
Abstract
Spermatogenesis is a multifactorial process that forms differentiated sperm cells in a complex microenvironment. This process involves the genome, epigenome, transcriptome, and proteome to ensure the stability of the spermatogonia and supporting cells. The identification of signaling pathways linked to infertility has been hampered by the inherent complexity and multifactorial aspects of spermatogenesis. Systems biology is a promising approach to unveil underlying signaling pathways and genes and identify putative biomarkers. In this study, we analyzed thirteen microarray libraries of infertile humans and mice, and different classes of male infertility were compared using differentially expressed genes and functional enrichment analysis. We found regulatory processes, immune response, glutathione transferase and muscle tissue development to be among the most common biological processes in up-regulated genes, and genes involved in spermatogenesis were down-regulated in maturation arrest (MArrest) and oligospermia cases. We also observed the overexpression of genes involved in steroid metabolism in post-meiotic and meiotic arrest. Furthermore, we found that the infertile mouse model most similar to human MArrest was the Dazap1 mutant mouse. The results of this study could help elucidate features of infertility etiology and provide the basis for diagnostic markers.
Similar content being viewed by others


Introduction
Infertility is defined as the inability to have children after one year of unprotected sexual intercourse1. Ten to fifteen percent of couples face infertility, which is related to male factors in almost 50% of cases2. The most common causes of male infertility are varicocele (37–40%), endocrine disorders (>20%), genital duct infection (8–35%), testicular defects (9%), genetic problems (15–30%), antisperm antibodies (8–19%), and idiopathic male infertility (15–25%)3,4,5.
Semen deficiencies in male infertility are often characterized as 1) oligospermia, in which there are fewer than 15 million sperm cells per milliliter, 2) azoospermia, which is the absence of sperm in ejaculate and which can be categorized into two major classes, obstructive azoospermia (OA) and non-obstructive azoospermia (NOA), 3) teratospermia, a condition in which less than 4% of sperm are morphologically normal, and 4) asthenospermia, which is when sperm have motility problems6. Idiopathic male infertility is a complicated condition with abnormal semen parameters that probably has a genetic basis7,8. Some cases are classified as “unexplained male infertility,” in which all characteristics of routine semen analysis and sexual history are normal9. Despite abundant studies, the origins of many infertility cases are still not known because spermatogenesis is a multifactorial and complex process. The cause of 21–29% of azoospermia cases is related to genetic factors, and 12–41% of azoospermia cases are idiopathic azoospermia10. The genetic basis of azoospermia involves numerous causes, such as abnormality, single and multiple gene disorders and epigenetics, and Y chromosome defects have a major role in male infertility10.
NOA patients go through four stages, such as pre-meiotic arrest (PreMA), meiotic arrest (MA), post-meiotic arrest (PostMA) and sertoli cell only syndrome (SCOS)11. Unlike NOA cases, we do not have enough information about the transcriptome of testis tissue for oligospermia and teratospermia cases because they are not a candidate for testis biopsy. Our knowledge about these cases should be based on the genome, the transcriptome of sperm cells, the genome of somatic cells of infertile men and the testis tissue of infertile mouse mutants12. Furthermore, unlike oligospermia and teratospermia cases, NOA cases are unable to create sperm, so we do not know about the transcriptome and proteome of NOA sperm13.
Based on current knowledge, the spermatogenesis process is regulated by 1500 to 2000 genes, and every alteration in these genes may disturb fertility12,14. Several studies have investigated the biology of spermatogenesis and identified many key genes involved in spermatogenesis pathways. There are some comprehensive reviews about the dependency among the genome, epigenome, transcriptome and proteome15 and the genes and pathways involved in male infertility8,12,16. High throughput technologies, such as gene expression profiling assays, have been extensively applied to investigate the molecular mechanisms associated with male infertility17. In 2006, in one of the first microarray experiments on SCOS and MA cases, 10 novel genes were identified that had been down-regulated in male infertility cases18. In 2008, Okada et al. revealed differentially expressed genes (DEGs) in NOA cases, investigated the top 10 biological processes (BP) of gene ontology (GO) terms for separate up- and down-regulated gene lists, and suggested some novel therapeutic targets for NOA treatment19. A transcriptome analysis of NOA and hypospermatogenesis (HS) (with and without AZFc [azoospermia factor c] region deletion) by Gatta et al. revealed that the transcripts of all cases with AZFc deletion were clustered together independently from the phenotype of testes (SCOS or HS). Furthermore, the transcripts of half of the idiopathic HS cases were clustered with the AZFc deletion cases, and many of the genes with post-meiotic functions were down-regulated in AZFc deletion cases20. In 2010, Saito et al. studied the microarray data of Ing2 knockdown (KD) mouse testes and showed that Ing2 plays a crucial role in spermatogenesis21. In 2013, Malcher et al. extracted the DEGs of PreMA, MA, PostMA and SCOS and focused on the expression of genes involved in the immune system22,23. In another study in 2015, Bansal et al. analyzed the GO of mixed up- and down-regulated genes in sperm gene expression profiles of idiopathic oligospermia and asthenospermia24. A bioinformatics analysis of four microarray datasets of NOA testes was conducted by Ansari-Pour et al. in 2016. They reconstructed a protein-protein interaction network of spermatogenic failure genes with a Y-centric focus base of DEGs25. A gene set enrichment analysis (GSEA) establishes whether an a priori defined set of genes shows statistically significant differences between two biological states or phenotypes26.
In this study, we elicited the DEGs involved in each type of human male infertility and multiple genes involved in certain infertile mouse mutants and several stages of arresting in meiosis and SCOS. Then GO and KEGG pathway analyses were performed on the DEGs. Furthermore, we performed a GSEA for each type of male infertility for humans and mice to discover the most important gene sets in male infertility. This study is a step toward finding a diagnostic biomarker for male infertility and could help explain the etiology of male infertility.
Results
DEGs and pathway analysis in maturation arrest azoospermia (MArrest), oligospermia and teratospermia
We extracted 597, 154 and 283 up-regulated genes and 525, 144 and 292 down-regulated genes from the libraries of MArrest, oligospermia and teratospermia (MArrest-oligo-terato-spermia), respectively (Supplementary Table S1). We found 26 up-regulated miRNAs for MArrest (miR-15A, miR-18a, miR-21, miR-23b, miR-27b, miR29c, miR-30e, miR-31, miR-32, miR-99a, miR-99AHG, miR-128-1, miR-107, miR-145, miR-154, miR-186, miR-197, miR-199a-2, miR-214, miR-218-1, miR-503, miR-509, miR-LET7A2, miR-LET7C, miR-LET7F1 and miR-LET7G), two up-regulated miRNAs for teratospermia (miR-9-2 and miR-181A2HG) and one down-regulated miRNA for teratospermia (miR-6805). There was no common up-regulated gene among MArrest-oligo-terato-spermia disorders, but five down-regulated genes (CLGN, KRT23, PCYT2, SMCP and SPINK2) were common among MArrest-oligo-terato-spermia disorders.
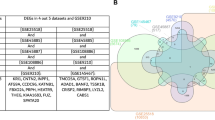
In up-regulated genes, the maximum similarity was between MArrest and teratospermia, with 12 common genes among 880 genes (0.014%) (MLC1, LSP1, GRK5, BCORL1, DICER1, CCNG1, RBMS3, DCN, RPL6, ADAMTS5, HNRNPU and PDE1A). Five genes (SPARCL1, ABI3BP, PMP22, DTNA and RPS6KA3) were common between MArrest and oligospermia among 751 genes (0.007%), and three genes (POU2F3, SRGAP2C and TERF1) were common between oligospermia and teratospermia among 437 genes (0.007%), as shown in Fig. 1a.

Venn diagram of similarities of DEGs, GO, KEGG pathway enrichment analysis between MArrest-oligo-terato-spermia. (a) Common up-regulated genes which the most similarity was between MArrest-terato-spermia (12 mutual genes). (b) Common down-regulated genes. The most similarity was between MArrest-oligo-spermia (27 mutual genes). (c) Common up-regulated BP that the common genes between MArrest-oligo-spermia were about regulatory process, immune system process and muscle tissue development. (d) Common down-regulated BP which mostly was between MArrest- oligo-spermia by spermatogenesis process. (e) Mutual up-regulated CC which common CC between MArrest- oligo-spermia related to plasma membrane. (f) Common down-regulated CC. (g,h) Common up- and down-regulated MF. (i,j) Common up- and down-regulated KEGG pathways.
In down-regulated genes, five genes among 960 genes (0.005%) were common in all three MArrest-oligo-terato-spermia disorders. The maximum similarity was between MArrest and oligospermia, with 27 common genes among 668 genes (0.04%) (ACSBG2, ACTL7B, ACTRT2, ALS2CR11, ANKRD7, ARMC12, C11orf71, C22orf23, CCDC54, CENPU, CFAP74, CRISP2, DMRTB1, FSCB, GK2, GKAP1, IRGC, LINC00467, MS4A6E, OLAH, PCSK4, PLCZ1, RIMBP3, SLC25A51, SOX5, SPATA22 and TSSK2). Eight genes among 817 genes (0.01%) (ADAM32, DYNLRB2, KLHL10, RIMBP3C, SEPT12, TIMD4, TSACC and TTC25) were common between MArrest and teratospermia, and three genes among 435 genes (0.007%) (HRASLS, LIMS3 and MRPL42) were common between oligospermia and teratospermia. The common down-regulated genes are listed in Fig. 1b.
We found 541 BP terms for MArrest up-regulated genes, 122 BP terms for oligospermic up-regulated genes and 98 BP terms for teratospermic up-regulated genes. There were 155, 44 and 147 common BP terms for MArrest, oligospermia and teratospermia down-regulated genes, respectively (p-value ≤ 0.01) (Supplementary Table S2).
In the up-regulated gene lists for BPs, one BP was common among all MArrest-oligo-terato-spermia disorders, 18 BPs were common between MArrest and oligospermia, 20 BP terms were common between MArrest and teratospermia, and four BPs were common between oligospermia and teratospermia, as can be seen in Fig. 1c.
As observed in Fig. 1d, in the down-regulated gene lists for BPs, one BP term was common among all MArrest-oligo-terato-spermia disorders, 10 BP terms were common between MArrest and oligospermia, eight BPs were common between MArrest and teratospermia, and two BP terms were common between oligospermia and teratospermia.
We investigated 66, 9 and 14 cellular component (CC) terms for up-regulated genes and 42, 8 and 57 CC terms for down-regulated genes of MArrest, oligospermia and teratospermia, respectively (Supplementary Table S2). In the up-regulated gene lists for CC, as depicted in Fig. 1e, one CC term was common between MArrest and oligospermia, one CC was common between MArrest and teratospermia, and one CC was common between oligospermia and teratospermia. In the down-regulated gene lists for CC, two CC terms were common between MArrest and oligospermia, three CCs were common between MArrest and teratospermia, and one CC term was common between oligospermia and teratospermia (see Fig. 1f).
We found 62, 7 and 26 molecular function (MF) terms for up-regulated genes and 33, 8 and 39 MF terms for down-regulated genes of MArrest, oligospermia and teratospermia, respectively (Supplementary Table S2). In the up-regulated gene lists for MF,as shown in Fig. 1g, one MF term was common among MArrest-oligo-spermia disorders. In the down-regulated gene lists for MF, one MF term was common between MArrest and teratospermia (see Fig. 1h). In the up-regulated gene lists for the KEGG pathway, as depicted in Fig. 1i, one pathway was common between MArrest and oligospermia (type I diabetes mellitus). In the down-regulated gene lists for the KEGG pathway, as illustrated in Fig. 1j, two pathways (fatty acid biosynthesis and metabolic pathways) were common between MArrest and oligospermia, one pathway was common between oligospermia and teratospermia (Propanoate metabolism), and one pathway was common between MArrest and teratospermia (Huntington’s disease).
DEGs and pathway analysis in PostMA, MA and SCOS
We extracted 152, 255 and 310 top up-regulated genes and 163, 255 and 309 top down-regulated genes from the libraries of different stages of NOA: PostMA, MA and SCOS, respectively (Supplementary Table S3). For the up-regulated genes, 21 genes among 717 genes (0.029%) were common among PostMA, MA and SCOS, and half of these genes were miRNA (LOC100130428, LOC100131541, MALAT1, MGC24103, miR-145, miR-199a-2, miR-21, miR-27b, miR-30e, miR-32, miR-99a, miR-LET7A2, miR-LET7C, miR-LET7G, PP12719, PWAR6, SNX2, TET2, ZEB2, ZNF189 and ZNF737). The maximum number of common genes was found between PostMA and MA, with 45 up-regulated genes among 407 genes (0.11%). Fourteen up-regulated genes among 565 genes (0.025%) were common between MA and SCOS, and five up-regulated genes among 462 genes (0.011%) were common between PostMA and SCOS. The common up-regulated genes have been shown in Fig. 2a. In down-regulated genes, 17 genes among 727 genes (0.023%) were common among PostMA, MA and SCOS, 45 genes among 564 genes (0.08%) were common between MA and SCOS, and 42 genes among 418 genes (0.1%) were common between PostMA and MA, as shown in Fig. 2b. We found 82 BP terms for PostMA up-regulated genes, 152 BP terms for MA up-regulated genes and 360 BP terms for SCOS up-regulated genes. We also discovered 59, 24 and 117 BP terms in PostMA, MA and SCOS for down-regulated genes, respectively (p-value ≤ 0.01) (Supplementary Table 4). In the up-regulated gene lists for BPs, 10 BP terms were common among PostMA, MA and SCOS, 14 BP terms were common between PostMA and MA, six BP terms were common between MA and SCOS, and six BP terms were common between PostMA and SCOS. The common BP terms for up-regulated genes are illustrated in Fig. 2c. As shown in Fig. 2d, in the down-regulated gene lists for BPs, eight BP terms were common among PostMA, MA and SCOS, four BP terms were common between PostMA and MA, six BP terms were common between MA and SCOS, and three BP terms were common between PostMA and SCOS. We investigated 9, 16 and 48 CC terms for up-regulated genes and 18, 16 and 37 CC terms for down-regulated genes of PostMA, MA and SCOS, respectively (Supplementary Table S4). In the up-regulated gene lists for CC, we found one common CC term between PostMA and MA and one common CC term between MA and SCOS (see Fig. 2e). As Fig. 2f indicates, in the down-regulated gene lists for CC, 5 CC terms were common among PostMA, MA and SCOS, seven CC terms were common between MA and SCOS, three CC terms were common between PostMA and SCOS, and one CC term was common between PostMA and MA. We found 13, 20 and 40 MF terms for up-regulated genes and 13, 10 and 15 MF terms for down-regulated genes of PostMA, MA and SCOS, respectively (Supplementary Table S4). In the up-regulated gene lists for MF, we observed three common MF terms among PostMA, MA and SCOS, and one MF term was common between PostMA and MA (see Fig. 2g). In the down-regulated gene lists for MF, two MF terms were common between PostMA and MA, and two MF terms were common between MA and SCOS, as shown in Fig. 2h. We found 6, 9 and 47 KEGG pathways for up-regulated genes and 2, 1 and 10 KEGG pathways for down-regulated genes (Supplementary Table S4). In up-regulated genes, we observed common miRNAs among PostMA, MA and SCOS that are involved in cancer. Three pathways were common between MA and SCOS for up-regulated genes (antigen processing and presentation, Epstein-Barr virus infection and longevity regulating), and the pathways of steroid biosynthesis and taste transduction were common between PostMA and MA up-regulated genes (see Fig. 2i). As can be seen in Fig. 2j, we did not find any common pathways for down-regulated genes.

Venn diagram of similarities of DEGs, GO, KEGG pathway enrichment analysis between PostMA, MA and SCOS. (a) Common up- regulated genes with 21 mutual genes between all three groups. (b) Common down-regulated genes which 17 mutual genes were between all groups and 42 common genes were between PostMA and MA. (c,d) Common up and down-regulated BP, 10 BP terms were common between all PostMA, MA and SCOS, 14 BP terms were common between MA and PostMA, and 6 BP term was common between PostMA and SCOS. In down-regulated gene lists for BP, 8 BP terms were common between all PostMA, MA and SCOS. (e,f) In up-regulated CC, no common terms found. In down-regulated gene lists for CC, 5 CC terms were common between all PostMA, MA and SCOS and 7 CC terms were common between MA and PostMA. (g,h) Common up- and down-regulated MF. (i,j) Common up and down-regulated KEGG pathways.
Comparison of infertile human and mouse
We compared nine types of infertility in male humans and mice, including MArrest, oligospermia and teratospermia in humans and Ing2 Knockout (KO), Bcl6 KD, Etv5 KD, Pou3f1 KD, Ikbkap KO and Dazap1 mutant in mice. As shown in Table 1, the higher number of common up-regulated genes was found between Etv5 KD and Pou3f1 KD, with 53 genes, Bcl6b KD and Etv5 KD, with 19 genes, Bcl6b KD and Pou3f1 KD, with 14 genes, MArrest and teratospermia, with 12 genes, and MArrest and Ing2 KO, with nine common genes. Additionally, the Arpc1b gene was up-regulated in Bcl6b, Etv5, Pouf31 and Dazap1 KD, and Alcam was up-regulated in Bcl6b, Etv5, Pouf31 and Ing2 KD infertile mice. The highest number of common down-regulated genes was found between MArrest and oligospermia, with 32 genes, MArrest and the Dazap1 mutant, with 21 genes, MArrest and teratospermia, with 13 genes, and MArrest and Ikbkap KO, with 8 common genes. Three down-regulated genes (PLCZ1, TSSK2 and ANKRD7) were common between MArrest, oligospermia and the Dazap1 mouse mutant. DMRTB1 was a down-regulated common gene among MArrest, oligospermia and Ikbkap KO, GPR137B was common among teratospermia, Bcl6b and Pou3f1 KD, and LMNB2 was common among Bcl6b, Etv5 and Pou3f1 KD infertile mice (Table 1 and Table 2).
GSEA
We investigated gene sets based on all DEGs for MA, PostMA and SCOS (Fig. 3). In up-regulated gene sets, one gene set was common among PostMA, MA and SCOS, one gene set was common between PostMA and MA, and two gene sets were common between MA and SCOS. In the down-regulated gene sets, 10 gene sets were common between MA and PostMA, and one gene set was common between MA and SCOS. Common gene sets between each type of NOA are shown in Table 3.

Gene set enrichment analysis (GSEA) of PostMA, MA and SCOS. (a) Common gene sets for up-regulated genes. (b) Common gene sets for down-regulated genes with 10 gene sets common between PostMA and MA.
Principal component analysis (PCA)
We found that in three teratospermia libraries, infertile samples were completely distinguished from normal samples. In three libraries from the stages of before and after meiotic arrest and SCOS, the clusters of normal samples and SCOS samples were separated, but there was an overlap between some samples of PostMA, MA and control cases (Fig. 4).

Principal component analysis (PCA) of eight human microarray libraries that show the visualization of quality, similarity and overlapping of library samples.
Discussion
Understanding similarities among male infertility diseases could facilitate disease classification, help reveal hidden etiologies, and pave the way for new diagnostic tests and drugs. Toward this goal, we showed that in silico analyses are in good agreement with previous experimental results. Several studies have shown a direct association of an increase in steroid levels with azoospermia and oligospermia19, and male hormonal contraceptive trials use steroids to induce azoospermia and oligospermia27,28. Furthermore, steroid sex hormones regulate the spermatogenesis process and the development of skeletal muscles29. In this study, we observed that one of the major common BPs of up-regulated genes in MArrest and oligospermia was the development of muscle tissue and its regulation, and half of the common BPs of up-regulated genes in PostMA and MA were related to the steroid process. The miR-145 regulates the development of smooth muscles30, and its high level of expression leads to the inhibition of cell-cell adhesion and cell motility31. We observed the overexpression of miR-145 in NOA, and there were also up-regulated BPs related to muscle development in MArrest and oligospermia and down-regulated BPs related to sperm-egg recognition and sperm motility in NOA and teratospermia. Fu et al. found that several BPs of spermatogenesis-related genes were involved in sperm-egg recognition and fusion, and a protein-protein interaction analysis showed that these genes were down-regulated genes in teratospermia32. Moreover, a study on teratospermia suggested that the binding capacity of sperm to oocytes is low because of a lower expression of adhesion molecules in teratospermic spermatozoa33. In our study, we showed that half of the common down-regulated BPs in MArrest and teratospermia were related to sperm-egg adhesion.
Male germ cells are extremely sensitive to stress34. Glutathione is an important intracellular antioxidant, and several studies have indicated that decreased glutathione and glutathione transferase null genotypes lead to oligospermia and azoospermia34,35,36,37. We observed an up-regulation in glutathione transferase genes, which reduces the glutathione level in oligospermia and NOA (PostMA, MA and SCOS). Moreover, there are various results confirming the direct association between abnormal spermatogenesis due to the response to a stimulus38,39,40 and immune response41. We observed that one of the major similarities in MArrest, oligospermia and teratospermia was indeed overexpression of immune response, stimulus response and their regulation related genes. Bansal et al. revealed that idiopathic male infertility and asthenospermia are associated with changes in the expression of BPs, such as response to a stimulus, the immune system process, reproduction and the multicellular organismal process24. In this study, we showed the same BPs in MArrest, oligospermia and teratospermia. Noveski et al. determined that miR-23b, miR-32, miR-154 and miR-99 in MArrest and SCOS were up-regulated42, and we found that these genes were also up-regulated in NOA. SOX9 is an essential protein for the maturation of sertoli cells and normal spermatogenesis, and it is a possible target of miR-145 43,44. Furthermore, we observed that mir-145 is one of the common up-regulated genes. Approximately half of the common up-regulated genes in PostMA, MA and SCOS were miRNAs.
In 17 common down-regulated genes among PostMA, MA and SCOS, 9 genes were involved in spermatogenesis (ADAM29 45 , DNAH17 46 , FAM166A 47 , FAM71F1 22 , GAPDHS 48 , GGN 22 , LYZL6 49 , SMCP 50 and SPACA4 22), two genes were non-coding, and six genes (ABHD1, ARMC12, FAM205A, PRR30, TEX38 and TPPP2) did not have specific and direct roles in spermatogenesis. GGN has a high level of expression in the late pachytene stage and primary spermatogenesis51. ABHD1 is a member of the ABHD family, which has a role in spermatogenesis52. TPPP2 has a high expression level in testes and has a role in testicular cancer53. In addition, in down-regulated genes common among PostMA, MA and SCOS, there were eight BPs that were classified into three clusters, including development and differentiation of spermatogenesis, sperm motility and sperm-egg recognition. Three common down-regulated CCs among PostMA, MA and SCOS and four CCs between MA and SCOS were related to the flagellum, which matches observations made by Fu et al.32.
Okada et al., Zhuang et al., Fu et al. and Noveski et al. identified meaningful BPs by using separate enrichment analyses for up- and down-regulated genes19,32,42,54.
In human and mouse male infertility, we observed the highest similarity between Etv5 and Pou3f1 KD in the up-regulated gene lists. Furthermore, in the down-regulated gene lists, we observed the highest similarity between MArrest and oligospermia among other types of male infertility, such as teratospermia and infertile mouse mutants. DAZ is one of the most important genes, and its deletion leads to NOA8. Dazap1 is one of the isoforms of DAZ, and we observed the highest number of common genes between Dazap1 mouse mutants and MArrest cases, with 21 common down-regulated genes, in comparison to other infertile mice. IKAP protein is encoded by the Ikbkap gene, which is a subunit of the Elongator complex and plays a role in chromatin remodeling55. Lin et al. revealed that a loss of function of Ikbkap in mice was the cause of defects in synapsis and meiotic recombination, leading to apoptosis and spermatogenesis arrest. In the present study, we observed that Ikbkap KO mice were highly similar to MArrest cases, with eight common down-regulated genes in which the process of meiosis was disrupted (Table 1 and Table 2). Tanespimycin or 17-allylamino-17-demethoxygeldanamycin (17AAG) is an antitumor drug that works by inhibiting HSP90 (heat shock protein 90)56. In GSEA analysis, the low level of expression of a gene list in MA, PostMA and SCOS is similar to a gene set in ovarian cancer cells when treated with tanespimycin. However, there were no significant expression changes in HSP90 in MA, PostMA and SCOS, although there were 17 gene sets related to spermatogenesis and 32 gene sets related to cancer in several stages of spermatogenesis arrest (Fig. 3).
In conclusion, we revealed that when comparing MArrest and oligospermia, the genes associated with immune response processes, muscle tissue development, and glutathione transferase and regulatory genes were up-regulated, and the genes related to spermatogenesis were down-regulated. When comparing PostMA, MA and SCOS, we found several common DEGs. Ten up-regulated miRNAs were common among all three NOA types, and the expression of genes associated with the spermatogenesis process was down-regulated. The proteins of these down-regulated genes have a function in sperm motility and flagellum development. Further work is needed to investigate the epigenomics and proteomics of male infertility to complement gene expression studies. Our study indicates which pathways one should focus on in future studies.
Methods
In this study, we emphasized on unveiling underlying genes and signaling pathways and identifying putative biomarkers that are differentially expressed in male infertility microarray datasets. For this purpose, we used of functional enrichment analysis approaches including pathway enrichment analysis and GSEA. Figure 5 depicts the workflow used for this study.

The workflow of this study.
Microarray datasets and analysis
The microarray datasets related to male infertility were collected from the gene expression omnibus (GEO) repository57. Table 4 presents detailed information on the microarray datasets used. GEO2R (http://www.ncbi.nlm.nih.gov/geo/geo2r/), which employs a linear-based model for microarray analysis (limma), was used to obtain DEGs between male infertile and control samples. The top 100 DEGs were extracted in each infertile group against the normal group (p-value ≤ 0.01). Our expression study consists of three comparison steps, including (i) MArrest, oligospermia and teratospermia, (ii) PostMA, MA and SCOS, and (iii) nine types of human and mouse infertility.
Pathway enrichment analysis
After extracting up- and down-regulated genes from each library, the enriched GO terms (BP, CC and MF) and KEGG pathways were determined. Up- and down-regulated genes were then separately submitted to the Enrichr tool58. The common enriched GO terms and KEGG pathways for each comparison between an infertile group and a control group [such as NOA (PostMA, MA and SCOS), oligospermia and teratospermia] were extracted (p-value ≤ 0.01). We applied Venn diagrams (http://bioinformatics.psb.ugent.be/webtools/Venn/) for GO and KEGG pathway terms between three kinds of male infertility, MArrest, oligospermia and teratospermia, and three types of NOA (PostMA, MA and SCOS).
There are two strategies for GO and pathway enrichment analysis of DEGs: the analysis of all DEGs together or the split analysis of up- and down-regulated genes separately59,60,61. In this study, we used the second strategy, as suggested by other recent works62,63,64,65,66,67,68. Hong et al. compared the two types of GO and pathway enrichment analysis strategies using gene expression profiles of microarray and RNA-Seq, and they indicated that the separate strategy is more powerful and accurate59. When all DEGs are integrated together, the results might differ from when up-regulated and down-regulated genes are analyzed separately. For example, if a pathway has a considerable number of up-regulated genes and few down-regulated genes, the complete number of differentially regulated genes in the pathway might lead to statistically non-significant results, while computing the enrichment of over-represented genes separately might highlight an implication of the pathway in the system under investigation61. Therefore, we used the separated strategy to interpret the results.
GSEA
GSEA is a powerful analytical method for interpreting gene expression data. We used software from the Broad institute69. All curated gene sets (C2.all.v 5.0 curated) were downloaded from the Molecular Signatures Database (MSigDB) and used to select significant gene sets based on the measurement of expression data69. A false discovery rate (FDR) less than 0.25 and p-values less than 0.01 were considered significant.
PCA
The quality of eight human microarray libraries was examined with PCA. PCA was applied to eight normalized and log-transformed libraries of human male infertility using the R package70. All samples of each library were placed in a specific two-dimensional scatter plot without selection or weighting.
References
Gnoth, C. et al. Definition and prevalence of subfertility and infertility. Hum Reprod 20, 1144–1147, https://doi.org/10.1093/humrep/deh870 (2005).
Massart, A., Lissens, W., Tournaye, H. & Stouffs, K. Genetic causes of spermatogenic failure. Asian journal of andrology 14, 40–48, https://doi.org/10.1038/aja.2011.67 (2012).
Tahmasbpour, E., Balasubramanian, D. & Agarwal, A. A multi-faceted approach to understanding male infertility: gene mutations, molecular defects and assisted reproductive techniques (ART). Journal of assisted reproduction and genetics 31, 1115–1137, https://doi.org/10.1007/s10815-014-0280-6 (2014).
Cavallini, G. Male idiopathic oligoasthenoteratozoospermia. Asian journal of andrology 8, 143–157, https://doi.org/10.1111/j.1745-7262.2006.00123.x (2006).
Poongothai, J., Gopenath, T. S. & Manonayaki, S. Genetics of human male infertility. Singapore medical journal 50, 336–347 (2009).
Cooper, T. G. et al. World Health Organization reference values for human semen characteristics. Human reproduction update 16, 231–245, https://doi.org/10.1093/humupd/dmp048 (2010).
Hotaling, J. M. Genetics of male infertility. The Urologic clinics of North America 41, 1–17, https://doi.org/10.1016/j.ucl.2013.08.009 (2014).
Krausz, C., Escamilla, A. R. & Chianese, C. Genetics of male infertility: from research to clinic. Reproduction 150, R159–174, https://doi.org/10.1530/REP-15-0261 (2015).
Esteves, S. C. A clinical appraisal of the genetic basis in unexplained male infertility. Journal of human reproductive sciences 6, 176–182, https://doi.org/10.4103/0974-1208.121419 (2013).
Hamada, A. J., Esteves, S. C. & Agarwal, A. A comprehensive review of genetics and genetic testing in azoospermia. Clinics (Sao Paulo) 68(Suppl 1), 39–60 (2013).
Shaha, C., Tripathi, R. & Mishra, D. P. Male germ cell apoptosis: regulation and biology. Philosophical transactions of the Royal Society of London. Series B, Biological sciences 365, 1501–1515, https://doi.org/10.1098/rstb.2009.0124 (2010).
Matzuk, M. M. & Lamb, D. J. The biology of infertility: research advances and clinical challenges. Nature medicine 14, 1197–1213, https://doi.org/10.1038/nm.f.1895 (2008).
Wosnitzer, M., Goldstein, M. & Hardy, M. P. Review of Azoospermia. Spermatogenesis 4, e28218, https://doi.org/10.4161/spmg.28218 (2014).
De Braekeleer, M., Nguyen, M. H., Morel, F. & Perrin, A. Genetic aspects of monomorphic teratozoospermia: a review. Journal of assisted reproduction and genetics 32, 615–623, https://doi.org/10.1007/s10815-015-0433-2 (2015).
Carrell, D. T., Aston, K. I., Oliva, R., Emery, B. R. & De Jonge, C. J. The “omics” of human male infertility: integrating big data in a systems biology approach. Cell and tissue research 363, 295–312, https://doi.org/10.1007/s00441-015-2320-7 (2016).
Ferlin, A. et al. Male infertility: role of genetic background. Reproductive biomedicine online 14, 734–745 (2007).
Rallapalli, G. et al. EXPRSS: an Illumina based high-throughput expression-profiling method to reveal transcriptional dynamics. BMC genomics 15, 341, https://doi.org/10.1186/1471-2164-15-341 (2014).
Lin, Y. H. et al. Identification of ten novel genes involved in human spermatogenesis by microarray analysis of testicular tissue. Fertility and sterility 86, 1650–1658, https://doi.org/10.1016/j.fertnstert.2006.04.039 (2006).
Okada, H. et al. Genome-wide expression of azoospermia testes demonstrates a specific profile and implicates ART3 in genetic susceptibility. PLoS genetics 4, e26, https://doi.org/10.1371/journal.pgen.0040026 (2008).
Gatta, V. et al. Testis transcriptome analysis in male infertility: new insight on the pathogenesis of oligo-azoospermia in cases with and without AZFc microdeletion. BMC genomics 11, 401, https://doi.org/10.1186/1471-2164-11-401 (2010).
Saito, M. et al. Targeted disruption of Ing2 results in defective spermatogenesis and development of soft-tissue sarcomas. PloS one 5, e15541, https://doi.org/10.1371/journal.pone.0015541 (2010).
Malcher, A. et al. Potential biomarkers of nonobstructive azoospermia identified in microarray gene expression analysis. Fertility and sterility 100(1686–1694), e1681–1687, https://doi.org/10.1016/j.fertnstert.2013.07.1999 (2013).
Malcher, A. et al. The gene expression analysis of paracrine/autocrine factors in patients with spermatogenetic failure compared with normal spermatogenesis. Am J Reprod Immunol 70, 522–528, https://doi.org/10.1111/aji.12149 (2013).
Bansal, S. K., Gupta, N., Sankhwar, S. N. & Rajender, S. Differential Genes Expression between Fertile and Infertile Spermatozoa Revealed by Transcriptome Analysis. PloS one 10, e0127007, https://doi.org/10.1371/journal.pone.0127007 (2015).
Ansari-Pour, N., Razaghi-Moghadam, Z., Barneh, F. & Jafari, M. Testis-Specific Y-Centric Protein-Protein Interaction Network Provides Clues to the Etiology of Severe Spermatogenic Failure. Journal of proteome research 15, 1011–1022, https://doi.org/10.1021/acs.jproteome.5b01080 (2016).
Croken, M. M., Qiu, W., White, M. W. & Kim, K. Gene Set Enrichment Analysis (GSEA) of Toxoplasma gondii expression datasets links cell cycle progression and the bradyzoite developmental program. BMC genomics 15, 515, https://doi.org/10.1186/1471-2164-15-515 (2014).
Nieschlag, E. & Vorona, E. MECHANISMS IN ENDOCRINOLOGY: Medical consequences of doping with anabolic androgenic steroids: effects on reproductive functions. European journal of endocrinology 173, R47–58, https://doi.org/10.1530/EJE-15-0080 (2015).
Nieschlag, E. Clinical trials in male hormonal contraception. Contraception 82, 457–470, https://doi.org/10.1016/j.contraception.2010.03.020 (2010).
Sato, K. et al. Resistance training restores muscle sex steroid hormone steroidogenesis in older men. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 28, 1891–1897, https://doi.org/10.1096/fj.13-245480 (2014).
Cordes, K. R. et al. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 460, 705–710, https://doi.org/10.1038/nature08195 (2009).
Gotte, M. et al. miR-145-dependent targeting of junctional adhesion molecule A and modulation of fascin expression are associated with reduced breast cancer cell motility and invasiveness. Oncogene 29, 6569–6580, https://doi.org/10.1038/onc.2010.386 (2010).
Fu, G., Wei, Y., Wang, X. & Yu, L. Identification of candidate causal genes and their associated pathogenic mechanisms underlying teratozoospermia based on the spermatozoa transcript profiles. Andrologia 48, 576–583, https://doi.org/10.1111/and.12484 (2016).
Szczygiel, M. & Kurpisz, M. Teratozoospermia and its effect on male fertility potential. Andrologia 31, 63–75 (1999).
Xiong, D. K., Chen, H. H., Ding, X. P., Zhang, S. H. & Zhang, J. H. Association of polymorphisms in glutathione S-transferase genes (GSTM1, GSTT1, GSTP1) with idiopathic azoospermia or oligospermia in Sichuan, China. Asian journal of andrology 17, 481–486, https://doi.org/10.4103/1008-682X.143737 (2015).
Atig, F. et al. Impact of seminal trace element and glutathione levels on semen quality of Tunisian infertile men. BMC urology 12, 6, https://doi.org/10.1186/1471-2490-12-6 (2012).
Wu, Q. F. et al. Genetic polymorphism of glutathione S-transferase T1 gene and susceptibility to idiopathic azoospermia or oligospermia in northwestern China. Asian journal of andrology 10, 266–270, https://doi.org/10.1111/j.1745-7262.2008.00347.x (2008).
Bhardwaj, A., Verma, A., Majumdar, S. & Khanduja, K. L. Status of vitamin E and reduced glutathione in semen of oligozoospermic and azoospermic patients. Asian journal of andrology 2, 225–228 (2000).
Agarwal, A. et al. Comparative proteomic network signatures in seminal plasma of infertile men as a function of reactive oxygen species. Clinical proteomics 12, 23, https://doi.org/10.1186/s12014-015-9094-5 (2015).
Hu, H. et al. RNA-Seq identifies key reproductive gene expression alterations in response to cadmium exposure. BioMed research international 2014, 529271, https://doi.org/10.1155/2014/529271 (2014).
Agarwal, A., Mulgund, A., Sharma, R. & Sabanegh, E. Mechanisms of oligozoospermia: an oxidative stress perspective. Systems biology in reproductive medicine 60, 206–216, https://doi.org/10.3109/19396368.2014.918675 (2014).
Hussein, M. R. et al. Phenotypic characterization of the immune and mast cell infiltrates in the human testis shows normal and abnormal spermatogenesis. Fertility and sterility 83, 1447–1453, https://doi.org/10.1016/j.fertnstert.2004.11.062 (2005).
Noveski, P. et al. MicroRNA expression profiles in testicular biopsies of patients with impaired spermatogenesis. Andrology 4, 1020–1027, https://doi.org/10.1111/andr.12246 (2016).
Lian, J. et al. Altered microRNA expression in patients with non-obstructive azoospermia. Reproductive biology and endocrinology: RB&E 7, 13, https://doi.org/10.1186/1477-7827-7-13 (2009).
Zhang, L. et al. Association of hsa-miR145 overexpression in human testicular cells with male infertility. Molecular medicine reports 11, 4365–4372, https://doi.org/10.3892/mmr.2015.3273 (2015).
Cerretti, D. P., DuBose, R. F., Black, R. A. & Nelson, N. Isolation of two novel metalloproteinase-disintegrin (ADAM) cDNAs that show testis-specific gene expression. Biochemical and biophysical research communications 263, 810–815, https://doi.org/10.1006/bbrc.1999.1322 (1999).
Neesen, J. et al. Identification of dynein heavy chain genes expressed in human and mouse testis: chromosomal localization of an axonemal dynein gene. Gene 200, 193–202 (1997).
Wang, S. et al. Proteomic characteristics of human sperm cryopreservation. Proteomics 14, 298–310, https://doi.org/10.1002/pmic.201300225 (2014).
Wang, Y., Stary, J. M., Wilhelm, J. E. & Newmark, P. A. A functional genomic screen in planarians identifies novel regulators of germ cell development. Genes & development 24, 2081–2092, https://doi.org/10.1101/gad.1951010 (2010).
Wei, J. et al. Characterisation of Lyzls in mice and antibacterial properties of human LYZL6. Asian journal of andrology 15, 824–830, https://doi.org/10.1038/aja.2013.93 (2013).
Cullinane, D. L., Chowdhury, T. A. & Kleene, K. C. Mechanisms of translational repression of the Smcp mRNA in round spermatids. Reproduction 149, 43–54, https://doi.org/10.1530/REP-14-0394 (2015).
Lu, B. & Bishop, C. E. Mouse GGN1 and GGN3, two germ cell-specific proteins from the single gene Ggn, interact with mouse POG and play a role in spermatogenesis. The Journal of biological chemistry 278, 16289–16296, https://doi.org/10.1074/jbc.M211023200 (2003).
Grimaldi, P. et al. The endocannabinoid system and pivotal role of the CB2 receptor in mouse spermatogenesis. Proceedings of the National Academy of Sciences of the United States of America 106, 11131–11136, https://doi.org/10.1073/pnas.0812789106 (2009).
Liu, M. et al. Scanning of novel cancer/testis proteins by human testis proteomic analysis. Proteomics 13, 1200–1210, https://doi.org/10.1002/pmic.201200489 (2013).
Zhuang, X. et al. Integrated miRNA and mRNA expression profiling to identify mRNA targets of dysregulated miRNAs in non-obstructive azoospermia. Scientific reports 5, 7922, https://doi.org/10.1038/srep07922 (2015).
Cuajungco, M. P. et al. Cloning, characterization, and genomic structure of the mouse Ikbkap gene. DNA and cell biology 20, 579–586, https://doi.org/10.1089/104454901317094990 (2001).
Modi, S. et al. HSP90 inhibition is effective in breast cancer: a phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clinical cancer research: an official journal of the American Association for Cancer Research 17, 5132–5139, https://doi.org/10.1158/1078-0432.CCR-11-0072 (2011).
Edgar, R., Domrachev, M. & Lash, A. E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic acids research 30, 207–210 (2002).
Chen, E. Y. et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC bioinformatics 14, 128, https://doi.org/10.1186/1471-2105-14-128 (2013).
Hong, G., Zhang, W., Li, H., Shen, X. & Guo, Z. Separate enrichment analysis of pathways for up- and downregulated genes. Journal of the Royal Society, Interface 11, 20130950, https://doi.org/10.1098/rsif.2013.0950 (2014).
Shigemizu, D. et al. Using functional signatures to identify repositioned drugs for breast, myelogenous leukemia and prostate cancer. PLoS computational biology 8, e1002347, https://doi.org/10.1371/journal.pcbi.1002347 (2012).
D’Andrea, D., Grassi, L., Mazzapioda, M. & Tramontano, A. FIDEA: a server for the functional interpretation of differential expression analysis. Nucleic acids research 41, W84–88, https://doi.org/10.1093/nar/gkt516 (2013).
Feng, C. et al. Subpathway-CorSP: Identification of metabolic subpathways via integrating expression correlations and topological features between metabolites and genes of interest within pathways. Scientific reports 6, 33262, https://doi.org/10.1038/srep33262 (2016).
Golla, U., Joseph, D. & Tomar, R. S. Combined Transcriptomics and Chemical-Genetics Reveal Molecular Mode of Action of Valproic acid, an Anticancer Molecule using Budding Yeast Model. Scientific reports 6, 35322, https://doi.org/10.1038/srep35322 (2016).
Li, M. et al. Identifying Reproducible Molecular Biomarkers for Gastric Cancer Metastasis with the Aid of Recurrence Information. Scientific reports 6, 24869, https://doi.org/10.1038/srep24869 (2016).
Synnergren, J. et al. Molecular signature of cardiomyocyte clusters derived from human embryonic stem cells. Stem Cells 26, 1831–1840, https://doi.org/10.1634/stemcells.2007-1033 (2008).
Bao, X. et al. Cell adhesion molecule pathway genes are regulated by cis-regulatory SNPs and show significantly altered expression in Alzheimer’s disease brains. Neurobiology of aging 36(2904), e2901–2907, https://doi.org/10.1016/j.neurobiolaging.2015.06.006 (2015).
Stenz, L., Escoffier, J., Rahban, R., Nef, S. & Paoloni-Giacobino, A. Testicular Dysgenesis Syndrome and Long-Lasting Epigenetic Silencing of Mouse Sperm Genes Involved in the Reproductive System after Prenatal Exposure to DEHP. PloS one 12, e0170441, https://doi.org/10.1371/journal.pone.0170441 (2017).
Talkhabi, M., Razavi, S. M. & Salari, A. Global transcriptomic analysis of induced cardiomyocytes predicts novel regulators for direct cardiac reprogramming. Journal of cell communication and signaling 11, 193–204, https://doi.org/10.1007/s12079-017-0387-5 (2017).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102, 15545–15550, https://doi.org/10.1073/pnas.0506580102 (2005).
Jolliffe, I. T. & Cadima, J. Principal component analysis: a review and recent developments. Philosophical transactions. Series A, Mathematical, physical, and engineering sciences 374, 20150202, https://doi.org/10.1098/rsta.2015.0202 (2016).
Pacheco, S. E. et al. Integrative DNA methylation and gene expression analyses identify DNA packaging and epigenetic regulatory genes associated with low motility sperm. PloS one 6, e20280, https://doi.org/10.1371/journal.pone.0020280 (2011).
Platts, A. E. et al. Success and failure in human spermatogenesis as revealed by teratozoospermic RNAs. Human molecular genetics 16, 763–773, https://doi.org/10.1093/hmg/ddm012 (2007).
Wu, X., Goodyear, S. M., Tobias, J. W., Avarbock, M. R. & Brinster, R. L. Spermatogonial stem cell self-renewal requires ETV5-mediated downstream activation of Brachyury in mice. Biology of reproduction 85, 1114–1123, https://doi.org/10.1095/biolreprod.111.091793 (2011).
Chen, H. Y., Yu, Y. H. & Yen, P. H. DAZAP1 regulates the splicing of Crem, Crisp2 and Pot1a transcripts. Nucleic acids research 41, 9858–9869, https://doi.org/10.1093/nar/gkt746 (2013).
Acknowledgements
S.M.R. was funded by Royan Institute, Tehran. Iran.
Author information
Authors and Affiliations
Contributions
M.S. and A.S.Y. designed the study. S.M.R., M.J., O.W. and A.S.Y. implemented the methods, and analyzed the data. S.M.R., A.D. and M.S. contributed to the interpretation of the results. S.M.R. and M.S. drafted the manuscript and prepared all figures and tables. All authors participated in improving the writing of the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Razavi, S.M., Sabbaghian, M., Jalili, M. et al. Comprehensive functional enrichment analysis of male infertility. Sci Rep 7, 15778 (2017). https://doi.org/10.1038/s41598-017-16005-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-16005-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
