
Abstract
The phylogeographic structure of the land snail Camaena cicatricosa was analyzed in this study based on mitochondrial gene (COI and 16srRNA, mt DNA) and internal transcribed spacer (ITS2) sequences in 347 individuals. This snail is the vector of the zoonotic food-borne parasite Angiostrongylus cantonensis and one of the main harmful snails distributed exclusively in China. The results revealed significant fixation indices of genetic differentiation and high gene flow between most populations except several populations. An isolation-by-distance test showed no significant correlation between genetic distance and geographical distance among C. cicatricosa populations, which suggested that gene flow was not restricted by distance. The levels of haplotype and nucleotide diversity of C. cicatricosa were generally high, except those in some special populations, according to the mt DNA and ITS2 data. Furthermore, the phylogenetic trees and asteroid networks of haplotypes indicated nonobvious genetic structure, the same as results got based on the synonymous and non synonymous sites of 347 sequences of the COI gene. All lines of evidence indicated that climatic changes and geographical and human barriers do not substantially affect the current population structure and distribution of the investigated snails.
Similar content being viewed by others

Introduction
Molecular phylogeographic analyses can provide valuable information on specific genetic structures, genetic variation and population formation1. However, the population phylogeography of an organism can be affected by various factors, such as climate, geographical conditions, the ecological environment, and historical processes as well as human activities etc.2,3,4. The influence of physical barriers and environmental variations on genetic structure has been investigated in many organisms, such as Sula leucogaster, Alectoris chukar and Adelphocoris suturalis 5,6,7. An increasing number of researchers are now studying population genetics using cytoplasmic and nuclear genomes, due to the large amount of evolutionary information they provide7,8,9. The genetic structure of camaenids is currently poorly understood because of the difficulty of collecting specimens. In land snails, gene flow among different populations appears to be limited because of their poor migration ability10,11. Additionally, the morphology of the shell shows great intra-specific variation in the family Camaenidae, in terms of shell size, shape, color, spiral bands, growth lines, aperture, and the umbilicus, and these aspects may be affected by both the local environment and genetic evolution9. Therefore, investigation of the population genetic structure and phylogeography of camaenids will significantly enhance the current understanding of the geographic differentiation and historical biogeography of land snails.
The snail Camaena cicatricosa (Müller, 1774) (Stylommatophora, Camaenidae) is an important and harmful terrestrial mollusk found in southern China. It not only damages crops, leading to reductions in yield and quality, but also spreads zoonotic food-borne parasitic disease, and causes substantial damage to human and animal health12. The classification of this species is rather confusing, and its scientific name has been repeatedly revised. Different scholars present diverse viewpoints13,14,15,16,17,18,19,20. Shell morphological characteristics of C.cicatricosa are variable, and four synonymous names, which are C. c. ducalis (Ancey, 1885), C. c. inflata (Mllendorff, 1885), C. c. obtecta (Fischer, 1898) as well as C. c. connectens (Dautzenberg & Fischer, 1906) have been proposed. However, these subspecies are different each other in shell shape and size, openness of umbilicus, sharpness of peripheral angel, convexity of whorls and the presence of a hump beside umbilicus. These taxa previously were treated as synonyms, varieties or subspecies of C.cicatricosa based on only comparative shell morphology13,14,15,16,17,18,19,20. In 2016, the sinistral were revised and upgraded to species by Ding et al. according to shell morphology, anatomical as well as molecular characteristics21. Some studies have indicated that this snail is mainly distributed in Guangdong, Guangxi, Yunnan, Guizhou, Hunan, Hainan and Vietnam17,22,23. However, researchers recently clarified the phylogeny and taxonomy of sinistral camaenids, and found that this camaenid snail is distributed only in the provinces of Guangdong and Guangxi21. Previous studies on this snail have mainly focused on its taxonomy and mitochondrial genome21,23, but the phylogeography and population structure of C. cicatricosa have not been thoroughly understood.
In the present study, the genetic structure of this species was assessed based on mitochondrial cytochrome c oxidase subunit 1 (COI), 16srRNA and nuclear internal transcribed spacer II region (ITS2) sequences. The main goals of this study were to (i) analyze the genetic diversity and structure of C. cicatricosa and (ii) examine the geographical pattern of its haplotypes. Genetic variation and gene flow were also evaluated, and factors influencing genetic variation were investigated.
Results
Genetic diversity
A final combined dataset consisting 861 bp of mitochondrial gene sequences was obtained from 347 individuals, including fragments of the COI (559 bp) and 16srRNA (302 bp) genes, with the following nucleotide content: 37.56% T, 13.80% C, 29.79% A and 18.85% G. A total of 46 polymorphic sites were identified including 7 singleton variables and 39 parsimony-informative sites. No insertions or deletions were detected among these fragments. The average number of nucleotide differences ranged from 0.000 to 6.725, with an average of 2.513. Haplotype diversity ranged from 0.000 to 0.848, with an average of 0.454, and nucleotide diversity from 0.000 to 0.008, with an average of 0.003 (Table S1). Thirty-nine haplotypes, including 13 shared haplotypes and 26 unique haplotypes, were derived from all of the individuals. The haplotype with the highest frequency, Hap1, was found in populations CH, GM, QY, ST, TH, ZS, NN and ZP, which accounted for 23.34%. The other frequent haplotypes included Hap13 (accounting for 8.36%), Hap3 (accounting for 7.78%), Hap5 and Hap20 (accounting for 7.2% each) (Table S2). The pairwise Fst values between all populations were significantly different (P < 0.01; P < 0.05). The maximum value between HY and GM, HJ and GM, HZ and GM, HJ and HY, HZ and HY, WZ and HY, HZ and HJ, WZ and HJ, and WZ and HZ was 1.000 uniformly, followed by values between populations ST and GM, TH and GM, ST and HY, TH and HY, ST and HJ. The smallest Fst value, between ZS and CH, was −0.048, followed by −0.015 between populations SZ and HJ (Table S3). An isolation-by-distance (IBD) test showed no significant correlation between genetic distance and geographical distance (R2 = 0.0423) among all of the populations, and the results indicated that gene flow was not restricted by distance (Fig. 1).

Scatter plot showing the correlation of genetic distance and geographical distance (km) based on mt DNA data.
For the ITS2 region, 347 sequences were successfully acquired, with a length of 549 bp and nucleotide content of 22.22% T, 25.93% C, 18.12% A, and 33.73% G. Forty-six polymorphic sites, including 27 singleton variable and 19 parsimony-informative sites, were obtained. The average number of nucleotide differences ranged from 0.000 to 2.788, with an average of 0.775. Haplotype diversity ranged from 0.000 to 0.824, with an average of 0.373, and nucleotide diversity from 0.000 to 0.005, with an average of 0.002 (Table S4). Twenty-one shared haplotypes and eight unique haplotypes were derived from all individuals. Hap3 was dispersed in all populations, except YD and appeared 215 times, accounting for 61.96% of the haplotypes. Other frequent haplotypes included Hap8 (accounting for 13.26%), Hap16 (accounting for 6.34%), Hap6 and Hap7 (accounting for 4.03% respectively) (Table S5). The pairwise Fst values between most populations indicated significant differences, especially between WZ and YC, YD and YC, and HJ and YC. The smallest Fst value, between HJ and CH, was −0.096 (Table S6). An IBD test showed no significant correlation between genetic distance and geographical distance (R2 = 0.0098) among all of the populations (Fig. 2). The results were in accordance with the mt DNA data.

Scatter plot showing the correlation of genetic distance and geographical distance (km) based on ITS2 data.
Gene flow
The pairwise Nm values between all populations were acquired based on mitochondrial gene. The largest value between CH and SH was 3.538, what followed were 2.726 between ZQ and GP, 2.087 between YD and NN, 2.044 between ZQ and ZP. The smallest Nm value was −16.917 between SZ and HJ, and the second smallest was −5.459 between CH and ZS (Table S7). For the ITS2, five populations (LFS, NN, WZ, CH, YF) showed much higher levels of gene flow than the rest of studied populations. The largest value was 35.464, but the smallest value was merely −125.250 (Table S8).
Phylogenetic analyses and network construction
Molecular phylogenetic analyses were conducted based on the mt DNA sequences of thirty-nine haplytypes as well as sequences from two additional specimens, C. jinpingensis and C. menglunensis, which were used as outgroup to root the tree because of their close genetic relationship. The synonymous and non synonymous sites of 347 sequences from a single COI gene were used to build phylogenetic trees as well. Based on the tree using the NJ method, it was observed that populations from Guangdong and Guangxi provinces were mixed together and did not present an evident structure (Figs 3, 4 and 5). In the phylogenetic tree based on synonymous sites, certain individuals from the same population showed sister relationships firstly, and then gathered together with other individuals. While in the phylogenetic tree based on non synonymous sites, all individuals except ZQ16 and ZQ20 have equal relationships.

Phylogenetic tree inferred via the ML and NJ methods based on mt DNA data. Red represents the Guangxi populations; Blue represents the Guangdong populations. Numbers on and below the nodes represent ML and NJ bootstrap values respectively.

Phylogenetic tree inferred via the NJ method based on synonymous sites of the COI gene.

Phylogenetic tree inferred via the NJ method based on non synonymous sites of the COI gene.
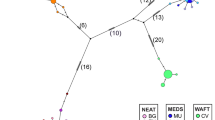
In a haplotype network, the ancient haplotype should generally be located at the center of the network, being widely distributed among populations, while, other more recent haplotypes should be located at the tips of the network. In this asteroid mitochondrial haplotype network, the most frequent haplotype (Hap1) was located at the center of the network, being distributed in populations CH, GM, QY, ST, TH, ZS, NN and ZP, including 81 individuals. Therefore, Hap1 was considered the major haplotype. The other haplotypes, including Hap3, Hap5, Hap13 and Hap20, were comprised of 27, 25, 28 and 25 individuals, respectively. One exclusive haplotype (Hap33) and two missing haplotypes had mutated from Hap1, and other haplotypes had mutated from these two missing haplotypes. Hap15, which appeared 20 times, had mutated into 10 other haplotypes and 1 missing haplotype. The star-like network suggested that Hap33, Hap29 and Hap19 were different from most other haplotypes by only few mutations (Fig. 6). Furthermore, all of the haplotypes in this network could not be divided into effective groups.

Haplotype network of C. cicatricosa based on mt DNA.
Molecular phylogenies were also analyzed based on ITS2 sequences from 29 haplytypes using NJ and ML methods. The snails C. jinpingensis and C. menglunensis were chosen as outgroup here as well. In these two trees, populations from Guangdong and Guangxi provinces were again mixed together and did not exhibit an evident structure (Fig. 7). This result was in accordance with the mt DNA data.

Phylogenetic tree inferred via the ML and NJ methods based on ITS2 sequences. Red represents the Guangxi populations; Blue represents the Guangdong populations. Numbers on and below the nodes represent ML and NJ bootstrap values respectively.
Anther asteroid network was acquired based on ITS2 haplotypes. Hap3 was located at the center of this network and was considered the major haplotype. Two other haplotypes (Hap8 and Hap16) were also common and were distributed in additional populations. The major haplotype Hap3 appeared 215 times and generated 12 other haplotypes as well as 4 kinds of missing haplotypes. The rest of the haplotypes were likely generated from the major haplotype two or three times. Additionally, 21 exclusive haplotypes were connected with the major haplotypes through a mutation, 15 of which were located at the end of the network. The haplotype Hap3 was found in the most individuals, except the YD population from Guangdong. The populations in Guangdong included all of the shared haplotypes except Hap26. Only five haplotypes were distributed in Guangxi populations, which included only five individuals, four of which were located at the edge of the network (Fig. 8). Similar to the results based on mtDNA data, different populations of C. cicatricosa could not be divided into groups, indicating an inconspicuous genetic structure.

Haplotype network of C. cicatricosa based on ITS2 sequences.
Discussion
Genetic diversity is the basis of ecosystem diversity and species diversity, and any species has its unique gene pool or genetic organization form. Generalized genetic diversity refers to the sum of the genetic information carried by all organisms on the planet. In a narrow sense, genetic diversity refers to analyses within the species, namely genetic variation between different populations within species or different individuals in a population. Genetic diversity includes not only the level of variation, but also the distribution pattern of variation, that is, the genetic structure of population24,25. Up to now, there are lots of studies on species genetic diversity, including plants, insects, fish, aquatic mollusk and others7,26,27,28,29. However, there is no study on camaenids in this field. What factors affect the level of genetic diversity of camaenids? Which factors have great influence and which having small influence? How they impact on the weak migrating snails? Above questions are confusing. In 2012, Leffer et al. published a comprehensive and novel comment on the level of genetic diversity within species30. They pointed out that in addition to the geographic range, other complex factors such as ecological factors, life history traits and genome architecture of closely related species are expected to have discernable effects on genetic diversity, which remains incompletely understood by the present paradigm30,31. In the present study, we have analyzed the genetic diversity of the land snail C. catricosa and related influencing factors based on mitochondrial genes and ITS2 region, which is the type species of the genus Camaena. Although these data are limited, they can provide reference basis and valuable resources for the subsequent study.
Nucleotide transition usually appears in classification orders that show close relationship, and nucleotide transversion is evident in classification orders that show distant relationships32,33. In this study, nucleotide transition was the main variation observed among species of C. cicatricosa, which is consistent with previous research. In the genetic diversity analyses, populations GM, HZ, HJ, HY and ZP exhibited lower haplotype and nucleotide diversity (Table S1); four of these populations were distributed in Guangdong, while one was distributed in Guangxi. The landscape of Guangdong is comprised of low mountain ranges, rolling country and plains, extending from north to south. The mountains of Guangdong generally exhibit a northeast – southwest orientation. The southern border of this province faces the South China Sea, and disasters such as floods, typhoons and droughts occur often34. Therefore, the terrain as well as the climate may play an important role in these populations with lower genetic diversity, as observed in other organism, such as Adelphocoris suturalis and Helix aspersa 26,27. The ZP population in Guangxi is located in the northern area and is isolated by mountains, implying that physical barrier might also cause lower diversity level.
The values of the fixation indices between populations are among the important parameters used to measure the degree of genetic differentiation28. A low level of genetic differentiation exists between populations when Fst is between 0 and 0.05. Whereas, a high level of genetic differentiation exists among populations when Fst is greater than 0.1529. In this study, the obtained pairwise Fst values showed significant genetic differentiation among 20 populations based on the analyses of mt DNA. (Table S3). However, the phylogenetic analyses and network construction showed a lack of genetic structure. It is suggested that the high level of gene flow could result in homogeneity39. Almost all pairwise Fst values based on mtDNA among populations were over 0.15 (Table S3). But the data based on ITS2 sequences were not significant as those of mt DNA. It is indicated that the nuclear gene had a relatively low variability and a slow evolutionary rate compared to mt DNA1,40. Environment, climate and physical barriers factors may play important roles in genetic differentiation. For example, the Nanling Mountains, Pearl River, Guijiang and Xijiang River might have acted as geographical barriers41. On the other hand, there are many primitive forests in Guangxi including Shiwan mountain National Preserve, Yulin mountain Forest Park and so on, which served as ecological environmental and climate barriers42.
Gene flow is one of the main factors that are used to estimate population genetic structure. Populations with a high level of gene flow exhibit fewer genetic differentiations than populations with low gene flow43. When Nm > 1, high gene flow and low genetic differentiation generally exist in populations, whereas Nm < 1 indicates that populations are differentiated because of genetic drift43. In the present study, relatively large Nm values existed between the most populations, and values between the least populations were small (Tables S7, S8). Although this particular snail has low dispersal ability, the anthropochory, wind, water and other factors can lead to a wider distribution, especially human activities19. In the two examined provinces, C. cicatricosa is used to produce food and multi- functional medicines, and can be carried far away from its home territory44. Further more, trade in Guangdong province is frequent, and snails could be spread through cargo transportation too.
There are many factors that affect genetic structure and population distribution. An isolation-by-distance test showed no significant correlation between genetic distance and geographical distance among C. cicatricosa populations, which suggested that gene flow is not restricted by distance. Height, microclimate, host species and other factors in different locations could affect genetic structure. Recently, Huang reviewed a new hypothesis about the cause of genetic diversity in species referred to as the maximum genetic diversity (MGD) hypothesis32. An important novel point of the MGD hypothesis is its emphasis on the internal system or physiology of a species45. Because of its adaptation to mankind-disturbed environments, such as farmland and forest ecotone, this species has a large distributional range. The probability to be passively transported far distances and to establish a new population through human activities is also very high46,47. Besides, the snail C. cicatricosa is hermaphroditic creature mating with other conspecific individuals, which can laid more eggs (10–25 eggs each clutch), and has shorter gestation period (5–36 days) between the last copulation and the first egg-laying44,48. Organisms having higher fecundity and abundance tend to be more competitive49. The complex topography and geomorphology, varied physical conditions and a wide diversity of ecosystems in mountainous areas may result in allopatric and sympatric speciation50,51. Based on the dominance of C. cicatricosa in these areas, we must analyze physiological and ecological sections of this species in future research.
In the present study, the phylogenetic trees and networks of haplotypes based on the obtained datasets showed a lack of genetic structure among C. cicatricosa populations. These findings were confirmed by phylogenetic trees based on synonymous and non synonymous sites. Though the populations in Guangdong and Guangxi still can not be separated in the phylogenetic tree based on synonymous sites, individuals from the same population or province have closer relationships than those from different populations or provinces. The phylogenetic tree based on non synonymous sites suggested equal relationships except ZQ16 and ZQ20. That is to say the phylogenetic tree based on synonymous sites could reflect further information and make more sense. The results of this study suggested that the cause of homogeneity is the high level of gene flow, as demonstrated in other animals39. It is inferred that climatic changes and geographical barriers do not substantially affect the current population structure and distribution of this snail. In the future, more samples and a broader distribution of sampling locations are the necessary first step to study the genetic distribution patterns of C. cicatricosa. Most importantly, physiology, biology and ecology should not be overlooked.
Methods
Sample collection
This study was based on 347 individuals collected by the authors from 20 locations in China in 2013–2015 years (Table 1, Fig. 9)21,52,53. The geographic coordinates were recorded using a GPS. Live adults were drowned in water for 12–24 hours and then euthanized in hot water to ensure their death. Their soft bodies were preserved in 75% or 95% ethanol and stored at −20 °C. The empty shells were cleaned, dried and preserved at room temperature. Samples were deposited in the State Key Laboratory of Molluscan Quarantine and Identification, FJIQBC.

Map of sampling locations for the snail C. cicatricosa.
DNA extraction, amplification and sequencing
Approximately 0.02–0.04 g of foot muscle tissue was used for DNA extraction. The muscle tissue was bathed in sterile water for 3–6 hours to remove residual alcohol. Total genomic DNA was isolated using the Qiagen DNeasy Blood and Tissue kit (QIAGEN), and stored at −20 °C for further use. Partial fragments of the mitochondrial COI and 16srRNA genes, and the total sequence of ITS2 were amplified via PCR using the primer pairs (Table S9), reaction systems and amplification conditions listed in Table S9. The PCR products were analyzed through 1.2% agarose gel electrophoresis.
After sequencing, the raw sequences were proof-read in chromatograms and aligned into contigs using BioEdit 7.254. ITS2 sequences were annotated using HMMer55 and the ITS2 Database56. The alignment of mitochondrial protein-coding genes was inferred from the amino acid alignment and examined for the presence of stop codons and other indicators. Sequence alignments were generated using ClustalW implemented in MEGA 657. Sixty-seven haplotype sequences were generated and deposited in GenBank under accession numbers KU927017–KU927046 for COI, KX365248–KX365255 for 16srRNA and KU958515–KU958543 for ITS2.
The nucleotide composition, mutation sites and base substitution were analyzed in MEGA 657. The number of polymorphic sites (S), haplotype diversity (Hd), nucleotide diversity (Pi), average number of nucleotide differences (K) and number of haplotypes (Hap) were calculated using DnaSP 5.058 or Arlequin 3.559. The gene flow was estimated using DnaSP 5.058 too.
The genetic differentiation among populations was further estimated based on pairwise FST values (the values of fixation indices of genetic differentiation between populations). Arlequin 3.559 was employed to calculate the genetic distance matrix to test for the presence of IBD in the dataset. Google Earth (http://earth.google.com) was used to estimate the linear geographic distance (km) between the sampling locations. Significance was tested with the Mantel test employing 1000 randomizations in IBDWS 3.2360.
Phylogeographic analyses and network construction
Two datasets of mitochondrial and nuclear haplotypes were analyzed through both neighbor-joining (NJ) and maximum likelihood (ML) analyses using MEGA6 with default settings. Furthermore, the synonymous and non synonymous sites of 347 sequences of the COI gene were employed to build phylogenetic trees. The node support values were assessed via bootstrap resampling using 1000 replicates. C. jinpingensis and C. menglunensis were employed as outgroup due to their close relationship with C. cicatricosa. The haplotype networks of the ITS2 and mt DNA data were constructed in Network 4.661 with the median-joining algorithm.
References
Avise, J. Phylogeography: the history and formation of species. Harvard university press (2000).
Bohonak, A. J. Dispersal, gene flow, and population structure. Q. Rev. Biol. 74, 21–45 (1999).
Byrne, M. Evidence for multiple refugia at different time scales during Pleistocene climatic oscillations in southern Australia inferred from phylogeography. Quat. Sci. Rev. 27, 2576–2585 (2008).
Jin, Y. T., Brown, R. P. & Liu, N. F. Cladogenesis and phylogeography of the lizard Phrynocephalus vlangalii (Agamidae) on the Tibetan Plateau. Mol. Ecol. 17, 1971–1982 (2008).
Huang, Z., Liu, N., Zhou, T. & Ju, B. Effects of environmental factors on the population genetic structure in chukar partridge (Alectoris chukar). J. Arid. Environ. 62, 427–434 (2005).
MorrisPocock, J. A., Steeves, T. E., Estela, F. A., Anderson, D. J. & Friesen, V. L. Comparative phylogeography of brown (Sula leucogaster) and red-footed boobies (S. sula): the influence of physical barriers and habitat preference on gene flow in pelagic seabirds. Mol. Phylogenet. Evol. 54, 883–896 (2010).
Zhang, L. J. et al. Phylogeographic structure of cotton pest Adelphocoris suturalis (Hemiptera: Miridae): strong subdivision in China inferred from mtDNA and rDNA ITS markers. Sci. Rep. 5, 14009 (2015).
Ye, Z., Zhu, G. P., Chen, P. P., Zhang, D. L. & Bu, W. J. Molecular data and ecological niche modeling reveal the Pleistocene history of a semi-aquatic bug (Microvelia douglasi douglasi) in East Asia. Mol. Ecol. 23, 3080–3096 (2014).
Nishi, H. & Sota, T. Geographical divergence in the Japanese land snail Euhadra herklotsi inferred from its molecular phylogeny and genital characters. Zool Sci. 24, 475–485 (2007).
Gittenberger, E., Piel, W. H. & Groenenberg, D. S. J. The Pleistocene glaciations and the evolutionary history of the polytypic snail species Arianta arbustorum (Gastropoda, Pulmonata, Helicidae). Mol. Phylogenet. Evol. 30, 64–73 (2004).
Rundell, R. J., Holland, B. S. & Cowie, R. H. Molecular phylogeny and biogeography of the endemic Hawaiian Succineidae (Gastropoda: Pulmonata). Mol. Phylogenet. Evol. 31, 246–255 (2004).
Zhou, W. C. et al. The intermediate host of Angiostrongylus cantonensis Molluscan. Chinese J of Zoon. 23, 401–408 (2007).
Pilsbry, H. A. Manual of Conchology. Academy of Natural Sciences, Philadelphia, USA (1891).
Pilsbry, H.A. Manual of Conchology. Academy of Natural Sciences, Philadelphia, USA (1894).
Fischer, H. & Dautzenberg, P. Catalogue des mollusques terrestres et fluviatiles de l’Indo-Chine orientale cites jusqu’à ce jour. Mission Pavie, Etudes diverses. 3, 390–442 (1904).
Dautzenberg, P. H. & Fischer, H. Liste de mollusques récoltés par M. H. Mansuy en Indo-chine et au Yunnan, et description d’espèces nouvelles. J. de Conchylogie. 53, 343–471 (1906).
Yen, T. C. Die chinesischen land-und Sü.wasser-Gastropoden des Natur-Museums Senckenberg. Abhandlungen der Senckenbergischen Naturforschenden Gesellschaft 444, 1–235 (1939).
Zilch, A. Die Typen und Typoid des Natur-Museums Senckenberg, 29: Mollusca, Camaenidae (3). Arch. Molluskenkund. 93, 243–262 (1964).
Chen, D. N. & Gao, J. X. Economic Fauna Sinica of China, Terrestrial Mollusca. Science Press, Beijing (1987).
Schileyko, A. A. Check-list of land pulmonate molluscs of Vietnam (Gastropoda: Stylommatophora). Ruthenica 21, 1–68 (2011).
Ding, H. L. et al. Revision of sinistral land snails of the genus Camaena (Stylommatophora, Camaenidae) from China based on morphological and molecular data, with description of a new species from Guangxi, China. ZooKeys 584, 25–48 (2016).
Müller, O. F. Vermivm terrestrium et fluviatilium, v.2. Havni., apud Heineck et Faber (1774).
Wang, P. et al. The mitochondrial genome of the land snail Camaena cicatricosa (Müller, 1774) (Stylommatophora, Camaenidae): the first complete sequence in the family Camaenidae. ZooKeys 451, 33–48 (2014).
Nei, M. Molecular evolutionary genetics. Columbia University Press, NewYork (1987).
McNeely, J. A., Miller, K. R., Reid, W. V., Mittermeier, R. A. & Werner, T. B. Conserving the world’s biological diversity. Gland, IUCN (1990).
López, A. & Bonasora, M. G. Phylogeography, genetic diversity and population structure in a Patagonian endemic plant. Aob Plants 9, (2017)
Wieczorek, K. & Osek, J. Prevalence, genetic diversity and antimicrobial resistance of Listeria monocytogenes isolated from fresh and smoked fish in Poland. Food Microbiology 64, 164–171 (2017).
Song, J., Li, Q., Zhong, X., Kong, L. & Yu, H. Genetic diversity and outlier loci detecting of shell color variation in the Pacific oyster (Crassostrea gigas) by SNP markers. Aquatic Living Resources 30, 10 (2017).
Jones, J. S. Ecological genetics and natural selection in molluscs. Science 182, 546–552 (1973).
Leffler, E. M. et al. Revisiting an old riddle: what determines genetic diversity levels within species? PLoS Biol 10, e1001388 (2012).
Nordborg, M. Linkage disequilibrium, gene trees and selfing: an ancestral recombination graph with partial self-fertilization. Genetics 154, 923–929 (2000).
Simon, C. et al. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 87, 651–701 (1994).
Frati, F., Simon, C., Sullivan, J. & Swofford, D. Evolution of the mitochondrial cytochrome oxidase II gene in Collembola. J. Mol. Evol. 44, 145–158 (1997).
Wu, Y. W. Geography of Guangdong. Guangdong people’s press, Guangzhou (1973)
Ting, Y. Q. Studies on the ecological characteristics of cotton mirids-effect of temperature and humidity on the development and distribution of the pests. Plant Prot. 2, 285–296 (1963).
Arnaud, J. F. Metapopulation genetic structure and migration pathways in the land snail Helix aspersa: influence of landscape heterogeneity. Landscape Ecol. 18, 333–346 (2003).
Rousset, F. Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics. 145, 1219–1228 (1997).
Weight, S. Evolution and the genetics of population variability within and among natural population. University of Chicago Press, Chicago (1978).
Szalanski, A. L., Austin, J. W., McKern, J. A., Steelman, C. D. & Gold, R. E. Mitochondrial and ribosomal internal transcribed spacer 1 diversity of Cimex lectularius (Hemiptera: Cimicidae). J. Med. Entomol. 45, 229–236 (2008).
Avise, J. C. Molecular markers, natural history and evolution. Chapman and Hall, New York (1994).
You, Q. J., Lu, W., Jiang, Z. H., Chen, Y. Y. & Meng, C. H. Outline of insect ecogeography in Guangxi. Guangxi Sciences 1, 37–42 (1994).
Wei, Y. L., Wang, G. Z., Chen, W. H., Luo, S. W. & Luo, Q. K. Characteristics and formation and evolution of granite landscape of the five emperor Mountain National Geopark in Guangxi, Pubei. Tropical geography 37, 66–81 (2017).
Millar, C. L., Libby, W. J., Falk, D. A. & Holsinger, K. E. Strategies for conserving clinal, ecotypic, and disjunct population diversity in widespread species. Oxford University Press, New York (1991).
Zhang, L. Researches on the individual development and isozyme of Camaena cicatricose. Guizhou Normal University, Guizhou (2008).
Huang, S. New thoughts on an old riddle: What determines genetic diversity within and between species. Genomics 108, 3–10 (2016).
Aubry, S., Labaune, C., Magnin, F., Roche, P. & Kiss, L. Active and passive dispersal of an invading land snail in Mediterranean France. J. of Anim. Ecol. 75, 802–813 (2006).
Guiller, A. & Madec, L. Historical biogeography of the land snail Cornu aspersum: a new scenario inferred from haplotype distribution in the Western Mediterranean basin. BMC Evol. Biol. 10, 18 (2010).
Xiao, W. L. Study on the bionomics of the Camaena cicatricosa. Journal of Jinan University 1, 46–52 (1989).
Stearns, S. C. The evolution of life histories. Oxford University Press, Oxford (1992).
Harl, J. et al. Phylogeography of the land snail genus Orcula (Orculidae, Stylommatophora) with emphasis on the Eastern Alpine taxa: speciation, hybridization and morphological variation. BMC Evol. Biol. 14, 223 (2014).
Criscione, F. & Khler, F. Snails in the desert: assessing the mitochondrial and morphological diversity and the influence of aestivation behavior on lineage differentiation in the Australian endemic Granulomelon Iredale, 1933 (Stylommatophora: Camaenidae). Mol. Phylogenet. Evol. 94, 101–112 (2016).
Folmer, O., Black, M., Hoeh, W., Lutz, R. & Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Marine. Bio. 3, 294–299 (1994).
Palumbi, S. et al. The Simple Fool’s Guide to PCR. University of Hawaii, Honolulu (1991).
Hall, T. A. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98 (1999).
Eddy, S. Profile hidden markov models. Bioinformatics. 14, 755–763 (1998).
Koetschan, C. et al. The ITS2 database III - sequences and structures for phylogeny. Nucleic. Acids. Res. 38, 275–279 (2010).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Librado, P. & Rozas, J. DnaSPv5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 25, 1451–1452 (2009).
Excoffier, L. & Lischer, H. L. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567 (2010).
Jensen, J. L., Bohonak, A. J. & Kelley, S. T. Isolation by distance web service. Bmc. Genet. 6, 13 (2005).
Bandelt, H. J., Macaulay, V. & Richards, M. Median networks: speedy construction and greedy reduction, one simulation, and two case studies from human mtDNA. Mol. Phylogenet. Evol. 16, 8–28 (2000).
Acknowledgements
This research is supported by the National Natural Science Foundation of China (31372162), the Agricultural Science and Technology Major Project Funds of Fujian (2017NZ0003-1) and the National Key R&D Program of China (2017YFF0210304).
Author information
Authors and Affiliations
Contributions
W.C. Zhou, H.F. Yang and P. Wang designed the study and conducted the analyses. S.P. Yang and W.C. Zhou collected samples. P. Wang, H.F. Yang and W.C. Zhou wrote the manuscript. H.L. Ding and J.H. Lin performed the molecular analyses. All authors discussed results and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhou, W., Yang, H., Ding, H. et al. Population genetic structure of the land snail Camaena cicatricosa (Stylommatophora, Camaenidae) in China inferred from mitochondrial genes and ITS2 sequences. Sci Rep 7, 15590 (2017). https://doi.org/10.1038/s41598-017-15758-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-15758-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
