
Abstract
Cigarette smoking is undoubtedly a risk factor for lung cancer. Moreover, smokers with genetic mutations on chromosome 3p21.3, a region frequently deleted in cancer and notably in lung cancer, have a dramatically higher risk of aggressive lung cancer. The RNA binding motif 5 (RBM5) is one of the component genes in the 3p21.3 tumour suppressor region. Studies using human cancer specimens and cell lines suggest a role for RBM5 as a tumour suppressor. Here we demonstrate, for the first time, an in vivo role for RBM5 as a tumour suppressor in the mouse lung. We generated Rbm5 loss-of-function mice and exposed them to a tobacco carcinogen NNK. Upon exposure to NNK, Rbm5 loss-of-function mice developed lung cancer at similar rates to wild type mice. As tumourigenesis progressed, however, reduced Rbm5 expression lead to significantly more aggressive lung cancer i.e. increased adenocarcinoma nodule numbers and tumour size. Our data provide in vivo evidence that reduced RBM5 function, as occurs in a large number of patients, coupled with exposure to tobacco carcinogens is a risk factor for an aggressive lung cancer phenotype. These data suggest that RBM5 loss-of-function likely underpins at least part of the pro-tumourigenic consequences of 3p21.3 deletion in humans.
Similar content being viewed by others

Introduction
Late stage detection makes lung cancer one of the most fatal forms of cancer, with a five-year survival rate of 17% overall, or below 2% for those with stage IV disease at diagnosis1. A 2012 World Health Organization report estimated that of the 1.8 million people diagnosed with lung cancer worldwide, 1.6 million would die from the disease2. However, if diagnosed in its earliest stages, surgery, chemotherapy and radiation therapy present a likely cure.
RNA binding motif 5 (RBM5) is one of the genes located within the tumour suppressor region 3p21.3; a region containing 19 genes that is frequently deleted in lung cancer and other types of carcinomas3. Moreover, the 3p21.3 deletion is detected in pre-neoplastic lesions in smokers4, indicating that it is an early change in the multistep pathogenesis of lung cancer. Despite this, a role for 3p21.3 deletions, and its component genes, in in vivo tumour development is still a matter of conjecture.
Human lung cancers can be divided into two main histopathological subtypes: non-small-cell lung cancer (NSCLC) and small-cell lung cancer (SCLC). NSCLC accounts for approximately 85% of lung cancers and can be divided into adenocarcinoma and squamous cell carcinoma (SCC). About 40% of human lung cancers are adenocarcinomas. Decreased RBM5 expression, at the mRNA and protein levels, have been reported in primary NSCLC specimens compared to normal adjacent tissues5. Further, RBM5 is one of nine genes down-regulated in metastases of primary tumours6. RBM5 is also included in the 17 common gene signatures associated with metastasis identified in multiple solid tumour types. Solid tumours carrying this gene expression signature have higher rates of metastasis and poor clinical outcomes6. Decreased RBM5 expression in primary lung tumours has been shown to correlate with lymph node metastasis7. In addition, RBM5 has been implicated in breast cancer development8,9, vestibular schwannomas10 and renal carcinomas11. Collectively, these data suggest that reduced RBM5 expression is associated with increased cancer risk and that RBM5 is a tumour suppressor.
In support of this hypothesis, in vitro over-expression of RBM5 was shown to inhibit the growth of human lung cancer cell lines by increasing apoptosis and inducing cell cycle arrest in G112. The inhibition of cell growth was associated with decreased cyclin A and phosphorylated retinoblastoma (RB) and an increase in the expression of the proapototic protein Bax12. RBM5 has been shown to suppress anchorage-dependent and anchorage-independent growth in A9 mouse fibrosarcoma cells and to inhibit their tumour forming activity in nude mice5.
RBM5 is an RNA binding protein that has previously been shown to regulate the splicing of apoptosis-related pre-mRNAs, including Caspase 213, FAS receptor and c-FLIP 14, B-lymphocyte cytidine deaminase enzyme AID (activation-induced cytidine deaminase)15, Notch pathway regulator Numb in HeLa cells16 and numerous transcripts required for male germ cell development17. Its role as a splicing regulator is conserved in Arabidopsis 18.
Since the cloning of the RBM5 gene19, a growing body of literature strongly suggests a role for RBM5 as a tumour suppressor12,13,14,16. However, the in vivo tumour suppression activity of RBM5 has not been tested. Within this study, we have generated Rbm5 heterozygous knockout mice, i.e. analogous to the reduced expression seen in lung cancer patients, and have used them to demonstrate a role for RBM5 in in vivo tumour suppression function in the lung.
Results and Discussion
To test the in vivo function of RBM5, we generated an Rbm5 knockout mouse line using a gene trapped ES cell line. The gene-trapped cassette was inserted into intron 1 of the Rbm5 gene. This produced a truncated Rbm5 mRNA containing exon 1, resulting in no protein production (null allele) (Fig. 1a). Upon the establishment of a heterozygous knockout colony, the colony was backcrossed onto a C57BL6/J background for 10 generations. Mice heterozygous for the Rbm5 knockout allele (referred to as HET, Rbm5 +/−) were viable, fertile and survived to adulthood with no detectable developmental defects. By contrast, and although a small number of homozygous Rbm5 knockout mice (referred to as KO, Rbm5 −/−) were found, there was a pronounced deficit in Rbm5 −/− pups on the day of birth i.e. 8%, compared to an expected frequency of 25%. There was an absence of Rbm5 −/− mice at weaning age (0% at 3 weeks postnatal) (Fig. 1b). Indeed, virtually all Rbm5 −/− pups were dead by 3 days post-natal. These data show that Rbm5 is essential for either embryonic or early post-natal development. To define the time point when the embryo/foetus loss occurred, we collected pups from heterozygous time matings at E18.5. At this age, the ratio of each genotype was as expected according to the Mendelian rule (Fig. 1b). This finding indicates that Rbm5 is absolutely required for the survival of newborn pups. The pathology underlying Rbm5 −/− pup death is currently unknown.

Generation and characterisation of the Rbm5 gene trap mouse line. (a) The Rbm5 gene trap mouse line: The U3neoSVFS gene-trapped cassette was inserted into intron 1 of the Rbm5 gene (ENSMUSG00000032580). This produced a truncated Rbm5 mRNA containing exon 1 (ENSMUSE00000371436), resulting in the production of truncated Rbm5 mRNA and no protein production (null allele). SA: Splicing acceptor site; Neo: Neomycin resistance gene; Poly A: Polyadenylation signal. (b) Distribution of genotypes of progeny from heterozygous knockout breeding pairs at embryonic day 18.5 (E18.5), day of birth (Day 0) and at 3 weeks. WT: wild type; HET: heterozygous knockout; KO: homozygous knockout. (c) Verification of gene trapping efficient by RT-PCR on E18.5 lung using primers Ex1-Fw (5′-CTCCTGCTTTGTTCCCTCTG-3′) and Ex4-Rev (5′-CCATCTTCAGACCGGTCACT-3′). The WT allele expected PCR product is 298 bp and no products for the competed gene trap KO allele. -RT: negative control (no reverse transcriptase). (d,e) Quantitative PCR (qPCR) was performed to measure Rbm5 mRNA expression levels in adult lung (D) and adult testis (E) samples. n = 3 per genotype, 8 weeks old. Data is expressed as mean ± SD. Statistical significance for all analyses was determined using a two-tailed student t-test.
In order to ascertain the degree of gene trapping efficiency, we collected lung tissues from wild type (WT, Rbm5 +/+) and Rbm5 −/− foetuses at E18.5 and performed RT-PCR using primers flanking exons 1 and 4 (Fig. 1c). No Rbm5 transcript was detected in the Rbm5 −/− lungs compared to that of Rbm5 +/+ lungs. Furthermore, using quantitative PCR (qPCR) we showed that there was a significant reduction in the levels of Rbm5 mRNA in the Rbm5 +/− lung (64% reduction, Fig. 1d) and testis (76% reduction, Fig. 1e) collected from 8 week old mice. This data was mirrored using western blotting of adult lungs (Supplementary Fig. 1). This data indicate that the gene trap cassette was efficiently interrupting Rbm5 gene expression as expected and that Rbm5 +/− mice contained reduced Rbm5 expression, analogous to the situation observed in many lung cancer patients.
Based on mRNA expression pattern, Rbm5 is a ubiquitously expressed gene with the highest expression level found in the testis17. Using immunofluorescence labelling, we showed that RBM5 protein was widely localized in the adult mouse lung. RBM5 was detected in almost all cells within the conducting airway epithelium, as shown by double-labelling with the secretory cell marker CC10 (also known as SCGB1A1) (Fig. 2a,b). RBM5 expression in CC10-positive and CC10-negative cells indicated localisation in both secretory and ciliated cells, respectively. In the distal lung, RBM5 expression was also detected in many cell lineages, including type II alveolar epithelial cells (AECs), as shown by double-labelling with the type-II AEC marker Pro-surfactant protein C (ProSPC) (Fig. 2c,d). A strong nuclear localisation was observed in both cell types. This result was consistent with previous studies demonstrating nuclear RBM5 localisation in male germ cells17 and HeLa cells13.

RBM5 localised to type II alveolar epithelial cells (AECs) and Clara cells. The localisation of RBM5 in the adult lung (8 weeks old) was determined by immunofluorescence using a RBM5 mouse monoclonal antibody as described previously17. (a,b) RBM5 localised to Clara cells as indicated by double staining for RBM5 (red) and CC10 (green). (c,d ) RBM5 localised to type II alveolar epithelial cells (AECs) as indicated by double staining of RBM5 (red) and Pro-surfactant protein C (SPC, in green). Insets = negative controls (no primary antibody). Scare bars = 20 μm.
To date, the cellular origin of most types of lung cancers remained largely unknown. However, studies using a knock-in mouse model carrying a codon 12 K-Ras mutant gene indicate that type II AECs are the cells of origin of K-Ras-induced adenocarcinomas20. Given the localization of RBM5 to type II cells, and previous studies in cell lines and human tissue suggesting reduced RBM5 expression is associated with increased lung cancer risk7,12, we decided to test the susceptibility of reduced Rbm5 expression (Rbm5 +/−) on lung cancer progression i.e. is ascertain if RBM5 is a tumour suppressor in vivo.
Lung cancer, like many other cancers, is thought to conform to the 2-hit model i.e. genetic and environmental hits. Cigarette smoking is the greatest risk factor for lung cancer21 and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) is the most potent carcinogen identified in cigarette smoke and has been widely used as accepted means of lung cancer induction22. As such, in this study, we utilized NNK to induce lung cancer in Rbm5 heterozygous knockout (carrying one copy of the Rbm5 gene) and wild type mice. The purpose of using NNK was to accelerate carcinogenesis into a time frame that would allow an in-depth analysis of lung cancer initiation and progression. Although there is a strong correlation between cigarette smoking and lung cancer, susceptibility to lung cancer among smokers is not uniform23. Smokers who carry particular genetic mutations have a dramatically higher risk of developing lung cancer23. Studies have shown that the KRAS activating mutation (G12V) is often associated with smoking-related NSCLC24. Similarly, different strains of mice have been shown to have differential susceptibility to lung cancer induction upon exposure to NNK25. The A/J mouse strain, that carries a naturally occurring KRAS G12V mutation, is one of the most susceptible strains for NNK-induced lung cancer. Importantly, RBM5 has been found to be down-regulated in rat embryonic fibroblast cells that have been constitutively over-expressed RAS G12V protein26. These studies suggest a correlation between RBM5 and KRAS activating mutation in the pathogenesis of lung cancer.
Given this association, to investigate the role of RBM5 in lung cancer, Rbm5 +/− mice were backcrossed onto the A/J mouse strain for 10 generations. Six week-old Rbm5 +/− (HET) and Rbm5 +/+ (WT) littermates were injected with NNK to induce lung cancer as previously described27. The use of Rbm5 +/− mice (rather than Rbm5 −/− mice) is reflective of the situation in patients wherein chromosome 3p21.3 loss occurs in heterozygosity. Lung tissues were analysis for the presence of tumours and the progression of tumour formation at 16, 20 and 48 weeks post-NNK injection.
At 16 and 20 weeks post-NNK injection, a small number of lung tumours were observed in both Rbm5 +/+ and Rbm5 +/− mice, however no significant difference in tumour number or tumour area was observed (Supplementary Fig. 2a–d). The control group, which received saline injection, showed no sign of lung tumours (n = 10). This finding suggested that RBM5 dosage does not affect the initiation of NNK-induced lung cancer and is consistent with previously published data showing that NNK in isolation can induce lung cancer28. While not examined at molecular detail here, the histopathological progression of NNK-induced lung cancer progression was consistent with previous publications28.
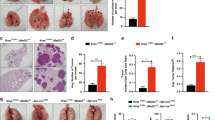
At 48 weeks post-NNK injection, however, Rbm5 +/− mice displayed a significant increase in both the number of individual tumours, and tumour area, compared to Rbm5 +/+ mice (Fig. 3). An independent, and blinded, analysis by a pathologist confirmed that the Rbm5 +/− lungs contained an increased numbers of adenocarcinoma nodules with increased tumour nodule sizes compared to that in Rbm5 +/+ lungs (148% and 153% increase on control wild type tissue respectively) (Fig. 3, Supplementary Table 1). These data indicate that reduced Rbm5 expression leads to more aggressive progression of lung adenocarcinomas in the Rbm5 +/− mice.

Rbm5 haploinsufficiency leads to accelerated lung cancer progression. (a–d) H&E staining of lungs collected from mice 48 weeks post-NNK injection. Number of tumours (e) and tumour area (f) in mice 48 weeks post-NNK injection. Data are expressed as mean +/− S.E.M. (standard error of mean). n = 13 WT and n = 16 HET. p < 0.05 was considered statistically significance. Statistical significance for all analyses was determined using a two-tailed student t-test.
Consistent with previous publications suggesting at AEC cells are the origin of many lung cancers20, tumours in both Rbm5 +/+ and Rbm5 +/− mice were Pro-SPC positive, and CC10 negative (Supplementary Fig. 3). Somewhat surprisingly, however, there was no significant difference in either the rates of apoptosis (TUNEL staining, data not shown) or Ki67 labelling with tumours between genotypes (Ki67: 186.5 +/− 59.92 in wild type versus 149.0 +/− 51.56 positive cells per mm2 in Rbm5 +/−). The rates of apoptosis per tumour area were too low to reliably quantitate in both genotypes. This data is perhaps, however, consistent with the long progression time required to see a difference in tumour mass between genotypes.
Lung cancer, like many other types of cancer, is believed to be initiated by over-activation of oncogenes and/or down-regulation of tumour suppressor genes. Although several studies using cell lines have provided evidence for RBM5 being a regulator of apoptosis and the cell cycle, direct in vivo evidence for its tumour suppressor activity remained elusive. Our data is the first to demonstrate the physiological role for RBM5 as a tumour suppressor in the lung. At 16–20 weeks post-exposure to NNK, we showed that reduced Rbm5 expression had no effect of the initiation of lung adenocarcinomas. As tumourigenesis progressed, however, reduced Rbm5 expression resulted in more aggressive tumour growth in Rbm5 +/− lungs. This data is consistent with previous studies showing that decreased RBM5 expression in human lung tumours is correlated with lymph node metastasis7, and ectopic over-expression of RBM5 inhibited tumour forming activity in nude mice5.
Metastatic lung cancer is a leading cause of mortality. Surgery, chemotherapy, and radiation therapy present a likely cure if the cancer is diagnosed in its earliest stages. The data presented herein suggests that a quantitative assessment of RBM5 expression in lung cancer biopsies, for example, may be a valuable prognostic marker for the prediction of high-risk cases for whom more aggressive post-surgical treatments may be warranted. Moreover, our data reveal that RBM5 is required for early post-natal survival and acts in vivo as a tumour suppressor that likely underpins at least part of the pro-tumourigenic outcomes resulting from 3p21.3 deletion in humans.
Methods
Mouse line production and genotyping
Animal procedures were conducted in accordance with the Australian National Health and Medical Research Council’s Guidelines on Ethics in Animal Experimentation and were approved by the Monash University Animal Experimentation Ethics Committee. The Rbm5 gene trap mouse line was generated at the Australian Phenomics Network (APN) Monash University Node using standard methods29 and a gene-trapped ES cell (PST20293-NR, on a 129Sv x C57BL/6J background) obtained from the Toronto Centre for Phenogenomics Centre for Modelling Human Disease. The U3neoSVFS gene-trapped cassette was inserted into intron 1 of the Rbm5 gene (ENSMUSG00000032580). This produced a truncated Rbm5 mRNA containing exon 1 (ENSMUSE00000371436), resulting in the production of truncated Rbm5 mRNA and no protein production (null allele).
Mouse genotypes were determined from tail biopsies using real time PCR with specific probes designed for each allele (Transnetyx, Cordova, TN). The Rbm5 KO allele was detected using the Neomycin probe and primers set which included: Forward primer: GGGCGCCCGGTTCTT; Reporter: ACCTGTCCGGTGCCC; and Reverse primer: CCTCGTCCTGCAGTTCATTCA. The Rbm5 WT allele was detected using the Rbm5 WT probe set which included: Forward primer: CATTACACCCCAGTGATTTTGCA; reporter: TTGGTGCTGTCCCTTAAGTC; and Reverse primer: CCTCTGGCGGCTGACA.
Verification of gene trapping efficient was performed by RT-PCR on E18.5 lung using primers Ex1-Fw (5′-CTCCTGCTTTGTTCCCTCTG-3′) and Ex4-Rev (5′-CCATCTTCAGACCGGTCACT-3′). The WT allele expected PCR product is 298 bp and no products for the competed gene trap KO allele.
Quantitative PCR (qPCR) was performed to measure Rbm5 mRNA expression levels in adult lung and adult testis samples (n = 3 per genotype, 8 weeks old) using TaqMan assays (Rbm5 exons 4-5: Mm00455721, Thermo Scientifics). The primers used in the as The levels of Rbm5 mRNA expression between Rbm5 +/+ and Rbm5 +/− lung and testis samples were normalised against mRNA levels of Ppia (Mm02342429) and Hprt (Mm00446968), respectively.
At the time of writing this manuscript, data contained within Ensembl indicated that the mouse Rbm5 gene (entry ENSMUSG00000032580) produced 24 transcripts, 7 of which are predicted to be protein coding and another 4 which are predicted to be subject to nonsense-mediated RNA decay. The remaining 13 splice variants contain retained introns and are of undefined significance. Primers against exons 4–5 will detect 5 of the 7 predicted protein coding isoforms and an additional 6 of the transcripts which are thought to undergo non-sense mediated decay.
Western blotting
In order to assess the degree of protein reduction in Rbm5 +/− mice, lung tissue was from wild type and Rbm5 +/− adult mice then processed for western blotting as described previously30. Blots were probed using the RBM5 monoclonal antibody A9 described in17. The A9 antibody is predicted to be able to bind to all 7 of the protein coding isoforms, and would also bind to 3 of the 4 transcripts/proteins predicted to be subject to nonsense-mediated RNA decay if protein was to be produced.
Immunochemical labelling of cells
The localisation of RBM5 in the adult lung (8 weeks old) was determined by immunofluorescence using an RBM5 mouse monoclonal antibody as described previously17. To distinguish different cell lineages in the lung, we co-labelled sections with antibodies against Pro-surfactant protein C (AB3786, Merck Millipore), as a marker for type II alveolar epithelial cells, and CC10 (CC10 (T-18), SC-9772, Santa Cruz Biotechnology), as a marker for secretory cells. Protein localisation was determined through confocal microscopy using an SP-8 microscope (Leica Microsystems).
Immunochemical labelling of cells
In order to label cells undergoing apoptosis sections were staining using the TUNEL kit (ApopTag Peroxidase In Situ Apoptosis Detection Kit, Merck,S71000) as per the manufacturer’s instructions. In order to label proliferating cells, additional sections were labelled for Ki67 (NCL-Ki67p, Novacastra, at a dilution of 1 in 1000) using using immunohistochemistry. Bound antibody was detected using the Dako polymer, anti-rabbit, HRP kit as per the manufacturer’s instructions. The number of proliferating cells was subsequently quantitated from scanned slides using an Aperio ePathology scanner (Leica Biosystems) and Imagescope software. The number of apoptotic cells per tumour was too low to reliably quantitate. N = 6 samples per genotype were analysed. For both sets of labelling wild type tissue was used as a positive control.
Lung cancer induction
NNK (Sapphire Biosciences, Cat. No. 000–01622) was used to induce lung tumours. Six-week old Rbm5 +/− (HET) and Rbm5 +/+ (wild type, WT) mice were administered three i.p. injections over one week (Monday, Wednesday, Friday) at a dose of 50 mg/kg body weight. Mice were separated into two groups and were either injected with NNK (in saline, 0.1 ml volume) or vehicle (saline, 0.1 ml volume). Mice were humanely killed at 16, 20 and 48 weeks after the final NNK injection. Mice were anaesthetised, an incision was made in the trachea then lungs were perfusion fixed via a tracheal cannula with 4% formaldehyde at exactly 200 mm H2O pressure. Fixed tissues were paraffin embedded, sectioned and used for histological analysis (hematoxylin and eosin (H&E) staining). Total tumour area was measured using Imagescope software (Apeiro).
Data analysis was performed using GraphPad Prism 6 software and p < 0.05 was considered statistically significance. Histopathology was independently assessed in a blinded manner by a qualified pathologist (BK).
References
Ettinger, D. S. et al. Non-small cell lung cancer, version 6.2015. Journal of the National Comprehensive Cancer Network: JNCCN 13, 515–524 (2015).
Ferlay, J. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. International journal of cancer. Journal international du cancer 136, E359–386, https://doi.org/10.1002/ijc.29210 (2015).
Angeloni, D. Molecular analysis of deletions in human chromosome 3p21 and the role of resident cancer genes in disease. Briefings in functional genomics & proteomics 6, 19–39, https://doi.org/10.1093/bfgp/elm007 (2007).
Kok, K., Naylor, S. L. & Buys, C. H. Deletions of the short arm of chromosome 3 in solid tumors and the search for suppressor genes. Advances in cancer research 71, 27–92 (1997).
Oh, J. J., West, A. R., Fishbein, M. C. & Slamon, D. J. A candidate tumor suppressor gene, H37, from the human lung cancer tumor suppressor locus 3p21.3. Cancer research 62, 3207–3213 (2002).
Ramaswamy, S., Ross, K. N., Lander, E. S. & Golub, T. R. A molecular signature of metastasis in primary solid tumors. Nature genetics 33, 49–54, https://doi.org/10.1038/ng1060 (2003).
Oh, J. J. et al. RBM5/H37 tumor suppressor, located at the lung cancer hot spot 3p21.3, alters expression of genes involved in metastasis. Lung cancer 70, 253–262, https://doi.org/10.1016/j.lungcan.2010.02.012 (2010).
Rintala-Maki, N. D. et al. Expression of RBM5-related factors in primary breast tissue. Journal of cellular biochemistry 100, 1440–1458, https://doi.org/10.1002/jcb.21134 (2007).
Rintala-Maki, N. D., Abrasonis, V., Burd, M. & Sutherland, L. C. Genetic instability of RBM5/LUCA-15/H37 in MCF-7 breast carcinoma sublines may affect susceptibility to apoptosis. Cell biochemistry and function 22, 307–313, https://doi.org/10.1002/cbf.1106 (2004).
Welling, D. B., Lasak, J. M., Akhmametyeva, E., Ghaheri, B. & Chang, L. S. cDNA microarray analysis of vestibular schwannomas. Otology & neurotology: official publication of the American Otological Society, American Neurotology Society [and] European Academy of Otology and Neurotology 23, 736–748 (2002).
Scanlan, M. J. et al. Antigens recognized by autologous antibody in patients with renal-cell carcinoma. International journal of cancer. Journal international du cancer 83, 456–464 (1999).
Oh, J. J. et al. 3p21.3 tumor suppressor gene H37/Luca15/RBM5 inhibits growth of human lung cancer cells through cell cycle arrest and apoptosis. Cancer research 66, 3419–3427, https://doi.org/10.1158/0008-5472.CAN-05-1667 (2006).
Fushimi, K. et al. Up-regulation of the proapoptotic caspase 2 splicing isoform by a candidate tumor suppressor, RBM5. Proceedings of the National Academy of Sciences of the United States of America 105, 15708–15713, https://doi.org/10.1073/pnas.0805569105 (2008).
Bonnal, S. et al. RBM5/Luca-15/H37 regulates Fas alternative splice site pairing after exon definition. Molecular cell 32, 81–95, https://doi.org/10.1016/j.molcel.2008.08.008 (2008).
Jin, W., Niu, Z., Xu, D. & Li, X. RBM5 promotes exon 4 skipping of AID pre-mRNA by competing with the binding of U2AF65 to the polypyrimidine tract. FEBS letters 586, 3852–3857, https://doi.org/10.1016/j.febslet.2012.09.006 (2012).
Bechara, E. G., Sebestyen, E., Bernardis, I., Eyras, E. & Valcarcel, J. RBM5, 6, and 10 differentially regulate NUMB alternative splicing to control cancer cell proliferation. Molecular cell 52, 720–733, https://doi.org/10.1016/j.molcel.2013.11.010 (2013).
O’Bryan, M. K. et al. RBM5 is a male germ cell splicing factor and is required for spermatid differentiation and male fertility. PLoS genetics 9, e1003628, https://doi.org/10.1371/journal.pgen.1003628 (2013).
Sugliani, M., Brambilla, V., Clerkx, E. J., Koornneef, M. & Soppe, W. J. The conserved splicing factor SUA controls alternative splicing of the developmental regulator ABI3 in Arabidopsis. The Plant cell 22, 1936–1946, https://doi.org/10.1105/tpc.110.074674 (2010).
Oh, J. J., Grosshans, D. R., Wong, S. G. & Slamon, D. J. Identification of differentially expressed genes associated with HER-2/neu overexpression in human breast cancer cells. Nucleic acids research 27, 4008–4017 (1999).
Xu, X. et al. Evidence for type II cells as cells of origin of K-Ras-induced distal lung adenocarcinoma. Proceedings of the National Academy of Sciences of the United States of America 109, 4910–4915, https://doi.org/10.1073/pnas.1112499109 (2012).
Walser, T. et al. Smoking and lung cancer: the role of inflammation. Proceedings of the American Thoracic Society 5, 811–815, https://doi.org/10.1513/pats.200809-100TH (2008).
Zheng, H. C. & Takano, Y. NNK-Induced Lung Tumors: A Review of Animal Model. Journal of oncology 2011, 635379, https://doi.org/10.1155/2011/635379 (2011).
Hecht, S. S. Tobacco smoke carcinogens and lung cancer. Journal of the National Cancer Institute 91, 1194–1210 (1999).
Pao, W. et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS medicine 2, e17, https://doi.org/10.1371/journal.pmed.0020017 (2005).
Razani-Boroujerdi, S. & Sopori, M. L. Early manifestations of NNK-induced lung cancer: role of lung immunity in tumor susceptibility. American journal of respiratory cell and molecular biology 36, 13–19, https://doi.org/10.1165/rcmb.2005-0330OC (2007).
Edamatsu, H., Kaziro, Y. & Itoh, H. LUCA15, a putative tumour suppressor gene encoding an RNA-binding nuclear protein, is down-regulated in ras-transformed Rat-1 cells. Genes to cells: devoted to molecular & cellular mechanisms 5, 849–858 (2000).
Miller, A., Brooks, G. D., McLeod, L., Ruwanpura, S. & Jenkins, B. J. Differential involvement of gp130 signalling pathways in modulating tobacco carcinogen-induced lung tumourigenesis. Oncogene 0, https://doi.org/10.1038/onc.2014.99 (2014).
Ge, G. Z., Xu, T. R. & Chen, C. Tobacco carcinogen NNK-induced lung cancer animal models and associated carcinogenic mechanisms. Acta biochimica et biophysica Sinica 47, 477–487, https://doi.org/10.1093/abbs/gmv041 (2015).
Cotton, L. M. et al. Utilising the resources of the International Knockout Mouse Consortium: the Australian experience. Mammalian genome: official journal of the International Mammalian Genome Society 26, 142–153, https://doi.org/10.1007/s00335-015-9555-1 (2015).
Okuda, H. et al. LRGUK1 is part of a multiprotein complex required for manchette function and male fertility. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 31, 1141–1152, https://doi.org/10.1096/fj.201600909R (2017).
Acknowledgements
This work was supported by grants from the Cancer Council of Victoria to DJ and MKOB. (#1059923) and the Lung Cancer Research Foundation to DJ. MKOB was supported in part by a NHMRC Principal Research Fellow (APP1058356). We thank Penelope Mitchell and Brett Clark for technical assistance.
Author information
Authors and Affiliations
Contributions
D.J., D.N.W., B.J.J. and M.K.O.B. - designed the experiments. D.J., D.N.W., A.E.O.C., D.J.M., S.G., A.D.B., B.K., A.M., T.J.C., B.J.J. and M.K.O.B. - conducted the experiments and analyzed data. D.J., T.J.C. and M.K.O.B. - wrote the manuscript. D.J. and A.E.O.C. - prepared the figures. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jamsai, D., Watkins, D.N., O’Connor, A.E. et al. In vivo evidence that RBM5 is a tumour suppressor in the lung. Sci Rep 7, 16323 (2017). https://doi.org/10.1038/s41598-017-15874-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-15874-9
This article is cited by
-
High resolution fluorescence imaging of the alveolar scaffold as a novel tool to assess lung injury
Scientific Reports (2024)
-
RNA splicing dysregulation and the hallmarks of cancer
Nature Reviews Cancer (2023)
-
Structural basis for specific RNA recognition by the alternative splicing factor RBM5
Nature Communications (2023)
-
A de novo paradigm for male infertility
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
