
Abstract
Metallic glasses are promising structural materials due to their unique properties. For structural applications and processing the coefficient of thermal expansion is an important design parameter. Here we demonstrate that predictions of the coefficient of thermal expansion for metallic glasses by density functional theory based ab initio calculations are efficient both with respect to time and resources. The coefficient of thermal expansion is predicted by an ab initio based method utilising the Debye-Grüneisen model for a Pd-based metallic glass, which exhibits a pronounced medium range order. The predictions are critically appraised by in situ synchrotron X-ray diffraction and excellent agreement is observed. Through this combined theoretical and experimental research strategy, we show the feasibility to predict the coefficient of thermal expansion from the ground state structure of a metallic glass until the onset of structural changes. Thereby, we provide a method to efficiently probe a potentially vast number of metallic glass alloying combinations regarding thermal expansion.
Similar content being viewed by others

Introduction
Metallic glasses are promising materials for micromechanical structural applications due to the combination of high strength and elasticity, hardness and toughness1,2,3, leading to an outstanding property combination compared to other material classes4. In addition to these property combinations, the absence of microstructural features2 makes metallic glasses ideal for micromechanical applications. The low viscosity in the supercooled liquid region allows the near net-shape production of structural components by thermoplastic forming, while volume shrinkage during casting is one magnitude lower compared to crystalline materials due to the lack of a first order phase transition during solidification5. For processing by near net shape production methods and application in micromechanical devices the coefficient of thermal expansion (CTE) is of critical importance in terms of dimensional accuracy and thermal stresses1,2,3,5.
Pd-based metallic glasses exhibit promising property combinations such as high strength and toughness, leading to damage tolerant materials6. Schnabel et al. reported a metalloid-free Pd57.0Al23.9Y7.7Cu11.4 metallic glass with extensive plastic deformation before fracture and a high damage tolerance4. Following the electronic structure based design proposal of Schnabel et al. that a low fraction of hybridised bonds is a fingerprint of damage-tolerance4, Pd62.1Al27.3Y4.2Ni6.4 is predicted to exhibit damage tolerance. In addition to the damage tolerance, the knowledge of the CTE is crucial for potential micromechanical applications.
While knowledge based ab initio models to predict structure and mechanical, electrical and magnetic properties of metallic glasses are state of the art7,8,9,10, there are no reports on Debye-Grüneisen model to predict the CTEs of metallic glasses in literature. The CTEs of cubic11,12,13,14,15,16,17, tetragonal16,17, hexagonal and trigonal materials17 have been calculated by the Debye-Grüneisen model. To our knowledge, the Debye-Grüneisen model has not been applied to amorphous materials yet. Here it is demonstrated that the ab initio based prediction of CTEs is efficient with respect to time and resources. As a model system we have evaluated a Pd54.7Al23.5Y7.8Ni11.3 thin film metallic glass by the ab initio based Debye-Grüneisen model and validated the theoretical predictions by high energy X-ray diffraction experiments.
Methods
In this work, ab initio molecular dynamic simulations were carried out using the density functional theory18 based openMX code19,20. Electronic potentials with the generalised gradient approximation were employed21. Basis functions were linear combinations of localised pseudoatomic orbitals22. The following basis functions were applied: Pd5.0-s2p1d1, Al6.0s1p2, Y6.5-s3p2d1 and Ni6.0-s2p2d2f1. The first symbol designates the chemical element followed by the cutoff radius. The last set of symbols defines the primitive orbitals. An N-point grid of 85 × 85 × 85 and a cutoff energy of 150 Ry has been used. For volume relaxation at 0 K the Vienna Ab-initio Simulation Package was utilised23,24. Thereby, the ultrasoft pseudopotentials were employed and the Brillouin zone was integrated on a 3 × 3 × 3 Monkhorst-Pack k-point grid25.
To model both short- and medium-ranged order, the structural model introduced by Hostert et al.8 was modified in terms of the size of the supercell and of relaxation time. Here, a larger supercell containing 389 atoms and 43 vacancies was employed. In order to obtain an amorphous structure, the supercell was heated to 4000 K for 4000 fs and quenched to 0 K with infinite cooling rate until the volume change between two subsequent cycles was <2%. As described previously by Hostert et al.8, the application of an infinite cooling rate is appropriate compared to cooling rates accessible in thin film synthesis. The bulk modulus B was obtained from the ground state by fitting the volume-energy data with the Birch-Munarghan equation of state26. Shear and Young’s modulus were calculated by means of volume conservative distortions27. In order to allow a medium range order to develop, relaxation of the atomic positions was allowed at 300, 400, 700 and 800 K for 4400 fs. Taking the scattering powers of the constituents into account, the pair distribution function (PDF) g(r) was calculated from the atomic positions8.
To estimate the CTE, a Debye-Grüneisen theory based method described by Söderlind et al.11 was used. Thereby the Helmholtz free energy (eq. (1)) was calculated for different temperatures.
Ee(r) is the temperature independent electron energy, N the number of atoms in the supercell, kB Boltzmann’s constant and T the temperature. From the mechanical properties obtained in the ab initio calculations, namely bulk modulus B and Poisson’s ration ν, the Debye temperature Θ was calculated by eq. (2)
h is the Planck constant and M the atomic mass. The dependence of the free energy on the Wigner-Seitz radius r was introduced via the change of Debye Θ temperature with r (eq. (3)), whereby γ is the Grüneisen parameter and r0 the equilibrium radius.
From the free energy curve calculated for several temperatures the equilibrium radii at each temperature were calculated. From the change of the equilibrium radius r0 with temperature T, the linear and volumetric thermal expansion coefficients αlin and αvol, respectively, were obtained by eq. (4)
To measure the thermal expansion by synchrotron X-ray diffraction Pd62.1Al27.3Y4.2Ni6.4 thin films were deposited on rotating NaCl substrates by magnetron sputtering of elemental targets in DC mode in an ultra-high vacuum chamber (base pressure <51 mPa). Floating bias was utilised, the power densities on the Pd, Al, Y and Ni targets were 4.2, 4.7, 1.7 and 1.3 W/cm2, respectively. Ar was employed as working gas with a pressure of 0.4 Pa during sputtering. For the synchrotron measurements, the NaCl substrate was dissolved in water to obtain thin film particles, which were subsequently cleaned in isopropanol and acetone.
High-energy X-ray diffraction was conducted at beamline P02.1 of the PETRA III electron storage ring at DESY, Hamburg. X-rays with a wavelength of 0.20701 Å were used and data collected with a Perkin Elmer XRD1621 fast detector. The sample was positioned in a Linkam THMS 600 heating stage and heated up with a heating rate of 10 K/min up to 863 K, hold at this temperature for 300 s and cooled down to room temperature with 10 K/min. Diffraction patterns were taken continuously with a measurement and detector read-out time of 12 s. The diffraction patterns were integrated over this time interval, which leads to a temperature resolution of 2 K. We used the FIT2D software28,29,30 to process the diffraction patterns.
The thermal expansion of an amorphous substance is connected to the shift of the principal peak position Q in reciprocal space as described in eq. (5)31, where Q is the principal peak position, V the atomic volume and αvol the volumetric CTE. T0 indicates the reference temperature, in this study room temperature.
The principal peak was fitted by a Pseudo-Voigt function in order to obtain the quantitative peak positions. Throughout this work, all CTEs are given as volumetric CTEs.
To study the structural changes during heating, the structure factor S(Q) and the reduced PDF G(r) were obtained using the PDFgetX3 software32. From the structure factor S(Q) the reduced PDF G(r) was calculated by a Fourier transformation. g(r) and G(r) are connected by
Results and Discussion
In this part, first the CTE of Pd54.7Al23.5Y7.8Ni11.3 metallic glass is predicted by the Debye-Grüneisen model and critically appraised by in situ X-ray diffraction. Thereafter, the calculated and experimentally observed structures are compared to validate the employed ab initio model. In the third part, a refined structural analysis is conducted to investigate the changes in the atomic structure observed in the experiments.
Thermal expansion
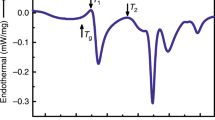
We evaluate the thermal expansion of Pd62.1Al27.3Y4.2Ni6.4 thin film metallic glasses by in situ X-ray diffraction and by ab initio methods utilising the Debye-Grüneisen model. Figure 1(a) shows the synchrotron X-ray diffraction results for the thermal expansion of Pd62.1Al27.3Y4.2Ni6.4 thin film powder. The ratio of the principal peak positions (Q(T0)/Q(T))3 of the structure factor S(Q) is depicted as a function of temperature since this ratio is directly related to volume changes in the glass31,33,34. Thus, the CTE is related to (Q(T0)/Q(T))3 below Tg provided that no structural change occurs31, as has been reported for several La-, Pd-, Sm-, Co- and Zr-based metallic glasses31,33,34,35,36,37,38,39,40. In the heating section, beginning at room temperature, the ratio (Q(T0)/Q(T))3 is increasing linearly. From 570 K onwards (this temperature is referred to as relaxation temperature Tr), the CTE decreases and eventually turns negative at Tn = 670 K. The (Q(T0)/Q(T))3 curve reaches a minimum at 775 K. According to Bednarcik et al.34 the minimum in the (Q(T0)/Q(T))3 curve corresponds to the glass transition temperature Tg. The continuing change in the (Q(T0)/Q(T))3 ratio due to homogenisation of the sample temperature at the maximum temperature of 863 K during a holding time of 5 min causes an infinite slope. In the cooling section, the (Q(T0)/Q(T))3 ratio decreases linearly. The linear regions of the heating and cooling sections are fitted in intervals of 50 K. The resulting CTEs are shown in Fig. 1(b). The average in CTE for the heating and cooling section are 3.5 ± 0.2 × 10−5 and 3.4 ± 0.1 × 10−5 K−1, respectively, which is consistent with CTE for other Pd-based metallic glasses41. In the supercooled region above Tg we measured a CTE of 2.7 × 10−4 K−1, which is 8 times larger than the CTEs in the glassy regions. This can be rationalized based on additional excitations of atomic oscillators at the glass transition33,34,42. In contrast to the thermal expansion while cooling, the thermal expansion while heating begins to deviate from a linear dependence at Tr. Throughout the whole cooling region a linear dependency of (Q(T0)/Q(T))3 to the temperature is observed. The non-linear dependency of (Q(T0)/Q(T))3 on temperature while heating is caused by the annihilation of free volume in the structure31,33,43. The CTEs in the heating and cooling section are consistent within the error of the measurement. This holds because the difference in free volume between the heating and cooling curves does not influence the relative shift of the principle maximum of S(Q) in the relaxation free temperature region33. While heating the metallic glass, excess free volume is annihilated. Hence, with increasing temperature the thermal expansion is superimposed by free volume annihilation. Above Tr the influence of free volume annihilation on the macroscopic thermal expansion is larger than the thermal volume expansion. Eventually, this causes a negative CTE at 670 K.

Thermal expansion of a Pd based metallic glass. (a) Ratio of the principle peak positions of the structure factor (Q(T0)/Q(T))3 of Pd62.1Al27.3Y4.2Ni6.4 as a function of temperature. The dashed lines are linear fits with the slopes as the volumetric coefficient of thermal expansion in the heating, cooling and supercooled region αh, αc and αsc, respectively. Dotted lines mark the relaxation and glass transition temperature Tr and Tg. Arrows indicate the direction of heating. (b) Coefficient of thermal expansion of Pd54.7Al23.5Y7.8Ni11.3 by ab initio based Debye-Grüneisen model. Debye temperature is 332 K. The red and blue triangles represent the experimental CTEs in the heating and cooling sections, respectively.
Figure 1(b) depicts the calculated CTEs from the Debye-Grüneisen model based on the ab initio calculations in the ground state and the experimental CTEs as a function of temperature. The calculated CTEs increase between 100 and 300 K from 1.5 × 10−5 to 3.4 × 10−5 K−1. Above the Debye temperature ΘD = 332 K, the CTE increases only slightly. Hence, we observe excellent agreement between the experimentally and theoretically obtained CTEs of the Pd54.7Al23.5Y7.8Ni11.3 thin film metallic glass, which is within the error of the measurement and the precision of the Debye temperature calculation. The Debye temperature depends on the temperature dependent equilibrium volume, elastic constants and composition, whereby the calculated CTE can deviate up to 30%17. The Debye-Grüneisen method models the acoustic mode of the phonons below Θ and the increased amplitudes of anharmonic vibrations at higher temperatures by the Grüneisen constant as long as structural changes such as relaxation or the approach of the melting point do not occur13. Deviations of the experimentally obtained CTEs from the predicted CTEs indicate regions with and without structural relaxation. For Pd62.1Al27.3Y4.2Ni6.4 the experimental CTE begins to decrease at Tr, marking the onset of structural relaxation. Therefore, we propose the Debye-Grüneisen model as a reliable ab initio based method to predict CTEs of metallic glasses, provided a realistic structural model is applied, which will be discussed in the next section.
The applicability of the Debye-Grüneisen model can be justified by the work of Safarik and Schwarz44. The signature of a metallic glass in terms of lattice vibrations are low frequency lattice vibrations that can be modelled by Einstein oscillators. These vibrations that deviate from the Debye model are highly anharmonic. However, the anharmonic modes do not influence the bulk modulus44. As the bulk modulus is one major input parameter for the Debye-Grüneisen model and is not influenced by an anomalous large density of low frequency vibrations in glasses, this model can be applied for metallic glasses without the addition of Einstein oscillators in the Debye-Grüneisen model.
Ab initio structural model vs. experiment
The CTE predicted by the Debye-Grüneisen model depends entirely on the atomic configuration obtained in the ab initio calculations. Hence, we will compare the simulated configuration with the experimentally observed one. Besides topology, glass transition temperature is a representative gauge to judge the validity of an atomic configuration. The glass transition temperature Tg is estimated by an empirical model introduced by Wang et al.45. They report the ratio of 1000Tg and MΘD 2 (the Lindemann criterion) is approximately constant for metallic glasses, considering Zr-based, Pd-based, Nd-based, La-based and Cu-based metallic glass systems, where M is the average atomic mass45. Based on this model, Tg of the calculated structure is 981 K compared to 775 K in the experiment (Fig. 1). This difference of 20% between theory and experiment is comparable with the deviations expected for the predicted CTE and even smaller compared to deviations of Tg calculated by classical molecular dynamics simulations for Ni-P45,46 and Zr-Al-Ni47,48 to the corresponding experimental values. Causes for this deviation might be the infinite quenching rate that increases Tg 46 and the small difference in composition caused by the discrete number of atoms in the supercell. Thus, the accuracy of our structural model with respect to Tg is within the temperature range expected from our model and literature.
Short and medium range order have been reported in literature to have different contributions to thermal expansion40. Hence, we compare the calculated and experimentally obtained PDF in Fig. 2. The PDFs calculated for 300, 400, 700 and 800 K, i.e. for whole temperature range encompassing Tr as well as Tg, resemble the first peak of the experimental PDF at 2.8 Å with a deviation of 0.1 Å. The second coordination shells of the calculations overlap with the experimental one. Similar to the experimental PDF, the calculated PDF for 300 K exhibits a distinct peak splitting with a peak at 4.8 Å and a shoulder at 5.3 Å, i.e. at the same positions as in the experimental case. The peak splitting is observed up to 800 K. In the third coordination shell, the calculated PDF at 300 K shows two distinct peaks which are different from the experimentally observed peak. However, the calculated third coordination shell covers the relevant r-range between 6.0 and 8.5 Å and exhibits the same asymmetric shape as the experimentally observed third coordination shell. For higher temperatures of 700 and 800 K the major peaks in the third coordination shell of the calculated PDFs are within the range of 7.5 and 7.0 Å. Despite computational constraints, the description of the short-range order up to the second coordination shell is feasible at room temperature, as the calculated PDF at 300 K resembles the experimental PDF despite the noise due to the low number of atoms. Therefore, the ab initio model introduced in ref.8 for a metalloid containing metallic glass produces a realistic atomic structure of a metalloid free metallic glass if the size of the supercell is adjusted to comprise higher coordination shells. Therefore, the increased size of the supercell compared to ref.8 is necessary. However, to account for structural relaxation an even larger supercell would be required. The number of 389 atoms used here, which corresponds to an increase of 338% in reference to ref.8, is still 1.5 times smaller than the number of atoms found in regions of correlative motion during structural relaxation in Pd-based metallic glasses49. To summarize the comparison of ab initio model and experimentally observed structure, the calculated PDF, glass transition temperature and CTE are consistent with the experimental values. Hence, the ab initio model represents an atomic configuration comparable to the atomic configuration obtained with magnetron sputtering.

Experimental and calculated pair distribution functions of Pd62.1Al27.3Y4.2Ni6.4 at 300, 400, 700 and 800 K.
Structural rearrangements
The deviation of (Q(T0)/Q(T))3 from a linear dependence on T and the deviation of the CTE from the theoretical prediction at Tr indicate the onset of structural rearrangements, which have implications on mechanical properties43. Temperature induced mechanical property and phase stability changes are of key importance for structural applications41. Thus, to investigate the atomic rearrangements, we employed a refined structural analysis. To understand the influence of structural rearrangements on the medium range order, we compare the structure factor obtained by high energy X-ray diffraction at room temperature; Tn = 670 K, at which the CTE changes from positive to negative values; Tg = 775 K; and the maximum heater temperature of 863 K (Fig. 3(a)). From the comparison of the structure factor at RT and 670 K we observe a slight peak shift towards lower wavevectors, while the peak width begins to decrease at Tr (Fig. 4(a)). This indicates an onset of structural rearrangements towards a more ordered structure, which is consistent with the comparison of predicted and measured CTE. Between 670 K and Tg = 775 K, the second peak of S(Q) (Fig. 3(a)) shifts significantly towards larger q values from 4.77 to 4.90 Å−1. Furthermore, the ratio of first and second peak positions q2/q1 (Fig. 4(b)) deviates from a constant value above 670 K. Hence, starting at Tn = 670 K inelastic structural changes occur that cannot solely be attributed to thermal expansion33. The changes in S(Q) (Fig. 3(a)) indicate that atomic rearrangements become more pronounced in the supercooled liquid region between Tg and 863 K. In the supercooled liquid region, the principal peak develops a shoulder at 3 Å−1 and decreases in height due to atomic rearrangements, while a peak sharpening of the main peak in the second coordination shell is observed, indicating the onset of the Bragg peaks and hence structural rearrangements towards the formation of a crystalline phase. These small Bragg peaks are irreversible and clearly visible in the cooled down sample, which consists of nanocrystals in an amorphous matrix after the heating cycle.

Changes in the structure of Pd62.1Al27.3Y4.2Ni6.4 during in situ heating and cooling. Structure factor S(Q) (a) and PDF (b) and difference between PDF at elevated temperature and room temperature (c) of Pd62.1Al27.3Y4.2Ni6.4 for room temperature (300 K), 670 K, glass transition temperature (775 K), the supercooled region (800, 826 and 849 K), maximum heating temperature and the cooled down sample. The inset in (a) shows the peak positions of the Bragg peaks of the fcc-phases of the main constituents Pd and Al.

Changes in the structure factor with temperature. (a) Full width half maximum (FWHM) of principal peak of the structure factor as a function of temperature in the heating section, the dashed line is a linear fit of the FWHM below Tr (b) second to first peak ratio q2/q1 of the structure factor as a function of temperature in the heating section, the dashed line marks a constant q2/q1 ratio.
Comparing the peak positions of S(Q) to the Bragg peaks of the crystalline phases of the main constituents Pd and Al (inset in Fig. 3(a)), the principle peak shifts towards the (111) peak of Pd, while the shoulder of the principal peak is near the Bragg peaks of the (200) plane. The main peak of the second coordination shell matches the (311) peak of Al, while its shoulders are between the (220) and (222) peaks, respectively, of both elements. Thus, the fcc crystalline structures appear to evolve via fcc-like short-range ordered clusters before the onset of crystallisation. Similar observations have been reported by Voyles et al. for the amorphous to crystalline transition of Si50.
Despite the nanocrystalline phase in the glass present in the cooling region, the CTE for the glass and a nanocrystal-glass composite material are equal within the error of the measurement. This equivalence of CTEs may be explained by the portion of the amorphous phase and the similarity of the CTE of the major constituent Pd. The rather broad principle peak of S(Q) of the cooled down sample in Fig. 3(a) and the small Bragg peak indicate a considerably large amorphous portion after the heating and cooling cycle. Additionally, the CTE of 3.4 × 10−5 K−1 for main constituent Pd13 is in agreement with the calculated CTE for the amorphous phase. Thus, the difference of CTE between crystalline and amorphous phase might be small, wherefore the CTE of the nanocrystal-glass composite is equal to the amorphous CTE.
Complementary to the medium-range order analysis, the development of short-range order is probed by employing real space PDFs. The experimentally obtained real space reduced PDF G(r) shown in Fig. 3(b) underlines the structural changes during heating. Corrugations at bond distances lower than 2.1 Å are artefacts from the Fourier transform of S(Q) to G(r)51. The first peaks of G(r) are at distances of 2.9, 4.8, 7.1 and 9.4 Å. The first peak indicates Pd-dominated bonding, as its position is at the distance of Pd-Pd bonds in icosahedral coordination. The G(r) at RT and 670 K resemble each other in terms of peak shape and position. Within the temperature range of RT to 863 K the shape of the nearest neighbour shell remains unchanged. However, the peak height is decreasing until Tg by 13%, while the full width half maximum (FWHM) is increasing until Tg by 8%. The difference between the PDF at elevated temperatures and PDF at 300 K (Fig. 4(c)) shows an increase of the peak flanks with a larger portion at the right flank, while the height at the peak centre decreases. Thus, thermal rejuvenation might occur, that broadens the atomic bond length distribution in the first coordination shell and thereby increases the disordering52 by thermally activated atomic movements. However, the thermal rejuvenation of the short range order does not affect the average atomic volume, as the FWHM of the principle peak of S(Q) begins to narrow and the (Q(T0)/Q(T))3 ratio begins to change already at 570 K. Since the average volume is represented by the principle peak of S(Q), the changing (Q(T0)/Q(T))3 ratio indicates free volume annihilation31 concurrent to thermal rejuvenation of the short range order. As can be derived by the continuously changing slope of Q(T0)/Q(T))3 towards a more negative value, the free volume annihilation rate in the medium range order increases up to Tg, where the average atomic volume begins to increase again. Thermal rejuvenation stops at the glass transition temperature, where structural relaxation sets on as the FWHM decreases and principle peak height increases53 strongly up to Tx, where the slope decreases and thus structural relaxation slows down. During structural relaxation, the right flank of the first coordination shell decreases (Fig. 4(c)), while the left flank is increased compared to the reference. Thus above Tg, free volume annihilation takes place in the short range order due to removal of large atomic bond distances53. This structural relaxation towards a more ordered structure at Tx is consistent with the continuous development of a more ordered (crystalline) structure, which has been already observed by the evolution of the Bragg peaks in S(Q).
While free volume is annihilated between the nearest neighbours above Tg, peak splitting is observed for the second to fifth coordination shell, indicating chemical rearrangement. Whereas the major subpeak of the second coordination shell below Tg is at 4.8 Å, the major peak at Tg is at 5.1 Å with shoulders at 4.3 and 5.6 Å. These three subpeaks become more pronounced with increasing temperature in the supercooled region. Also in the third coordination shell the subpeak at 7.4 Å, which was not dominating below Tg, evolves at Tg and eventually dominates the third coordination shell in the supercooled region. Shoulders evolve also in the fourth coordination shell in the supercooled region. This evolution of subpeaks in the second to fourth coordination shell hints to a rearrangement of local chemical environment53 of Pd, which is the major constituent in this metallic glass.
The different behaviour of short and medium range order in terms of free volume annihilation, thermal rejuvenation and relaxation hints to a heterogeneous atomic environment on the short range order level. To understand the relation of the individual coordination shells in the short range ordered region and the thermal expansion, we calculated the temperature induced relative shift of peak positions of the individual coordination shells. Figure 5 depicts the relative shift of the peak positions rij of the PDF (indices indicating the jth peak in the ith coordination shell) during heating. The average thermal expansion obtained from the shift of the principle peak of S(Q) is indicated by a solid line and supposed to be approximately the macroscopic thermal expansion40. The reference peak positons at T0 = RT for r11, r21, r22, r31, r32, r41 and r42 are 2.86, 4.75, 5.05, 6.95, 7.29 9.26 and 9.54 Å, respectively. From Fig. 5 we learn that the thermal expansion of the coordination shells is inhomogeneous. The first and third coordination shell contract compared to the macroscopic thermal expansion, while the second coordination shell expands more than by the macroscopic thermal expansion. The expansion of the fourth coordination shell matches the macroscopic thermal expansion. The peak shifts of the first to third coordination shells are due to rearrangements on the atomic level (Fig. 3(b,c)). Comparing the cooled down sample to the initial sample, the second peak at 3.4 Å forms in the first coordination shell, might be associated with the bonds involving Y, while the second coordination shell consists of three distinct peaks at 4.2, 5.0 and 5.7 Å instead of one peak containing two subpeaks. The difference of the PDF of the cooled down sample and the pristine sample (Fig. 3(c)) reveals a sharpening of the peak of the first coordination shell, indicating a narrower bond length distribution. The distinct peaks in the second coordination shell verify the increased ordering in the sample after the heating cycle. However, in Fig. 5 we observe that the higher coordination shells reflect the macroscopic thermal expansion obtained from the principal peak shift in S(Q)40. This can readily be understood based on the inverse relationship between wavenumber and bond distance. The principal peak of S(Q) contains the major information of the medium range order34,54. Thus, it is learned from the analysis of both S(Q) and G(r) that the macroscopic thermal expansion is reflected by the medium range order, while irreversible changes take place in the heterogeneous atomic environment of the short and near medium range order up to bond distances of 13.2 Å, which is in agreement to literature reports40 on CuZr metallic glasses. During structural relaxation, the exchange of atoms with different sizes between the heterogeneous coordination shells is possible55, allowing the contraction and expansion of coordination shells as is the case for the first to third coordination shell, leading to inhomogeneous thermal expansion on the atomic scale. Following the shifts of the peak positions in Fig. 5 we observe a significant change in slope around 745 K, inferring an increased rate of structural rearrangements. The inflection point at 775 K corresponds to the glass transition temperature Tg observed in (Q(T0)/Q(T))3. Further heating leads to a minimum at 800 K. This change of slope in real space is identified as crystallisation temperature Tx 34. This temperature coincides with the flattening of the width of the principle peak of the structure factor and of the q2/q1-ratio in Fig. 4. The onset of crystallisation at Tx is consistent with the observation of Bragg peaks in the structure factor (Fig. 3(a)) inferring nanocrystal formation within a glassy matrix.

Relative shift of peak positions rij of G(r) as a function of temperature of the jth peak in the ith coordination shell. The solid line marks the relative peak shift expected from the average CTE αc obtained from the structure factor S(Q). Empty symbols represent relative peak shifts of the calculated PDF from Fig. 2. Tg and Tx are the glass transition and crystallisation temperature, respectively.
The empty symbols in Fig. 5 show the relative peak shifts of the calculated PDFs in Fig. 2 and underline the lack of structural relaxation in the ab initio model. The peak positions of the calculated PDF do not shift in accordance with the experiments, but show a strong scattering. The scattering is likely caused by the small volume of the supercell, which introduces noise into the calculated PDF that is also observed for bond distance larger 4 Å (Fig. 2). Hence, a direct determination of CTEs from the shift of peak positions in real space is not feasible. However, as shown in Fig. 2 the description of the short-range order up to the third coordination shell by ab initio calculations is possible at room temperature. This enables the prediction of CTEs with the Debye-Grüneisen model, which is more efficient in terms of time and resources compared to experimental methods.
Conclusion
The average room temperature CTE of the Pd54.7Al23.5Y7.8Ni11.3 thin film metallic glass obtained from Debye-Grüneisen model is 3.4 × 10−5 K−1, while the experimental CTEs calculated from in situ heating and cooling cycles in synchrotron X-ray diffraction experiments are 3.5 ± 0.3 × 10−5 and 3.4 ± 0.1 × 10−5 K−1, respectively. Hence, we observe excellent agreement between the experimentally and theoretically obtained CTEs. Therefore, we propose the Debye-Grüneisen model as a reliable ab initio based method for predicting the CTE of metallic glasses. A comparison between the calculated and experimental PDF shows that the experimentally observed structure at room temperature is well resembled by the ab initio model. The temperature induced structural relaxation is not covered by our model containing 389 atoms, which is the cause for the deviation between theoretical and experimental CTE data for temperatures above Tr. The deviation originates in the rearrangement of atoms and the annihilation of free volume during structural relaxation within the experiment.
The in situ high energy X-ray diffraction structure analysis reveals an inhomogeneous expansion of the individual coordination shells. For the Pd62.1Al27.3Y4.2Ni6.4 thin film metallic glass the macroscopic thermal expansion is dominated by the medium range order, while thermal rejuvenation and atomic rearrangements are observed in the short range order. Structural relaxation of the average structure starts at 570 K. The glass transition temperature of Pd62.1Al27.3Y4.2Ni6.4 is 775 K, which we identified through a change of signs of the CTE. Above Tg, the peaks of S(Q) shift towards the positions of the Bragg peaks of the crystalline phases of the main constituents. Hence, before the onset of crystallisation a continuous evolution towards a fcc structure via a fcc-like arrangement of short-range ordered clusters is proposed, leading to the formation of a nanocrystal-glass composite material. The crystallisation temperature of 800 K is 25 K higher than Tg. The akin CTEs of the metallic glass and nanocrystal-glass composite material suggest thermal expansion based on the same mechanism of coordinated vibrations of strings of atoms next to highly defective areas in Pd-based metallic glasses and nanocrystal-glass composites.
Based on the average atomic volume, we observe free volume annihilation in the medium range order that exceeds the volume expansion at 670 K leading to a negative thermal expansion. Above Tg, free volume annihilation takes place in the short range order, while the average atomic volume increases in the supercooled region.
Data availability statement
The data generated and analysed during this study are available from the corresponding author on reasonable request.
References
Telford, M. The case for bulk metallic glass. Mater. Today 7, 36–43 (2004).
Ashby, M. & Greer, A. L. Metallic glasses as structural materials. Scripta Mater. 54, 321–326 (2006).
Inoue, A., Shen, B. & Takeuchi, A. Developments and Applications of Bulk Glassy Alloys in Late Transition Metal Base System. Mater. Trans. 47, 1275–1285 (2006).
Schnabel, V. et al. Electronic hybridisation implications for the damage-tolerance of thin film metallic glasses. Sci. Rep. 6, 36556 (2016).
Schroers, J. Processing of bulk metallic glass. Adv. Mater. 22, 1566–1597 (2010).
Demetriou, M. D. et al. A damage-tolerant glass. Nat. Mater. 10, 123–128 (2011).
Wang, X. D. et al. Atomic picture of elastic deformation in a metallic glass. Sci. Rep. 5, 9184 (2015).
Hostert, C. et al. Ab initio molecular dynamics model for density, elastic properties and short range order of Co-Fe-Ta-B metallic glass thin films. J. Phys. Condens. Mat. 23, 475401 (2011).
Schnabel, V., Evertz, S., Ruess, H., Music, D. & Schneider, J. M. Stiffness and toughness prediction of Co-Fe-Ta-B metallic glasses, alloyed with Y, Zr, Nb, Mo, Hf, W, C, N and O by ab initio molecular dynamics. J. Phys. Condens. Mat. 27, 105502 (2015).
Hostert, C., Music, D., Bednarcik, J., Keckes, J. & Schneider, J. M. Quantum mechanically guided design of Co43Fe20Ta5.5X31.5 (X = B, Si, P, S) metallic glasses. J. Phys. Condens. Mat. 24, 175402 (2012).
Söderlind, P., Nordström, L., Yongming, L. & Johansson, B. Relativistic effects on the thermal expansion of the actinide elements. Phys. Rev. B 42, 4544–4552 (1990).
Lu, X., Selleby, M. & Sundman, B. Theoretical modeling of molar volume and thermal expansion. Acta Mater. 53, 2259–2272 (2005).
Moruzzi, V. L., Janak, J. F. & Schwarz, K. Calculated thermal properties of metals. Phys. Rev. B 37, 790–799 (1988).
Lu, X.-G., Selleby, M. & Sundman, B. Calculations of thermophysical properties of cubic carbides and nitrides using the Debye–Grüneisen model. Acta Mater. 55, 1215–1226 (2007).
Tao, X. et al. Ab initio calculation of the total energy and elastic properties of Laves phase C15Al2RE (RE = Sc, Y, La, Ce–Lu). Comp. Mater. Sci. 44, 392–399 (2008).
Deligoz, E., Ozisik, H. B., Colakoglu, K. & Ciftci, Y. O. First principles prediction of structural stability, elastic, lattice dynamical and thermal properties of osmium carbides. Mater. Sci. Tech. 30, 842–849 (2014).
Music, D., Geyer, R. W. & Keuter, P. Thermomechanical response of thermoelectrics. Appl. Phys. Lett. 109, 223903 (2016).
Hohenberg, P. & Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 136, B864–B871 (1964).
Ozaki, T. & Kino, H. Numerical atomic basis orbitals from H to Kr. Phys. Rev. B 69 (2004).
Ozaki, T. & Kino, H. Efficient projector expansion for the ab initio LCAO method. Phys. Rev. B 72 (2005).
Perdew, B. & Ernzerhof. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Ozaki, T. Variationally optimized atomic orbitals for large-scale electronic structures. Phys. Rev. B 67 (2003).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Birch, F. Finite strain isotherm and velocities for single-crystal and polycrystalline NaCl at high pressures and 300°K. J. Geophys. Res. 83, 1257 (1978).
Music, D. et al. Elastic properties of Fe–Mn random alloys studied by ab initio calculations. Appl. Phys. Lett. 91, 191904 (2007).
Hammersley, A. P. FIT2D. An Introduction and Overview. ESRF, 1997.
Hammersley, A. P., Svensson, S. O., Hanfland, M., Fitch, A. N. & Hausermann, D. Two-dimensional detector software. From real detector to idealised image or two-theta scan. High Pressure Res. 14, 235–248 (1996).
Hammersley, A. P., Svensson, S. O. & Thompson, A. Calibration and correction of spatial distortions in 2D detector systems. Nucl. Instrum. Meth. A 346, 312–321 (1994).
Yavari, A. R. et al. Excess free volume in metallic glasses measured by X-ray diffraction. Acta Mater. 53, 1611–1619 (2005).
Juhás, P., Davis, T., Farrow, C. L. & Billinge, S. J. L. PDFgetX3. A rapid and highly automatable program for processing powder diffraction data into total scattering pair distribution functions. J. Appl. Crystallogr. 46, 560–566 (2013).
Mattern, N., Stoica, M., Vaughan, G. & Eckert, J. Thermal behaviour of Pd40Cu30Ni10P20 bulk metallic glass. Acta Mater. 60, 517–524 (2012).
Bednarcik, J. et al. Thermal expansion of a La-based bulk metallic glass. Insight from in situ high-energy x-ray diffraction. J. Phys. Condens. Mat. 23, 254204 (2011).
Georgarakis, K. et al. Variations in atomic structural features of a supercooled Pd–Ni–Cu–P glass forming liquid during in situ vitrification. Acta Mater. 59, 708–716 (2011).
Jing, G. et al. Formation and interesting thermal expansion behavior of novel Sm-based bulk metallic glasses. Intermetallics 15, 929–933 (2007).
Kato, H., Chen, H.-S. & Inoue, A. Relationship between thermal expansion coefficient and glass transition temperature in metallic glasses. Scripta Mater. 58, 1106–1109 (2008).
Mattern, N. et al. Structural behavior of Pd40Cu30Ni10P20 bulk metallic glass below and above the glass transition. Appl. Phys. Lett. 82, 2589–2591 (2003).
Qu, D. D., Mizuno, A., Watanabe, M., Bednarcik, J. & Shen, J. Undercooling behavior of Zr–Cu–Ni–Al bulk metallic glasses investigated by in situ synchrotron high energy X-ray diffraction. Mater. Sci. Eng. 555, 36–43 (2012).
Mattern, N., Bednarcik, J., Stoica, M. & Eckert, J. Temperature dependence of the short-range order of Cu65Zr35 metallic glass. Intermetallics 32, 51–56 (2013).
Liu, Y., Hata, S., Wada, K. & Shimokohbe, A. Thermal, Mechanical and Electrical Properties of Pd-Based Thin-Film Metallic Glass. Jpn. J. Appl. Phys. 40, 5382–5388 (2001).
Mattern, N. et al. Thermal behavior and glass transition of Zr-based bulk metallic glasses. Mater. Sci. Eng. 375-377, 351–354 (2004).
Chen, H. S. The influence of structural relaxation on the density and Young’s modulus of metallic glasses. J. Appl. Phys. 49, 3289–3291 (1978).
Safarik, D. J. & Schwarz, R. B. Evidence for highly anharmonic low-frequency vibrational modes in bulk amorphous Pd40Cu40P20. Phys. Rev. B 80, 94109 (2009).
Wang, W. H., Wen, P., Zhao, D. Q., Pan, M. X. & Wang, R. J. Relationship between glass transition temperature and Debye temperature in bulk metallic glasses. J. Mater. Res. 18, 2747–2751 (2003).
Lewis, L. J. Atomic dynamics through the glass transition. Phys. Rev. B 44, 4245–4254 (1991).
Guerdane, M. & Teichler, H. Structure of the amorphous, massive-metallic-glass forming Ni25Zr60Al15 alloy from molecular dynamics simulations. Phys. Rev. B 65, 177 (2001).
Inoue, A., Zhang, T. & Masumoto, T. Zr-Al-Ni Amorphous Alloys with High Glass Transition Temperature and Significant Supercooled Liquid Region. Mater. Trans. JIM 31, 177–183 (1990).
Qiao, J., Casalini, R., Pelletier, J.-M. & Kato, H. Characteristics of the structural and Johari-Goldstein relaxations in Pd-based metallic glass-forming liquids. J. Phys. Chem. B 118, 3720–3730 (2014).
Voyles, P. M., Gerbi, J. E., Treacy, M. M., Gibson, J. M. & Abelson, J. R. Absence of an abrupt phase change from polycrystalline to amorphous in silicon with deposition temperature. Phys. Rev. Lett. 86, 5514–5517 (2001).
Egami, T. & Billinge, S. J. L. Underneath the Bragg peaks. Structural analysis of complex materials (Elsevier, Amsterdam, 2012).
Tong, Y. et al. Structural rejuvenation in bulk metallic glasses. Acta Mater. 86, 240–246 (2015).
Dmowski, W., Fan, C., Morrison, M. L., Liaw, P. K. & Egami, T. Structural changes in bulk metallic glass after annealing below the glass-transition temperature. Mater. Sci. Eng. 471, 125–129 (2007).
Bednarcik, J., Michalik, S., Kolesar, V., Rütt, U. & Franz, H. In situ XRD studies of nanocrystallization of Fe-based metallic glass. A comparative study by reciprocal and direct space methods. Phys. Chem. Chem. Phys. 15, 8470–8479 (2013).
Tong, X. et al. Structural evolution and strength change of a metallic glass at different temperatures. Sci. Rep. 6, 30876 (2016).
Acknowledgements
We acknowledge financial support of the German Research Foundation within the SPP 1594 “Topological Engineering of Ultrastrong Glasses”. Ab initio calculations were performed on the JARA HPC cluster of RWTH Aachen University within the project JARA0131. Parts of this research were carried out at beamline P02.1 of the light source PETRA III at DESY, a member of the Helmholtz Association (HGF).
Author information
Authors and Affiliations
Contributions
All authors designed the research, discussed the result and wrote the manuscript; S.E. and D.M. performed the ab initio molecular dynamics simulations; S.E. synthesised the samples and analysed the synchrotron data; S.E. and V.S. conducted the synchrotron experiments with support of J.B.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Evertz, S., Music, D., Schnabel, V. et al. Thermal expansion of Pd-based metallic glasses by ab initio methods and high energy X-ray diffraction. Sci Rep 7, 15744 (2017). https://doi.org/10.1038/s41598-017-16117-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-16117-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
