - AGTTTGCTCACATGCCAGTTAC-3′). GAPDH (sense, 5′-GAAGGTGAAGGTCGGAGTC-3′; antisense 5′-GAAGATGGTGATGGGATTTC-3′; probe 5′-[Fl] CAAGCTTCCCGTTCTCAGCC [Fl-Q]-3′) expression was quantified in each sample in order to perform the normalization. All RT-qPCR experiments were performed in triplicate, including the template-omitted negative controls.
Public Available Dataset
Normalized TCGA RNA-sequencing data for hepatocellular carcinoma was downloaded from the Broad GDAC Firehose (http://gdac.broadinstitute.org/, accessed on November 1st, 2017). This dataset includes 360 HCC cases including seven HCCs mixed with hepatocholangiocarcinoma and two cases with fibrolamellar carcinoma. There are 266 cases with information of recurrence-free survival (RFS) and 336 cases with information of overall survival (OS) out of the 360 cases.
Statistical Analyses
Continuous variables were expressed as median (range) and expression of each target gene was compared by a Wilcoxon signed-rank test. Categorical variables were compared using the χ2 or Fisher’s exact tests, as appropriate. Correlation between each gene expression was analyzed with Spearman’s rank correlation coefficient. OS and RFS rate at each point of the follow-up time were estimated using the Kaplan-Meier method and compared using a log-rank test. The Cox proportional hazard regression model was used to perform univariate analysis for OS. All statistical analyses were performed using R (version 3.4.1; http//www.r-project.org/) and statistical significance was set at P < 0.05, which was obtained using two-tailed tests.
Results
Expression Profiling
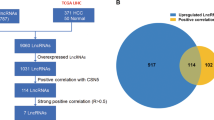
To identify novel tumor-related lncRNA in HCC, we compared the non-coding RNA expression profiles of a CN sample and the pooled SN samples. Microarray analysis revealed that lncRNAs upregulated in the CN sample (fold change higher than 1.2) were Urothelial Cancer Associated 1 (UCA1: fold change 7.98), HULC (fold change 3.49), MALAT1 (fold change 1.85), Growth Arrest Specific 5 (GAS5: fold change 1.28), and Taurine Up-Regulated 1 (TUG1: fold change 1.27) (Supplementary Table S1).
SN, CN, and HCC Tissue Sample Analysis
As determined by analyzing 11 SN and 48 paired CN and HCC, HULC and MALAT1 expression levels were significantly higher in the CN tissue samples (HULC: median, 0.019 [range, 0.0008 to 0.068]; MALAT1: 0.71 [0.12 to 6.8]; n = 48) than in the SN tissue samples (HULC: 0.0013 [0.0002 to 0.0076], P = 1.7e-06; MALAT1: 0.041 [0.019 to 0.45], P = 1.2e-06; n = 11) (Fig. 1a and b). However, no significant difference in HULC expression between CN and HCC was observed (0.012 [range, 0.0001 to 0.29], P = 0.12, n = 158) (Fig. 1a). MALAT1 expression levels in the HCC samples (0.19 [range, 0.011 to 3.4], n = 158) were shown to be significantly lower than those in the CN samples (P = 1.7e-10) (Fig. 1b). There was a weak positive correlation between HULC and MALAT1 expression in HCC tissue (correlation coefficient r = 0.46, P = 1.1e-06) (Fig. 1c).

HULC and MALAT1 expression levels in SN, CN, and HCC samples. (a) HULC expression levels in SN, CN, and HCC samples. (b) MALAT1 expression in SN, CN, and HCC samples. (c) Correlation between HULC expression and MALAT1 expression in HCC. CN, corresponding normal; HCC, hepatocellular carcinoma; HULC, Hepatocellular Carcinoma Up-Regulated Long Non-Coding RNA; MALAT1, Metastasis Associated Lung Adenocarcinoma Transcript 1.
When 158 curably resected HCC tissue samples were analyzed, HULC expression levels were shown to be higher in the HCC samples (0.047 [range, 0.00007 to 4.1], n = 158) in comparison with those in the CN tissues (0.036 [0.0005 to 1.9], P = 0.063; n = 58) (Fig. 2a). However, the expression of MALAT1 did not differ between CN (0.69 [0.026 to 19.1], n = 158) and HCC tissues (0.57 [0.032 to 23.1], P = 0.11; n = 158) (Fig. 2b). In the CN samples, no significant difference was found between other clinicopathological factors and HULC or MALAT1 expression. However, HULC expression levels in HCC cases where the capsular formation can be observed (0.059 [0.0003 to 4.0], n = 101) was significantly higher than in the HCC samples without capsular formation (0.039 [0.00007 to 4.1], P = 0.041, n = 45) (Fig. 2c). MALAT1 expression levels in HCC samples larger than 2 cm (0.51 [0.031 to 21], n = 116) were significantly lower than those in the HCC under 2 cm (0.94 [0.23 to 2.9], P = 0.019, n = 23) (Fig. 2d). The expression of HULC and MALAT1 in HCC cases with higher serum alpha-fetoprotein (AFP) levels (>20 ng/mL) (HULC: 0.030 [0.00007 to 1.7], n = 64; MALAT1: 0.36 [0.031 to 11.1, n = 64) were significantly lower than those in the HCC samples with lower serum AFP values (<20 ng/mL) (HULC: 0.061 [0.0007 to 4.1], P = 0.019, n = 80; MALAT1: 0.88 [0.058 to 23.1], P = 0.0036, n = 81) (Fig. 2e and f).

HULC and MALAT1 expression levels in 158 HCC samples. (a) HULC expression in CN and HCC samples. (b) MALAT1 expression levels in CN and HCC tissue samples. (c) HULC expression in the HCC cases accompanied by the formation of capsule (Fc) or without Fc. (d) MALAT1 expression in HCC samples larger or smaller than 2 cm. (e) HULC expression level in the HCC cases with higher serum AFP levels. (f) MALAT1 expression level in the HCC cases with higher serum AFP levels. CN, corresponding normal; HCC, hepatocellular carcinoma; Fc, formation of capsule; AFP, alpha-fetoprotein; HULC, Hepatocellular Carcinoma Up-Regulated Long Non-Coding RNA; MALAT1, Metastasis Associated Lung Adenocarcinoma Transcript 1.
HULC and MALAT1 Correlation with HCC Prognosis
Based on the results obtained by the real time RT-qPCR, 158 HCC cases were divided into two groups, according to the HULC and MALAT1 expression levels in HCC tissues (HULC: < 0.01 and ≥ 0.01; MALAT1: < median and ≥ median) and the effects of the expression levels on RFS and OS were evaluated. The distribution of each clinicopathological features stratified by HULC and MALAT1 expression levels in HCC is shown in Supplementary Table S2 and S3 (online). OS and RFS analysis stratified by HULC expression in HCC revealed high expression of HULC tended to have better RFS and OS but there was no significance between cases with high HULC and low HULC (RFS: P = 0.077; OS: P = 0.099) (Fig. 3a and b). In HCC tissue samples, no significant difference in RFS linked to the MALAT1 expression level was observed (P = 0.8) (Fig. 3c). However, the increased MALAT1 expression level were shown to be associated with the better OS (P = 0.028) (Fig. 3d), in comparison with that when low expression of these lncRNAs was detected. Furthermore, by univariate analysis, we detected significant correlations between OS and MALAT1 expression in HCC ≥ median (hazard ratio [HR], 0.58; 95% confidence interval (CI), 0.36 to 0.95; P = 0.03) as well as prothrombin time < 70% (HR, 2.03; 95% CI, 1.11 to 3.72; P = 0.02), B or C liver damage (HR, 2.09; 95% CI, 1.21 to 3.62; P = 0.01), multiple tumor (HR, 1.68; 95% CI, 1.00 to 2.81; P = 0.049), AFP ≥ 20 ng/mL (HR, 2.15; 95% CI, 1.35 to 3.45; P = 0.0 01) poorly differentiated HCC (HR, 2.29; 95% CI, 1.14 to 4.62; P = 0.02), serosal invasion (HR, 2.43; 95% CI, 1.44 to 4.11; P = 0.001), vascular invasion (HR, 2.39; 95% CI, 1.47 to 3.87; P = 0.0004), and positive surgical margin (HR, 1.94; 95% CI, 1.09 to 3.45; P = 0.02) (Table 2).

Survival analysis stratified by HULC and MALAT1 expression levels in HCC tissues. (a) RFS analysis stratified by HULC expression. (b) OS analysis stratified by HULC expression. (c) RFS analysis stratified by MALAT1 expression. (d) OS analysis stratified by MALAT1 expression. HULC, Hepatocellular Carcinoma Up-Regulated Long Non-Coding RNA; MALAT1, Metastasis Associated Lung Adenocarcinoma Transcript 1; OS, overall survival; RFS, recurrence-free survival.
The cohort used in this study is composed of cases with HBV 38 (24%), HCV 89 (56%), HBV + HCV 3 (2%), and None-virus 28 (18%). This proportion is similar to those in the EU, US, and Japan, as reported previously18. Then, using only HCV cases (81 informative cases with RFS and 82 informative cases with OS), a prognostic analysis was performed. Interestingly, the result revealed that the RFS and OS value, as stratified by the expression level of HULC was significantly different (RFS: HR, 0.30; 95% CI, 0.17 to 0.55; P < 0.0001; OS: HR, 0.32; 95% CI, 0.17 to 0.63; P = 0.0005, Fig. 4a and b). However the RFS and OS value, as stratified by the expression level of MALAT1 had the same tendency as seen in entire cohort without any significant difference (RFS: HR, 0.78; 95% CI, 0.48 to 1.3; P = 0.33, OS: HR, 0.62; 95% CI, 0.34 to 1.1; P = 0.12, Fig. 4c and d).

Survival analysis stratified by HULC and MALAT1 expression levels in HCC tissues from HCV patients. (a) RFS analysis stratified by HULC expression. (b) OS analysis stratified by HULC expression. (c) RFS analysis stratified by MALAT1 expression. (d) OS analysis stratified by MALAT1 expression. HULC, Hepatocellular Carcinoma Up-Regulated Long Non-Coding RNA; MALAT1, Metastasis Associated Lung Adenocarcinoma Transcript 1; OS, overall survival; RFS, recurrence-free survival.
HULC and MALAT1 Correlation with HCC Prognosis in a Public Available Dataset
Finally, we confirmed the impact of HULC and MALAT1 expression on HCC prognosis using TCGA dataset. Based on the normalized RNA-sequencing data, HCC cases were divided into two groups, according to the HULC and MALAT1 expression levels in HCC tissues (HULC: < 0.45 and ≥ 0.45; MALAT1: < 9.35 and ≥ 9.35). As a result, the prognosis of HCC cases from TCGA stratified by HULC and MALAT1 expression have the same tendency for HCC prognosis as seen in our collected 158 cases (Supplementary Figure S1).
Discussion
With the development of whole genome and transcriptome sequencing, many lncRNAs were shown to be highly expressed in tumor tissue19. Gene expression profile of the surrounding normal liver tissue is important for HCC prognosis and the potential for recurrence12. We attempted to identify lncRNAs that may be used as prognostic biomarkers for HCC, by comparing their expression levels in the non-tumorous background liver tissue of HCC patients and non-tumorous and non-hepatitis liver tissue from the metastatic hepatic cancer patient12. In this way, we found five lncRNAs, shown to be highly expressed in CN, and the expression profiles obtained using a small-sized cohort confirmed that HULC and MALAT1 show higher expression in the CN than SN samples. According to the cancer genome atlas (TCGA) dataset, HULC and MALAT1 are highly expressed in the HCC tissues (HULC: 371 tumor vs. 50 normal, fold change 2.04; MALAT1: 373 tumor vs. 50 normal, fold change 1.72)20.
HULC was first described as a lncRNA with induced expression in HCC compared with the non-neoplastic tissue21. In osteosarcoma and pancreatic cancer, an increased expression of HULC in tumor tissue was shown to indicate poor prognosis22,23. MALAT1 is highly conserved and it was originally reported as a biomarker of metastasis in the early lung adenocarcinoma24. Functionally, HULC has not been completely elucidated, and a previous report showed that HULC can act as an oncogene in HCC through the deregulation of lipid metabolism through a signaling pathway involving miR-9, PPARA, and ACSL125. MALAT1 acts as a proto-oncogene through the Wnt pathway activation and induction of the oncogenic splicing factor SRSF126.
In this study, we showed that the expression of HULC was significantly higher in HCC samples, but the expression of MALAT1 was not showed to be altered between CN and HCC. This may be caused by the presence of hepatitis in these cases. The underlying cause of hepatitis differed between the samples and in each study, the distribution of causes for hepatitis may vary. Furthermore, in breast cancer, the expression pattern of MALAT1 differs between transcript variants23, indicating that the transcript variant should be considered when determining the expression level of these lncRNAs in HCC27,28.
Unexpectedly, we did not observe the difference in lncRNA expression profiles between the background liver tissue and HCC tissue, and no correlation between HULC and MALAT1 levels in CN and other background factors was determined. However, we demonstrated a significant difference between the expression of these two lncRNAs in HCC and other clinicopathological tumor-related factors, indicating that the decreased aggressiveness of HCC may be associated with the expression level of both HULC and MALAT1. Additionally, we demonstrated that the induced HULC and MALAT1 expression in HCC correlates with a better prognosis. Few reports demonstrated that these two genes contribute to tumor development and recurrence26,29 and they are tumor indicators30. The preliminary HULC analysis with Gene Expression Omnibus (GEO) and in silico analysis showed that the higher expression of HULC is correlated with the better prognosis in HCC31. To the best of our knowledge, the relationship between these two lncRNAs and HCC prognosis, using clinical samples, has not been established previously. Our results indicate that these two lncRNAs may act as oncogenes, but they do not accelerate the progression of HCC.
This study is limited due to the use of a single-center cohort, without the possibility of the analysis of an independent cohort. In order to confirm these results further, they should be validated using an independent HCC cohort.
In conclusion, our findings suggest that the increased expression of HULC and MALAT1 in HCC tissue may represent a good prognostic biomarker for curatively resected HCC. Therefore, in combination with other tumor prognostic factors, the determination of the expression levels of these lncRNAs may lead to a more accurate prediction of HCC prognosis.
References
Parkin, D. M., Bray, F., Ferlay, J. & Pisani, P. Global cancer statistics, 2002. CA: a cancer journal for clinicians 55, 74–108 (2005).
Rahbari, N. N. et al. Hepatocellular carcinoma: current management and perspectives for the future. Annals of surgery 253, 453–469, https://doi.org/10.1097/SLA.0b013e31820d944f (2011).
Kobayashi, A. et al. Results of 404 hepatic resections including 80 repeat hepatectomies for hepatocellular carcinoma. Hepato-gastroenterology 53, 736–741 (2006).
Chen, M. S. et al. A prospective randomized trial comparing percutaneous local ablative therapy and partial hepatectomy for small hepatocellular carcinoma. Annals of surgery 243, 321–328, https://doi.org/10.1097/01.sla.0000201480.65519.b8 (2006).
Taura, K. et al. Implication of frequent local ablation therapy for intrahepatic recurrence in prolonged survival of patients with hepatocellular carcinoma undergoing hepatic resection: an analysis of 610 patients over 16 years old. Annals of surgery 244, 265–273, https://doi.org/10.1097/01.sla.0000217921.28563.55 (2006).
Morimoto, O. et al. Diagnosis of intrahepatic metastasis and multicentric carcinogenesis by microsatellite loss of heterozygosity in patients with multiple and recurrent hepatocellular carcinomas. Journal of hepatology 39, 215–221 (2003).
Chen, P. J. et al. Clonal origin of recurrent hepatocellular carcinomas. Gastroenterology 96, 527–529 (1989).
Imamura, H. et al. Risk factors contributing to early and late phase intrahepatic recurrence of hepatocellular carcinoma after hepatectomy. Journal of hepatology 38, 200–207 (2003).
Portolani, N. et al. Early and late recurrence after liver resection for hepatocellular carcinoma: prognostic and therapeutic implications. Annals of surgery 243, 229–235, https://doi.org/10.1097/01.sla.0000197706.21803.a1 (2006).
Cucchetti, A. et al. Comparison of recurrence of hepatocellular carcinoma after resection in patients with cirrhosis to its occurrence in a surveilled cirrhotic population. Annals of surgical oncology 16, 413–422, https://doi.org/10.1245/s10434-008-0232-4 (2009).
Matsuda, M., Fujii, H., Kono, H. & Matsumoto, Y. Surgical treatment of recurrent hepatocellular carcinoma based on the mode of recurrence: repeat hepatic resection or ablation are good choices for patients with recurrent multicentric cancer. Journal of hepato-biliary-pancreatic surgery 8, 353–359, https://doi.org/10.1007/s0053410080353 (2001).
Hoshida, Y. et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. The New England journal of medicine 359, 1995–2004, https://doi.org/10.1056/NEJMoa0804525 (2008).
Nomoto, S. et al. Expression analysis of THOP1 in background liver, a prognostic predictive factor in hepatocellular carcinoma, extracted by multiarray analysis. Annals of surgical oncology 21(Suppl 3), S443–450, https://doi.org/10.1245/s10434-014-3581-1 (2014).
Shi, X., Sun, M., Liu, H., Yao, Y. & Song, Y. Long non-coding RNAs: a new frontier in the study of human diseases. Cancer letters 339, 159–166, https://doi.org/10.1016/j.canlet.2013.06.013 (2013).
Gupta, R. A. et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071–U1148, https://doi.org/10.1038/nature08975 (2010).
Taft, R. J., Pang, K. C., Mercer, T. R., Dinger, M. & Mattick, J. S. Non-coding RNAs: regulators of disease. J. Pathol. 220, 126–139, https://doi.org/10.1002/path.2638 (2010).
Stangegaard, M. Gene expression analysis using agilent DNA microarrays. Methods in molecular biology (Clifton, N.J.) 529, 133–145, https://doi.org/10.1007/978-1-59745-538-1_9 (2009).
Llovet, J. M., Burroughs, A. & Bruix, J. Hepatocellular carcinoma. Lancet. 362(9399), 1907–17 (2003).
White, N. M. et al. Transcriptome sequencing reveals altered long intergenic non-coding RNAs in lung cancer. Genome Biol. 15, 16, https://doi.org/10.1186/s13059-014-0429-8 (2014).
FIREBROWSE(Version: 1. 1.35) http://firebrowse.org/ (2017)
Panzitt, K. et al. Characterization of HULC, a novel gene with striking up-regulation in hepatocellular carcinoma, as noncoding RNA. Gastroenterology 132, 330–342, https://doi.org/10.1053/j.gastro.2006.08.026 (2007).
Sun, X. H., Yang, L. B., Geng, X. L., Wang, R. & Zhang, Z. C. Increased expression of lncRNA HULC indicates a poor prognosis and promotes cell metastasis in osteosarcoma. International journal of clinical and experimental pathology 8, 2994–3000 (2015).
Peng, W., Gao, W. & Feng, J. Long noncoding RNA HULC is a novel biomarker of poor prognosis in patients with pancreatic cancer. Medical oncology (Northwood, London, England) 31, 346, https://doi.org/10.1007/s12032-014-0346-4 (2014).
Ji, P. et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 22, 8031–8041, https://doi.org/10.1038/sj.onc.1206928 (2003).
Cui, M. et al. Long noncoding RNA HULC modulates abnormal lipid metabolism in hepatoma cells through an miR-9-mediated RXRA signaling pathway. Cancer research 75, 846–857, https://doi.org/10.1158/0008-5472.can-14-1192 (2015).
Malakar, P. et al. Long noncoding RNA MALAT1 promotes hepatocellular carcinoma development by SRSF1 up-regulation and mTOR activation. Cancer research, https://doi.org/10.1158/0008-5472.can-16-1508 (2016).
Meseure, D. et al. Prognostic value of a newly identified MALAT1 alternatively spliced transcript in breast cancer. British journal of cancer 114, 1395–1404, https://doi.org/10.1038/bjc.2016.123 (2016).
Liu, Y. et al. A genetic variant in long non-coding RNA HULC contributes to risk of HBV-related hepatocellular carcinoma in a Chinese population. PloS one 7, e35145, https://doi.org/10.1371/journal.pone.0035145 (2012).
Lai, M. C. et al. Long non-coding RNA MALAT-1 overexpression predicts tumor recurrence of hepatocellular carcinoma after liver transplantation. Medical oncology (Northwood, London, England) 29, 1810–1816, https://doi.org/10.1007/s12032-011-0004-z (2012).
Konishi, H. et al. Plasma level of metastasis-associated lung adenocarcinoma transcript 1 is associated with liver damage and predicts development of hepatocellular carcinoma. Cancer science 107, 149–154, https://doi.org/10.1111/cas.12854 (2016).
Yang, Z. et al. HULC and H19 Played Different Roles in Overall and Disease-Free Survival from Hepatocellular Carcinoma after Curative Hepatectomy: A Preliminary Analysis from Gene Expression Omnibus. Disease markers 2015, 191029, https://doi.org/10.1155/2015/191029 (2015).
Acknowledgements
This study was partly supported by Japan Society for the Promotion of Science (JSPS) KAKENHI Grant-in-Aid for Scientific Research (C) (15K19848 to Fuminori Sonohara). We thank Yoko Nishikawa for collecting clinical samples and for helping to perform the experiments. We thank Dr. Daisuke Izumi (Kumamoto University, Japan) for discussing the analyses of this study. We thank Lauren J Patterson for providing English editing.
Author information
Authors and Affiliations
Contributions
Conception and design: F.S., S.N.; Financial support: Y.K., S.N.; Administrative support: Y.K., S.N.; Provision of study materials and patients: F.S., Y.I., M.H., S.Y., H.S., T.F.; Collection and assembly of data: F.S., Y.I., M.H., S.Y., H.S., T.F.; qPCR data analysis and interpretation: F.S., Y.I., S.N.; Manuscript writing: F.S., S.N.; Final approval of manuscript: all authors.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sonohara, F., Inokawa, Y., Hayashi, M. et al. Prognostic Value of Long Non-Coding RNA HULC and MALAT1 Following the Curative Resection of Hepatocellular Carcinoma. Sci Rep 7, 16142 (2017). https://doi.org/10.1038/s41598-017-16260-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-16260-1
This article is cited by
-
Serum long non-coding RNA SCARNA10 serves as a potential diagnostic biomarker for hepatocellular carcinoma
BMC Cancer (2022)
-
Lantern: an integrative repository of functional annotations for lncRNAs in the human genome
BMC Bioinformatics (2021)
-
Panel of potential lncRNA biomarkers can distinguish various types of liver malignant and benign tumors
Journal of Cancer Research and Clinical Oncology (2021)
-
Clinicopathological and prognostic significance of long noncoding RNA MALAT1 in human cancers: a review and meta-analysis
Cancer Cell International (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
