
Abstract
Botulinum neurotoxins (BoNTs) are highly potent toxins that cleave neuronal SNARE proteins required for neurotransmission, causing flaccid paralysis and death by asphyxiation. Currently, there are no clinical treatments to delay or reverse BoNT-induced blockade of neuromuscular transmission. While aminopyridines have demonstrated varying efficacy in transiently reducing paralysis following BoNT poisoning, the precise mechanisms by which aminopyridines symptomatically treat botulism are not understood. Here we found that activity-dependent potentiation of presynaptic voltage-gated calcium channels (VGCCs) underlies 3,4-diaminopyridine (3,4-DAP)-mediated rescue of neurotransmission in central nervous system synapses and mouse diaphragm neuromuscular junctions fully intoxicated by BoNT serotype A. Combinatorial treatments with 3,4-DAP and VGCC agonists proved synergistic in restoring suprathreshold endplate potentials in mouse diaphragms fully intoxicated by BoNT/A. In contrast, synapses fully intoxicated by BoNT serotypes D or E were refractory to synaptic rescue by any treatment. We interpret these data to propose that increasing the duration or extent of VGCC activation prolongs the opportunity for low-efficiency fusion by fusogenic complexes incorporating BoNT/A-cleaved SNAP-25. The identification of VGCC agonists that rescue neurotransmission in BoNT/A-intoxicated synapses provides compelling evidence for potential therapeutic utility in some cases of human botulism.
Similar content being viewed by others
Introduction
Botulinum neurotoxins (BoNTs) are a closely related family of protein neurotoxins expressed by members of the Clostridium genus of anaerobic bacteria1. Collectively, the BoNTs are the most poisonous substances known, with estimated human LD50 values as low as 0.1–1 ng/kg. The neurotoxins are categorized by antigenic properties into seven serotypes (BoNT/A-G), with highly conserved structural and functional properties2. All BoNT serotypes are initially expressed as 150 kDa proteins, which are activated by proteolytic cleavage into 100 kDa heavy chains (HC) and 50 kDa light chains (LC) that remain associated through a disulfide bond. The C-terminal of HC mediates highly selective and efficient binding to endosomal receptors on the presynaptic membrane of peripheral neurons. Following internalization via synaptic endocytosis, the N-terminal domain of HC forms a pore that facilitates translocation of LC through the endosomal membrane to the pre-synaptic cytosol3. The translocated LC refolds to form a zinc-dependent endoprotease that specifically targets and cleaves fusogenic SNARE proteins essential for neurotransmitter exocytosis. SNAP-25 is targeted and cleaved by BoNT/A, /C, and /E; synaptobrevin1/2 (SYB) is cleaved by BoNT/B, /D, /F, and /G; and syntaxin-1 (STX1) is cleaved by BoNT/C reviewed in2. BoNT proteolysis of neuronal SNARE proteins interferes with formation of the Ca2+-activated fusogenic complex required for fast neurotransmission, thereby blocking synaptic vesicle fusion. Symptoms of BoNT poisoning clinically manifest as a descending flaccid paralysis that results in death by asphyxiation once respiratory muscles are compromised. The only treatment options for clinical botulism are supportive care, such as mechanical ventilation and parenteral feeding, until neuromuscular function is restored. Some serotypes produce paralysis that persists for months, requiring sustained supportive care with increased risk of co-morbidities.
Although post-exposure prophylaxis with antitoxin can efficiently neutralize toxin prior to neuronal uptake4,5, antitoxin administration requires clinical evidence of progressive paralysis, and thus even patients that receive a timely administration of antitoxin are likely to suffer symptoms of botulism. Despite intensive research efforts over the past three decades, small molecule LC inhibitors with clinically suitable pharmacokinetics and pharmacodynamics have not been identified6,7. Consequently, there remains a critical need for treatments that reverse neuromuscular paralysis in botulism patients.
In vitro studies suggest that neuromuscular function of intoxicated tissues can be transiently improved by enhancing neuronal Ca2+ influx, such as through increased extracellular Ca2+, treatment with aminopyridines or exposure to cationic ionophores8,9,10. Of these, the aminopyridines are particularly intriguing because derivatives such as 4-aminopyridine and 3,4-diaminopyridine (3,4-DAP) are currently in clinical use for neuromuscular indications11. Aminopyridines block the intracellular domain of voltage-gated K+ channels (VGKC), extending the duration of action potential-induced presynaptic depolarization and increasing neurotransmitter release12,13. However, clinical evaluations of aminopyridines in patients with severe serotype A and B botulism have resulted in highly variable and conflicting reports of efficacy14,15,16. Because the precise mechanisms by which aminopyridines enhance muscle contraction in paralyzed tissues are not known, conditions under which they can mitigate botulism symptoms have not been established.
While augmenting presynaptic cytosolic Ca2+ influx is likely to increase release probability at non-intoxicated release sites in partially intoxicated NMJs17, it is unknown whether other mechanisms can also contribute to symptomatic rescue. Here, we investigated the cellular and molecular mechanisms of 3,4-DAP-mediated rescue of neurotransmission in networked cultures of primary rat neurons and isolated mouse diaphragms intoxicated by BoNT serotypes A, D or E (the serotypes most commonly associated with human disease). These studies were conducted under conditions in which neurotransmission was thoroughly blocked by BoNT intoxication, thereby enabling the evaluation of treatments that restore the ability of intoxicated synapses to undergo synaptic release. These data provide compelling evidence that enhancing presynaptic Ca2+ influx through agonism of voltage-gated Ca2+ channels (VGCCs) is sufficient to rescue neurotransmission in synapses intoxicated by BoNT/A. Furthermore, they reveal that drugs that synergize with 3,4-DAP to enhance phasic Ca2+ influx represent a potentially novel class of candidate botulism therapeutics with improved efficacy profiles.
Results
3,4-DAP rescue of evoked neurotransmission is unique to BoNT/A-poisoned synapses and dependent on VGCC activation
To characterize the effects of 3,4-DAP on action potential-elicited neurotransmission at intoxicated synapses, whole-cell patch-clamp electrophysiology was used to measure excitatory postsynaptic currents (EPSCs) in response to extracellular stimulation of proximal axons in primary neuron cultures18. These studies were done in the presence of GABA and NMDA receptor antagonists, permitting aggregate analysis of synaptic transmission at afferent glutamategic synapses. Field stimulation of primary rat hippocampal and cortical neurons at 18–24 d after plating produced EPSCs with amplitudes of 512.2 ± 77.1 pA (Fig. 1A). Spontaneous miniature EPSCs (mEPSCs) measured in the same neuron population had mean amplitudes of 14.8 ± 1.1 pA, indicating that field stimulation elicited release of approximately 35 quanta per trial. The elimination of EPSCs by addition of either tetrodotoxin (TTX) or the AMPA/kainic acid receptor antagonist CNQX confirmed that EPSCs constituted post-synaptic responses to presynaptic release (Fig. 1A). 3,4-DAP treatment of naïve cultures increased EPSC amplitudes by 166.7 ± 26.4% (p = 0.027; Fig. 1A), without altering spontaneous miniature EPSCs (mEPSC) amplitudes or frequencies (Supplementary Figure S1A), consistent with the known pharmacological role of 3,4-DAP as a voltage-gated potassium channel blocker.

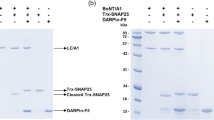
3,4-DAP restores evoked synaptic release in neuronal cultures intoxicated by BoNT/A but not BoNT/D or BoNT/E. (A) Representative whole-cell recordings of evoked neurotransmission in the presence of vehicle, TTX (5 µM), CNQX (20 µM), or 3,4-DAP (50 µM) and quantitation of normalized EPSC amplitudes versus vehicle (n = 6 each for TTX and CNQX; n = 16 for 3,4-DAP). (B) Representative Western blots of SNAP-25, STX1 and SYB2 from lysates harvested from primary neuron cultures treated with vehicle, BoNT/A, BoNT/D, or BoNT/E treatment and schematic of SNAP-25 and SYB structure with BoNT/A, /D and /E cleavage sites. Each panel representes a complete image from blots stained for STX1 and SNAP-25 (top) or SYB2 (bottom). Note that while proteolytic cleavage fragments of SNAP-25 can be immunologically detected, cleavage of SYB2 results in the rapid degradation of the N-terminal fragment, and therefore loss of reactivity is considered evidence of intoxication19. (C–E): Representative whole-cell recordings and of evoked neurotransmission and mean EPSC amplitudes in cultures intoxicated by BoNT/A, BoNT/D, or BoNT/E and treated with vehicle (n = 12), 3,4-DAP (50 µM; n = 12 for BoNT/A and n = 9 for BoNT/D or E) or 3,4-DAP plus VGCC antagonists (n = 8). Mean EPSC amplitudes are normalized to recordings from age-matched, non-intoxicated control cultures. Arrows represent stimulation times. Scale bars represent 400 ms (x-axis) and 100 pA (y-axis). All data presented as mean ± SEM. *Indicates p < 0.05, **Indicates p < 0.01, ns = not significant.
The effects of 3,4-DAP on neurotransmission were first tested in cultured neurons intoxicated with high concentrations of BoNT/A19. This approach allowed us to specifically test for treatments that restored synaptic function at fully intoxicated synapses. Western blot analysis of whole cell lysates revealed conversion of SNAP-25 to the BoNT/A cleavage product SNAP-25Δ9 in intoxicated cultures (Fig. 1B). BoNT/A intoxication reduced EPSC amplitudes to 4.0 ± 1.3% of naïve controls, demonstrating near-complete blockade of evoked release (Fig. 1C). 3,4-DAP treatment restored evoked release in 100% of evaluated neurons, increasing ESPC amplitudes to 45.1 ± 14.4% of naïve controls (p = 0.0012; Fig. 1C). As expected, 3,4-DAP had no effect on intoxicated neurons in the presence of TTX, indicating that synaptic rescue by 3,4-DAP was not mediated post-synaptically (Supplementary Figure S1B).
We next evaluated the molecular mechanisms involved in 3,4-DAP rescue of neurotransmission. A secondary neuropharmacological effect of 3,4-DAP is prolongation of voltage-gated Ca2+ channel (VGCC) activation, which increases presynaptic Ca2+ influx and vesicle release probability12. This suggested that enhanced Ca2+ influx through VGCCs in response to 3,4-DAP treatment contributed to the rescue effect. To test this hypothesis, BoNT/A-intoxicated cultures were treated with 3,4-DAP and L, N, R and P/Q-type VGCC antagonists (nimodipine, ω-agatoxin IVA and ω-conotoxin MVIIC). The presence of VGCC antagonists completely blocked rescue of neurotransmission by 3,4-DAP, demonstrating a requirement for VGCC activity (p = 0.029; Fig. 1C).
We next evaluated whether 3,4-DAP rescued EPSCs in cultures intoxicated by BoNT serotypes D and E. Unlike BoNT/A, which cleaves nine residues from the C-terminus of SNAP-25, BoNT/E removes an additional 17 amino acids, functionally eliminating the C-terminal SNARE domain. BoNT/D cleaves 56 residues from the N-terminal of rat synaptobrevin-2 (SYBΔ56), also eliminating the C-terminal SNARE domain (Fig. 1B)10. Intoxication with BoNT/D or BoNT/E resulted in proteolytic cleavage of SNAP-25 and SYB, respectively (Fig. 1B), and depressed EPSCs similar to BoNT/A (Fig. 1D,E). However, unlike BoNT/A-intoxicated synapses, 3,4-DAP had no effect on EPSCs in neurons intoxicated with serotype D (Fig. 1D; 7.5 ± 1.5% versus 5.0 ± 2.7%; p = 0.40) or E (Fig. 1E; 8.6 ± 3.0% versus 6.0 ± 4.1%, p = 0.61).
VGCC agonism is sufficient to rescue neurotransmission in synapses intoxicated by BoNT/A
The above data demonstrated that VGCC activation was necessary for 3,4-DAP rescue of neurotransmission in BoNT/A-intoxicated synapses. To more specifically determine whether VGCC currents were necessary for rescue, we recorded spontaneous synaptic release (mEPSCs) in the presence of elevated Ca2+ as well as VGCC agonists. In contrast to evoked release, spontaneous release is a probabilistic phenomenon, and consequently, mEPSC frequencies reflect the average release probability among the hundreds or thousands of afferent synapses on each neuron20,21. The ability to surveil the behavior of many synapses, with single-synapse resolution, facilitated a more specific evaluation of the effects of treatment conditions on release probabilities22.
To evaluate whether enhanced Ca2+ influx was sufficient to rescue spontaneous release from intoxicated synapses, mEPSCs were first recorded from intoxicated neurons in the presence of 2–16 mM extracellular Ca2+ (Fig. 2A,B). Consistent with previous studies, intoxication with BoNT/A reduced mEPSC frequencies by 99.98 ± 0.01% versus non-intoxicated controls in physiological Ca2+ (p < 0.001)19. Although 4 or 8 mM extracellular Ca2+ had no effect on mEPSC rates (p = 0.93 and 0.46, respectively), addition of 16 mM Ca2+ increased mEPSC frequencies to 7.8 ± 0.9% of controls (p < 0.01; Fig. 2B), without affecting mEPSC amplitudes as compared to naïve controls (16.4 ± 1.3 versus 15.4 ± 0.9 pA, p = 0.52). Addition of VGCC antagonists eliminated the recovery of mEPSCs in 16 mM Ca2+ (Fig. 2B; 0.4 ± 0.2%, p < 0.0001), confirming that the rescue effect required Ca2+ influx through VGCCs. In contrast, 16 mM Ca2+ did not restore spontaneous release in neuron cultures intoxicated with BoNT/D or BoNT/E (Fig. 2C).

Rescue of spontaneous synaptic activity in BoNT/A-silenced synapses by elevated extracellular Ca2+ requires VGCCs. (A) Representative whole-cell recordings and mean mEPSC frequencies from non-intoxicated cultures in the presence of 2, 4, 8, or 16 mM Ca2+. Representative whole-cell recordings and mean mEPSC frequencies from BoNT/A-intoxicated cultures, in the presence of 2, 4, 8, or 16 mM Ca2+ as well in 16 mM Ca2+ with VGCC antagonists (10 µM nimodipine, 0.5 µM ω-agatoxin IVA and 0.5 µM ω-conotoxin MVIIC). (C) Representative whole-cell recordings and mean mEPSC frequencies from neurons intoxicated by BoNT/D or BoNT/E and incubated in 2 or 16 mM Ca2+. mEPSC frequencies are normalized to recordings from age-matched, non-intoxicated control cultures. Scale bars represent 5 s (x-axis) and 40 pA (y-axis). All data presented as mean ± SEM and n ≥ 10 neurons for all conditions. **Indicates p < 0.01, ***Indicates p < 0.001.
While blockade of VGCCs proved to prevent rescue under multiple contexts, it remained possible that this was a reflection of the critical role that Ca2+ plays in neurotransmission, and not directly related to the rescue phenomenon. Thus, we next sought to establish whether agonists of VGCCs could rescue neurotransmission in the presence of physiological Ca2+. For these studies, the selective VGCC agonists GV-58 (N-, P/Q-types) or FPL 64176 (FPL; L-type) were added to BoNT/A-intoxicated neurons and spontaneous and evoked release were quantified. In non-intoxicated neurons, GV-58 and FPL increased mEPSC production by 469.4 ± 80.7% and 783.0 ± 60.4%, respectively (Fig. 3A) without affecting mEPSC amplitudes (Supplementary Figure S1B), confirming their presynaptic effects. In BoNT/A-intoxicated neurons, FPL increased mEPSC production from 0.15 ± 0.01% to 12.7 ± 2.9% (p < 0.0001), while GV-58 increased mEPSC production to 5.7 ± 1.6% (p = 0.03, Fig. 3B). Similarly, FPL increased EPSC amplitudes in BoNT/A-intoxicated neurons by 50.6 ± 8.9% (p < 0.05 versus vehicle-treated controls) while GV-58 increased EPSC amplitudes by 75.6 ± 14.3% (p < 0.001 versus vehicle-treated controls; Fig. 4A–D).

Use-dependent L-type or N/P/Q-type VGCC agonists restore spontaneous synaptic activity to neuronal cultures fully intoxicated by BoNT/A in physiological Ca2+. (A) Representative whole-cell recordings and mean mEPSC frequencies in non-intoxicated cultures treated with vehicle, FPL 64176 (10 µM) or GV-58 (50 µM). (B) Representative whole-cell recordings and mean mEPSC frequencies in BoNT/A-intoxicated cultures treated with vehicle, FPL 64176 (10 µM) or GV-58 (50 µM). mEPSC frequencies are normalized to recordings from age-matched, non-intoxicated control cultures. Scale bars represent 5 s (x-axis) and 40 pA (y-axis). All data presented as mean ± SEM and n ≥ 12 neurons for all conditions. *Indicates p < 0.05, ***Indicates p < 0.001.

Enhanced presynaptic Ca2+ influx rescues evoked release in BoNT/A-intoxicated cultures. (A) Representative whole-cell recordings of evoked neurotransmission in non-intoxicated cultures treated with vehicle, 16 mM Ca2+, FPL 64176 (10 µM), or GV-58 (50 µM). (B) Mean EPSC amplitudes from non-intoxicated cultures. (B) Representative whole-cell recordings of evoked neurotransmission in BoNT/A-intoxicated neurons treated with vehicle, 16 mM Ca2+, FPL 64176 (10 µM), or GV-58 (50 µM). (D) Mean EPSC amplitudes for BoNT/A-intoxicated cultures. EPSC amplitudes are presented as mean ± SEM and n ≥ 12 recordings for all conditions. Scale bars represent 400 ms (x-axis) and 100 pA (y-axis). Arrows represent stimulation times. *Indicated p < 0.05, **Indicates p < 0.01, ***Indicates p < 0.001, ns = not significant.
Combination treatment with 3,4-DAP and GV-58 restores EPSC amplitudes in neurons intoxicated by BoNT/A, but not BoNT/D or /E
We next evaluated whether combinatorial addition of 3,4-DAP and a VGCC agonist had additive effects on evoked release in BoNT/A-intoxicated cultures. GV-58 was chosen for these studies because it specifically targets VGCCs that are expressed at the synapse and associated with evoked release23,24. In BoNT/A-intoxicated neurons, combination treatment with 3,4-DAP and GV-58 restored EPSC amplitudes to levels that were indistinguishable from non-intoxicated cultures (Fig. 5A,B; 106.6 ± 21.0% versus 100 ± 15.1% respectively, p = 0.80), and 17-fold improved over intoxicated cultures treated with vehicle alone (6.0 ± 1.3%, p < 0.001). Combination treatment proved more effective than 3,4-DAP alone (p = 0.019), though there was no statistical improvement compared to GV-58 alone (p = 0.41).

3,4-DAP plus GV-58 restores evoked neurotransmission to baseline levels in cultured neurons intoxicated by BoNT/A, but not BoNT/D or BoNT/E. Representative EPSC and mean EPSC amplitudes from cultures intoxicated with: (A) vehicle; (B) BoNT/A; (C) BoNT/D; or (D) BoNT/E. Arrows represent stimulation times. Scale bars represent 400 ms (x-axis) and 100 pA (y-axis). Mean EPSC amplitudes are presented as mean ± SEM; n ≥ 13 recordings for 5A-B and n ≥ 9 recordings for 5C–D. *Indicates p < 0.05, ***Indicates p < 0.001, ns = not significant.
Combination treatment with 3,4-DAP and GV-58 had no effect on evoked release in BoNT/D-intoxicated cultures (p = 0.67; Fig. 5C) or BoNT/E-intoxicated cultures (p = 0.99; Fig. 5D), further illustrating that VGCC-mediated rescue of neurotransmission is specific to BoNT/A-intoxicated synapses.
3,4-DAP plus GV-58 restores suprathreshold release in mouse diaphragm endplates fully intoxicated by BoNT/A
Although these data demonstrated that enhanced VGCC activity could restore neurotransmission to CNS synapses severely intoxicated by BoNT/A, the physiological target of botulism is peripheral neurons. Thus, we used intracellular endplate recordings to evaluate the effects of treatment with 3,4-DAP and/or GV-58 on phrenic nerve-elicited endplate potentials (EPPs) in mouse hemidiaphragms preparations intoxicated ex vivo by BoNT/A or BoNT/E. The hemidiaphragm assay closely mimics the physiological progression of respiratory paralysis, and has been used extensively to study toxin mechanisms of action25,26.
To reproduce the comprehensive synaptic blockade used in CNS cultures, diaphragms were intoxicated with high doses of BoNT/A or /E and EPPs were serially monitored in sequential endplates until phrenic nerve stimulation failed to elicit EPPs in five consecutive endplates. This represented the statistical point where the majority of endplates are fully intoxicated, and few or no release sites remained competent to undergo vesicle fusion. Addition of either 3,4-DAP (2 µM) or GV-58 (50 µM; doses established in pilot dose-ranging studies) to BoNT/A-intoxicated preparations rapidly restored sub-threshold release, producing EPPs that were 3.49 ± 0.44 mV and 0.89 ± 0.11 mV, respectively (p < 0.01; Fig. 6). Surprisingly, combinatorial addition of 3,4-DAP plus GV-58 had a supra-additive effect in BoNT/A-intoxicated endplates, producing EPPs with amplitudes of 34.7 ± 5.19 mV (Fig. 6). In contrast, 3,4-DAP plus GV-58 had no apparent effect on diaphragms intoxicated by BoNT/E (Fig. 6), consistent with previous data suggesting that VGCC-mediated rescue was specific to BoNT/A-intoxicated synapses.

3,4-DAP plus GV-58 has superadditive effects in restoring robust EPP production in mouse diaphragm endplates fully intoxicated by BoNT/A. Representative evoked endplate recordings (top) and corresponding mean EPP amplitudes (in mV, bottom) from non-intoxicated and BoNT/A-intoxicated mouse phrenic nerve-hemidiaphragm preparations treated with vehicle, GV-58 (50 µM), 3,4-DAP (2 µM), or GV-58 plus 3,4-DAP (50 µM and 2 µM, respectively); and BoNT/E-intoxicated tissue treated with vehicle, or GV-58 plus 3,4-DAP. Representative traces are the average of six EPPs elicited from single endplates. Scale bars represent 25 ms (x-axis) and 5 mV (y-axis). Grouped data is presented as mean ± SEM (n = 4–6 diaphragm per condition; 5 endplates recorded per diaphragm). ***Indicates p < 0.001, ns = not significant.
Discussion
In this study, we investigated the cellular and molecular mechanisms responsible for 3,4-DAP-mediated rescue of neurotransmission with the goal of determining the conditions under which 3,4-DAP could be effective in restoring synaptic function. We found that 3,4-DAP rescue of synaptic release in intoxicated synapses was specifically mediated through VGCC activity. Furthermore, treatments that either increased Ca2+ influx through VGCCs or prolonged VGCC opening times were sufficient to rescue evoked and spontaneous release in BoNT/A-intoxicated neuron cultures. Functional rescue of neurotransmission was unique to BoNT/A and did not occur with serotypes D or E under any circumstance. The physiological relevance of these findings were confirmed in diaphragm NMJs, where combination treatment with 3,4-DAP and GV-58 proved synergistic, restoring putatively suprathreshold release to hemidiaphragms comprehensively paralyzed by BoNT/A.
The finding that rescue was unique to BoNT/A-intoxicated synapses is consistent with biochemical data suggesting that SNAP-25Δ9 remains competent to engage in or facilitate vesicle fusion27. In in vitro vesicle fusion studies, SNAP-25Δ9 directly engages with SYB1/2 and STX1 to mediate low-efficiency vesicle fusion. In contrast, vesicle fusion is abolished by substitution with the BoNT/E-cleavage product SNAP-25Δ26, illustrating that SNAP-25Δ9 and SNAP-25Δ26 have functionally distinct abilities to engage in neurotransmission. Deletion studies conducted in BoNT/A-intoxicated PC12 adrenal chromaffin cells found that overexpression of SNAP-25Δ9 restores low-level catecholamine release28. Collectively, these data suggest that SNAP-25Δ9 can directly participate in vesicle fusion, albeit with reduced efficiency compared to intact SNAP-25.
Although low-efficiency engagement of SNAP-25Δ9 in vesicle release provides a plausible mechanistic basis for rescue of neurotransmission in BoNT/A-intoxicated cultures, we cannot exclude the possibility that a compensatory release mechanism is activated in response to VGCC agonism. However, this putative mechanism must still rely on SNAP-25Δ9, otherwise rescue would have been observed in synapses intoxicated with BoNT/D or /E. Examples of such a mechanism could include direct stabilization of SNAP-25Δ9 in the fusogenic complex by coordination with Ca2+ ions or recruitment of an accessory protein. However, synthetic vesicles incorporating SYB, STX1 and SNAP-25Δ9 are capable of supporting vesicle fusion in the absence of Ca2+ or external protein factors, suggesting that neither Ca2+ nor Ca2+-regulated accessory proteins are required for fusion activity29. Alternatively, we propose that increased Ca2+ flux through activated VGCCs in the presence of 3,4-DAP, VGCC agonists or elevated extracellular Ca2+ promotes rescue of neurotransmission by prolonging the kinetics of release. Synaptic release is initiated by local increases in Ca2+, which cause the Ca2+ sensor synaptotagmin (SYT) to undergo a conformation change that permits assembly of SYB, STX1 and SNAP-25 into the fusogenic trans-SNARE configuration30,31. In the absence of Ca2+, SYT is a negative regulator of vesicle fusion, thus the duration of SYT permissiveness is determined by the magnitude and duration of local Ca2+ transients17. This leads us to hypothesize that 3,4-DAP and/or VGCC agonists effectively extend the temporal window during which SNARE-mediated fusion is molecularly permissible by increasing presynaptic Ca2+ transients. This would prolong the window during which less efficient fusion events can occur, such as those involving SNAP-25Δ9.
The ability of use-dependent VGCC agonists to rescue neurotransmission has therapeutic implications for stages of botulism other than complete paralysis. For example, most CNS neurons have 1–3 release sites per synaptic bouton32, whereas typical motor nerve terminals contain 800–1200 distinct release sites33. Continuous single-endplate recordings reveal the progressive depression of evoked release in response to BoNT intoxication34, illustrating that partially intoxicated endplates contain a mixture of intact and intoxicated release sites. Since 3,4-DAP treatment increases EPSC amplitudes in both non-intoxicated and BoNT/A-intoxicated synapses, we hypothesize that enhanced Ca2+ influx will increase neurotransmission from partially intoxicated motor nerve terminals through two distinct mechanisms: increased release probability at non-intoxicated release sites, which would occur in a serotype-independent fashion; and rescue of fusion at release sites associated with SNAP-25Δ9. If correct, Ca2+-mediated rescue of neurotransmission will then be influenced by factors such as the serotype involved and the extent of paralysis. These factors may contribute to the highly variable and conflicting results reported in the small number of clinical studies using aminopyridines as post-symptomatic treatments for serotype A and B botulism14,15,16,35.
Adult NMJs and CNS synapses are believed to predominantly rely on N-type (Cav2.2) and P/Q-type (Cav2.1) VGCCs for synaptic transmission23,24. These two VGCC subtypes incorporate synaptic protein interaction domains that interact with Stx1 and SNAP-25, placing the channels in close proximity to docked synaptic vesicles, and providing a structural association between voltage-gated Ca2+ influx and synaptic vesicle fusion36,37. Although N- and P/Q-type VGCCs are often co-located within the presynaptic compartment, they appear to have functional distinctions. For example, in detonator synapses N-type VGCCs contribute to multivesicular release at specific release sites, whereas P/Q-type VGCCs drive widespread synaptic release38. In contrast, neuronal L-type VGCCs (Cav1.2 through Cav1.4) have been proposed to gate Ca2+-release from internal Ca2+ stores, activating signaling processes that indirectly modulate presynaptic function39,40. L-type VGCCs are primarily believed to mediate somatic Ca2+ transients41,42, although several studies suggest that they also directly contribute to neurotransmission43,44. While our data indicate that L- or N/P/Q-type VGCC agonists can promote release from naïve as well as BoNT/A-intoxicated CNS synapses, we did not determine whether release is exclusively mediated by direct modulation of Ca2+ transients at release sites, or whether indirect Ca2+ signaling mechanisms are also involved. Moreover, since central synapses, motor endplates and autonomic synapses are likely to exhibit morphological and functional differences in VGCC activity, VGCC agonists may have synapse type-specific effects on rescue of neurotransmission.
Although aminopyridines have been studied as BoNT countermeasures for decades, their clinical potential has been compromised by adverse CNS effects at presumptive therapeutic doses45,46. Therefore, there is interest in reducing the central access of 3,4-DAP, or alternatively in identifying co-treatments that allow reduction in 3,4-DAP concentrations to tolerable levels while retaining or improving therapeutic efficacy47,48. While the combination of VGCC agonists and 3,4-DAP appeared very robust in rescuring synaptic release, direct modulation of Ca2+ influx carries the risk of neurotoxic responses. For example, excessive cytosolic Ca2+ levels can activate compensatory changes in presynaptic function that reduce Ca2+ sensitivity49 or cause presynaptic Ca2+ toxicity50. However, most VGCC agonists are use-dependent, enhancing Ca2+ influx predominantly during action potential firing51,52. This results in the temporally constrained influx of Ca2+ at specific subcellular compartments, as opposed to tonic Ca2+ currents. For similar reasons, combinatorial use of 3,4-DAP and GV-58 is under preclinical evaluation as a treatment for myasthenic diseases53. However, it should be noted that although both FPL and GV-58 produced encouraging in vitro results as BoNT/A treatments, in vivo use of both drugs is limited by their poor aqueous solubility and there is a need for derivatives with improved pharmacokinetic and/or pharmacodynamics profiles.
In conclusion, we demonstrate that enhanced Ca2+ influx through VGCCs restores neurotransmission to BoNT/A-intoxicated synapses. Our findings suggest that co-administration of use-dependent VGCC agonists with 3,4-DAP may increase the safety and efficacy of treatments to prolong survival during the acute phase of botulism, or alternatively, to accelerate recovery of muscle function during the chronic phase of botulism.
Experimental procedures
Animal use statement
The experimental protocol was approved by the Animal Care and Use Committee at the United States Army Medical Research Institute of Chemical Defense (USDA certificate number 51-F-0006). All procedures were conducted in accordance with the principles stated in the Guide for the Care and Use of Laboratory Animals and the Animal Welfare Act of 1966 (P.L. 89–544), as amended.
Reagents/Supplies
BoNT/A1 (2.5 × 108 MLD50/mg), BoNT/D1 (2.8 × 107 MLD50/mg) and BoNT/E1 (3.0 × 107 MLD50/mg) were purchased from Metabiologics (Madison, WI, USA). For BoNT/E, toxin was activated by a 60 min incubation at 37 °C with 0.3 mg/mL TPCK-treated trypsin in 0.05 M sodium phosphate buffer (pH 6.5)19. ω-agatoxin IVA, ω-conotoxin MVIIC and GV-58 were purchased from Alomone Labs (Jerusalem, Israel). FPL 64176 was purchased from Tocris Bioscience (Bristol, UK). All reagents for electrophysiology buffers were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Neuronal culture and BoNT intoxication
Fresh E18 rat hippocampal, cortical and ventricular zone tissues were obtained from BrainBits (Springfield, IL, USA), dissociated according to the manufacturer’s instructions, and plated at a density of 125,000 cells/cm2 on polyethylenimine/laminin-coated glass coverslips (Sigma-Aldrich). Neuronal cultures were maintained at 5% CO2, 37 °C, and 95% humidity in NbActiv4 medium (BrainBits). Experiments were performed 16 to 24 days after plating. For neuronal intoxication, BoNT/A, BoNT/D or BoNT/E were prepared at 100x final concentration in fresh NbActiv4 medium and added to neuron cultures at 100 pM. Neurons were analyzed 20–24 hours after addition of BoNT.
Whole-cell patch clamp electrophysiology
Experiments were performed to record spontaneous AMPAR-mediated miniature excitatory post-synaptic currents (mEPSCs) as previously described19. Briefly, neurons were bathed in extracellular recording buffer containing (in mM): 140 NaCl, 3.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, 10 Glucose, 10 HEPES, 5 TTX, 20 bicuculline, and 10 MK-801 (pH = 7.3; 315 ± 10 mOsm/Kg). Patch pipettes (5–10 MΩ) were fabricated from borosilicate glass (Sutter Instruments, Novato, CA) and filled with an intracellular recording buffer containing (in mM): 140 K-gluconate, 5 NaCl, 2 Mg-ATP, 0.5 Li-GTP, 0.1 CaCl2, 1 MgCl2, 1 EGTA and 10 HEPES (pH = 7.3; 315 ± 10 mOsm/Kg). Whole-cell −70 mV voltage-clamp recordings were collected at 20–22 °C with an EPC10 (Heka, Lambrecht/Pfalz, Germany) and Heka Patchmaster 2.53 software. mEPSC frequency were detected from whole-cell recordings using Mini-Analysis v6 (Synaptosoft Inc., Fort Lee, NJ).
For evoked AMPAR-medicated excitatory post-synaptic currents (eEPSCs), neurons were bathed in extracellular recording buffer without TTX. Recording electrodes were filled with an intracellular recording buffer containing (in mM): 140 CsCl, 5 NaCl, 2 Mg-ATP, 0.5 Li-GTP, 0.1 CaCl2, 1 MgCl2, 1 EGTA, 10 HEPES, and 10 QX-314 (pH 7.3; 315 ± 10 mOsm/Kg). After establishing whole-cell configuration, the intracellular solution was allowed to equilibrate for 2 min. A concentric bipolar stimulating electrode (FHC, Bowdoin, ME) was placed 100–150 µm from the neuron to be recorded and a train of 10 stimulations (1.0 ms, 0.9 mA, 0.1 Hz) was applied to stimulate presynaptic fibers. The mean amplitude of the first three EPSCs was calculated using Axograph X software (Axograph.com).
Endplate electrophysiology
For ex vivo diaphragm studies, Male C57BL/6 mice (6–10 wk; Jackson Labs, Bar Harbor, ME) were group-housed and provided a standard diet with regular enrichment and water ad libitum. Mice were thoroughly anesthetized using 5% isoflurane and euthanized by decapitation. Diaphragm muscles and corresponding phrenic nerves were isolated from euthanized mice by dissection at 22–24 °C in Tyrode’s solution (137 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 1 mM MgSO4, 24 mM NaHCO3, 1 mM NaH2PO4 and 11 mM D-glucose, pH 7.4). After dissection, muscles were attached with dissection pins to a 10-mm tissue culture dish containing Sylgard (Dow Corning; Midland, MI, USA) and the preparation was perfused with oxygenated Tyrode’s. Muscle viability was verified using a 0.2 ms squarewave of direct current at suprathreshold amplitudes to the phrenic nerve using a bipolar stimulating electrode driven by a stimulation isolation unit (Digitimer North America; Ft. Lauderdale, FL, USA).
Muscle potentials were recorded on a HEKA Elektronik EPC10 patch clamp amplifier using sharp glass electrodes (10–20 MΩs) pulled with a Sutter Instrument P1000. Muscles fibers were impaled close to endplate junctions and recordings with a starting resting membrane potential (RMP) > −60 mV were rejected. Muscle contraction was selectively blocked with 1 μM μ-conotoxin GIIIB, which preferentially blocks muscle-specific voltage-activated Na+ channels (Alamone Labs). For all experimental conditions, recordings were performed at 22–24 C in perfused oxygenated Tyrode’s. EPPs were elicited by suprathreshold phrenic nerve stimulation with a square wave of constant current for 0.2 ms at 0.05 Hz. EPP amplitudes were averaged from 6 consecutive stimulations per muscle fiber and corrected for non-linear summation using the following conversion: EPP /1-(0.8 * EPP /(Vm - Vrev))54. For this correction we used experimentally determined values of Vm (the resting membrane potential) and 0 mV for the Vrev (reversal potential). For each hemidiaphragm, corrected EPP amplitudes from 5 endplates were averaged to obtain a single value.
Naïve recordings were performed after 30 minutes of equilibration post-dissection. Hemidiaphragms were intoxicated by bath addition of 670 pM of BoNT/A, or 210 pM BoNT/E coupled with suprathreshold phrenic nerve stimulation at 0.5 Hz for 30 min, followed by persistent 0.05 Hz stimulation. These concentrations and conditions were sufficient to produce rapid and comprehensive synapse failure within 3–4 hours. Comprehensive intoxication was determined to be the point where consistent stimulation failure was observed in six stimulations (0.05 Hz) per endplate, observed across 5 consecutive endplates. To characterize the effects of 3,4-DAP and GV-58 on endplate function, the hemidiaphragm preparations were incubated with 2 µM of 3,4-DAP and/or 50 µM GV-58 in oxygenated Tyrode’s for 45 minutes prior to recordings.
For all electrophysiology experiments, current measurements were filtered online at 2.9 kHz and digitized at 10 kHz. Data analysis and graphing were performed in Prism v6.1 (GraphPad software, La Jolla, CA).
Western blot analysis
Protein from neuronal cultures was harvested, quantified by BCA (Pierce), separated by SDS-PAGE and transferred to PVDF. Blots were stained and imaged as previously described19. Primary antibodies used for Western blotting included SNAP-25 (Covance, Princeton, NJ), STX1, and SYB2 (Synaptic Systems, Gottingen, Germany). Blots were fluorescently probed using a goat anti-mouse secondary conjugated to Alexa Fluor 488 (Invitrogen, Carlsbad, CA) for detection of STX1 (33 kDa), SYB2 (12 kDa) and SNAP-25 (18, 24 or 25 kDa).
Statistical analyses
For each patched neuron, evoked amplitudes were averaged among the first three consecutive stimulations to produce the mean evoked current per neuron (in pA). Mean evoked amplitude values were then averaged among neurons within each condition and presented as percentages of age- and lot-matched control neurons. mEPSC frequencies were calculated by determining the mean frequency (in Hz) of events measured during 180 s of continuous voltage-clamp recording. Spontaneous release rates measured in BoNT-intoxicated neurons were normalized to mean frequencies observed in age- and lot-matched, vehicle-treated neurons and presented as percentages of control values. Statistical significance among means was calculated either using Student’s t-test analysis (for two-sample comparisons), 1-way ANOVA testing (significance from control determined using the Dunnett’s test against the vehicle control population), or 1-way ANOVA testing with Bonferroni’s multiple comparison test. Quantitative data are presented as mean ± the standard error of the mean, with statistical significance listed, or represented using the following indicators: * indicates p < 0.05; ** indicates p < 0.01; *** indicates p < 0.001.
Data availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information files).
References
Schiavo, G., Rossetto, O. & Montecucco, C. Clostridial neurotoxins as tools to investigate the molecular events of neurotransmitter release. Semin Cell Biol 5, 221–229 (1994).
Simpson, L. L. Identification of the major steps in botulinum toxin action. Annual review of pharmacology and toxicology 44, 167–193 (2004).
Montal, M. Translocation of botulinum neurotoxin light chain protease by the heavy chain protein-conducting channel. Toxicon: official journal of the International Society on Toxinology 54, 565–569, https://doi.org/10.1016/j.toxicon.2008.11.018 (2009).
Chertow, D. S. et al. Botulism in 4 adults following cosmetic injections with an unlicensed, highly concentrated botulinum preparation. Jama 296, 2476–2479, https://doi.org/10.1001/jama.296.20.2476 (2006).
Arnon, S. S., Schechter, R., Maslanka, S. E., Jewell, N. P. & Hatheway, C. L. Human botulism immune globulin for the treatment of infant botulism. N Engl J Med 354, 462–471, https://doi.org/10.1056/NEJMoa051926 (2006).
Larsen, J. C. Botulinum Neurotoxin (BoNT) therapeutics: Time to think outside the BoNT. The Botulinum Journal 1, 261–269 (2009).
Smith, T., Roxas-Duncan, V. & Smith, L. Botulinum Neurotoxins as Biothreat Agents. Journal of Bioterrorism and Biodefense S2–003 (2012).
Dreyer, F., Rosenberg, F., Becker, C., Bigalke, H. & Penner, R. Differential effects of various secretagogues on quantal transmitter release from mouse motor nerve terminals treated with botulinum A and tetanus toxin. Naunyn Schmiedebergs Arch Pharmacol 335, 1–7 (1987).
Capogna, M., McKinney, R. A., O’Connor, V., Gahwiler, B. H. & Thompson, S. M. Ca2 + or Sr2 + partially rescues synaptic transmission in hippocampal cultures treated with botulinum toxin A and C, but not tetanus toxin. J Neurosci 17, 7190–7202 (1997).
Keller, J. E. & Neale, E. A. The role of the synaptic protein snap-25 in the potency of botulinum neurotoxin type A. The Journal of biological chemistry 276, 13476–13482, https://doi.org/10.1074/jbc.M010992200 (2001).
Lundh, H., Leander, S. & Thesleff, S. Antagonism of the paralysis produced by botulinum toxin in the rat. The effects of tetraethylammonium, guanidine and 4-aminopyridine. J Neurol Sci 32, 29–43 (1977).
Thomsen, R. H. & Wilson, D. F. Effects of 4-aminopyridine and 3,4-diaminopyridine on transmitter release at the neuromuscular junction. J Pharmacol Exp Ther 227, 260–265 (1983).
Lin-Shiau, S. Y., Day, S. Y. & Fu, W. M. Use of ion channel blockers in studying the regulation of skeletal muscle contractions. Naunyn Schmiedebergs Arch Pharmacol 344, 691–697 (1991).
Davis, L. E., Johnson, J. K., Bicknell, J. M., Levy, H. & McEvoy, K. M. Human type A botulism and treatment with 3,4-diaminopyridine. Electromyogr Clin Neurophysiol 32, 379–383 (1992).
Dock, M. et al. Treatment of severe botulism with 3,4-diaminopyridine. Presse Med 31, 601–602 (2002).
Friggeri, A. et al. 3,4-Diaminopyridine may improve neuromuscular block during botulism. Crit Care 17, 449, https://doi.org/10.1186/cc12880 (2013).
Augustine, G. J. How does calcium trigger neurotransmitter release? Curr Opin Neurobiol 11, 320–326 (2001).
Maximov, A., Pang, Z. P., Tervo, D. G. & Sudhof, T. C. Monitoring synaptic transmission in primary neuronal cultures using local extracellular stimulation. J Neurosci Methods 161, 75–87, https://doi.org/10.1016/j.jneumeth.2006.10.009 (2007).
Beske, P. H. et al. Botulinum and Tetanus Neurotoxin-Induced Blockade of Synaptic Transmission in Networked Cultures of Human and Rodent Neurons. Toxicological sciences: an official journal of the Society of Toxicology 149, 503–515, https://doi.org/10.1093/toxsci/kfv254 (2016).
Del Castillo, J. & Katz, B. Quantal components of the end-plate potential. J Physiol 124, 560–573 (1954).
Bennett, M. R., Gibson, W. G. & Robinson, J. Probabilistic secretion of quanta: spontaneous release at active zones of varicosities, boutons, and endplates. Biophys J 69, 42–56, https://doi.org/10.1016/S0006-3495(95)79873-3 (1995).
Beske, P. H., Scheeler, S. M., Adler, M. & McNutt, P. M. Accelerated intoxication of GABAergic synapses by botulinum neurotoxin A disinhibits stem cell-derived neuron networks prior to network silencing. Front Cell Neurosci 9, 159, https://doi.org/10.3389/fncel.2015.00159 (2015).
Uchitel, O. D. et al. P-type voltage-dependent calcium channel mediates presynaptic calcium influx and transmitter release in mammalian synapses. Proc Natl Acad Sci USA 89, 3330–3333 (1992).
Protti, D. A., Reisin, R., Mackinley, T. A. & Uchitel, O. D. Calcium channel blockers and transmitter release at the normal human neuromuscular junction. Neurology 46, 1391–1396 (1996).
Dreyer, F. & Schmitt, A. Transmitter release in tetanus and botulinum A toxin-poisoned mammalian motor endplates and its dependence on nerve stimulation and temperature. Pflugers Archiv: European journal of physiology 399, 228–234 (1983).
Molgo, J. et al. Presynaptic actions of botulinal neurotoxins at vertebrate neuromuscular junctions. Journal de physiologie 84, 152–166 (1990).
Lu, B. The destructive effect of botulinum neurotoxins on the SNARE protein: SNAP-25 and synaptic membrane fusion. PeerJ 3, e1065, https://doi.org/10.7717/peerj.1065 (2015).
O’Sullivan, G. A., Mohammed, N., Foran, P. G., Lawrence, G. W. & Oliver Dolly, J. Rescue of exocytosis in botulinum toxin A-poisoned chromaffin cells by expression of cleavage-resistant SNAP-25. Identification of the minimal essential C-terminal residues. The Journal of biological chemistry 274, 36897–36904 (1999).
Holt, M., Riedel, D., Stein, A., Schuette, C. & Jahn, R. Synaptic vesicles are constitutively active fusion machines that function independently of Ca2+. Curr Biol 18, 715–722, https://doi.org/10.1016/j.cub.2008.04.069 (2008).
Sudhof, T. C. Calcium control of neurotransmitter release. Cold Spring Harb Perspect Biol 4, a011353, https://doi.org/10.1101/cshperspect.a011353 (2012).
Zhou, Q. et al. Architecture of the synaptotagmin-SNARE machinery for neuronal exocytosis. Nature 525, 62–67, https://doi.org/10.1038/nature14975 (2015).
Biro, A. A., Holderith, N. B. & Nusser, Z. Quantal size is independent of the release probability at hippocampal excitatory synapses. J Neurosci 25, 223–232, https://doi.org/10.1523/JNEUROSCI.3688-04.2005 (2005).
Meriney, S. D. & Dittrich, M. Organization and function of transmitter release sites at the neuromuscular junction. J Physiol 591, 3159–3165, https://doi.org/10.1113/jphysiol.2012.248625 (2013).
Gundersen, C. B., Katz, B. & Miledi, R. The reduction of endplate responses by botulinum toxin. Proc R Soc Lond B Biol Sci 213, 489–493 (1981).
Siegel, L. S. & Price, J. I. Ineffectiveness of 3,4-diaminopyridine as a therapy for type C botulism. Toxicon: official journal of the International Society on Toxinology 25, 1015–1018 (1987).
Neher, E. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron 20, 389–399 (1998).
Haydon, P. G., Henderson, E. & Stanley, E. F. Localization of individual calcium channels at the release face of a presynaptic nerve terminal. Neuron 13, 1275–1280 (1994).
Chamberland, S., Evstratova, A. & Toth, K. Short-Term Facilitation at a Detonator Synapse Requires the Distinct Contribution of Multiple Types of Voltage-Gated Calcium Channels. J Neurosci 37, 4913–4927, https://doi.org/10.1523/JNEUROSCI.0159-17.2017 (2017).
Berridge, M. J., Bootman, M. D. & Roderick, H. L. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4, 517–529, https://doi.org/10.1038/nrm1155 (2003).
Liu, Y., Dore, J. & Chen, X. Calcium influx through L-type channels generates protein kinase M to induce burst firing of dopamine cells in the rat ventral tegmental area. The Journal of biological chemistry 282, 8594–8603, https://doi.org/10.1074/jbc.M610230200 (2007).
Catterall, W. A. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol 3, a003947, https://doi.org/10.1101/cshperspect.a003947 (2011).
Simms, B. A. & Zamponi, G. W. Neuronal voltage-gated calcium channels: structure, function, and dysfunction. Neuron 82, 24–45, https://doi.org/10.1016/j.neuron.2014.03.016 (2014).
Flink, M. T. & Atchison, W. D. Passive transfer of Lambert-Eaton syndrome to mice induces dihydropyridine sensitivity of neuromuscular transmission. J Physiol 543, 567–576 (2002).
Losavio, A. & Muchnik, S. Spontaneous acetylcholine release in mammalian neuromuscular junctions. Am J Physiol 273, C1835–1841 (1997).
Lemeignan, M., Millart, H., Lamiable, D., Molgo, J. & Lechat, P. Evaluation of 4-aminopyridine and 3,4-diaminopyridine penetrability into cerebrospinal fluid in anesthetized rats. Brain Res 304, 166–169 (1984).
Sanders, D. B. 3,4-Diaminopyridine (DAP) in the treatment of Lambert-Eaton myasthenic syndrome (LEMS). Annals of the New York Academy of Sciences 841, 811–816 (1998).
Harris, T. L. et al. Lycopodium clavatum exine microcapsules enable safe oral delivery of 3,4-diaminopyridine for treatment of botulinum neurotoxin A intoxication. Chem Commun (Camb) 52, 4187–4190, https://doi.org/10.1039/c6cc00615a (2016).
Zakhari, J. S. et al. Formulating a new basis for the treatment against botulinum neurotoxin intoxication: 3,4-Diaminopyridine prodrug design and characterization. Bioorg Med Chem 19, 6203–6209, https://doi.org/10.1016/j.bmc.2011.09.019 (2011).
Scott, R. Use-dependent control of presynaptic calcium signalling at central synapses. J Anat 210, 642–650, https://doi.org/10.1111/j.1469-7580.2007.00728.x (2007).
Harris, R. J., Symon, L., Branston, N. M. & Bayhan, M. Changes in extracellular calcium activity in cerebral ischaemia. J Cereb Blood Flow Metab 1, 203–209, https://doi.org/10.1038/jcbfm.1981.21 (1981).
Fan, J. S., Yuan, Y. & Palade, P. Kinetic effects of FPL 64176 on L-type Ca2+ channels in cardiac myocytes. Naunyn Schmiedebergs Arch Pharmacol 361, 465–476 (2000).
Tarr, T. B. et al. Evaluation of a novel calcium channel agonist for therapeutic potential in Lambert-Eaton myasthenic syndrome. J Neurosci 33, 10559–10567, https://doi.org/10.1523/JNEUROSCI.4629-12.2013 (2013).
Tarr, T. B. et al. Complete reversal of Lambert-Eaton myasthenic syndrome synaptic impairment by the combined use of a K+ channel blocker and a Ca2+ channel agonist. J Physiol 592, 3687–3696, https://doi.org/10.1113/jphysiol.2014.276493 (2014).
McLachlan, E. M. & Martin, A. R. Non-linear summation of end-plate potentials in the frog and mouse. J Physiol 311, 307–324 (1981).
Acknowledgements
We thank Megan Lyman, Sydney Mason, Marc Cerasoli and Celinia Ondeck for technical assistance; Cindy Kronman for editorial review and the Geneva Foundation for contractual assistance. This work was conducted under funding from the Defense Threat Reduction Agency – Joint Science and Technology Office, Medical S&T Division (grant number CBM.THRTOX.01.10.RC.021); National Institute of Allergy and Infectious Diseases (IAA AOD12058-0001-0000 and R01 5R01AI093504). This research was performed while P.B. and K.H. held Defense Threat Reduction Agency-National Research Council Research Associateship awards; and J.M. and M.E. held Oak Ridge Institute of Science and Engineering Fellowship awards. The views expressed in this article are those of the authors and do not reflect the official policy of the Department of Army, Department of Defense, or the U.S. Government.
Author information
Authors and Affiliations
Contributions
P.B. and K.H. conducted all in vitro electrophysiology experiments, analyzed the results, prepared figures, and contributed to all sections of the manuscript. J.M. conducted diaphragm endplate recordings, analyzed the results, prepared figures, and contributed to all sections of the manuscript. M.E. conducted western blot studies, contributed to preparation of figures and wrote methods. P.M. conceived the idea for the project, designed experiments, and contributed to all section of the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Beske, P.H., Hoffman, K.M., Machamer, J.B. et al. Use-dependent potentiation of voltage-gated calcium channels rescues neurotransmission in nerve terminals intoxicated by botulinum neurotoxin serotype A. Sci Rep 7, 15862 (2017). https://doi.org/10.1038/s41598-017-16064-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-16064-3
This article is cited by
-
Antidotal treatment of botulism in rats by continuous infusion with 3,4-diaminopyridine
Molecular Medicine (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
