
Abstract
Coronary heart disease (CHD) is associated with complex metabolic disorders, but its molecular aetiology remains unclear. Using a novel nontargeted metabolomics approach, we explored the global metabolic perturbation profile for CHD. Blood samples from 150 patients with severe obstructive CHD and 150 angiographically normal controls were collected. Metabolic fingerprinting was performed by ultra-high performance liquid chromatography coupled to quadruple time-of-flight mass spectrometry (UHPLC-QTOF/MS) technique. After adjusting for CHD traditional risk factors and metabolic batch, a comprehensive list of 105 metabolites was found to be significantly altered in CHD patients. Among the metabolites identified, six metabolites were discovered to have the strongest correlation with CHD after adjusting for multiple testing: palmitic acid (β = 0.205; p < 0.0001), linoleic acid (β = 0.133; p < 0.0001), 4-pyridoxic acid (β = 0.142; p < 0.0001), phosphatidylglycerol (20:3/2:0) (β = 0.287; p < 0.0001), carnitine (14:1) (β = 0.332; p < 0.0001) and lithocholic acid (β = 0.224; p < 0.0001); of these, 4-pyridoxic acid, lithocholic acid and phosphatidylglycerol (20:3/2:0) were, to the best of our knowledge, first reported in this study. A logistic regression model further quantified their positive independent correlations with CHD. In conclusion, this study surveyed a broad panel of nontargeted metabolites in Chinese CHD populations and identified novel metabolites that are potentially involved in CHD pathogenesis.
Similar content being viewed by others

Introduction
Coronary heart disease (CHD) is one of the leading causes of mortality worldwide, but its pathogenic mechanism remains poorly understood. It is well-known that CHD is linked with complex metabolic disorders, such as obesity, insulin resistance, and diabetes1,2. And certain circulating metabolites, such as homocysteine, cholesterol and triglycerides, have been recognized for decades as being strongly associated with CHD3,4,5. Beyond these well-known associations, researchers believe that there might still be many other dysregulated metabolites that have yet to be described. Therefore, metabolomics, a powerful approach to detect a broad spectrum of small-molecule metabolites, is increasingly being applied to CHD studies and is leading to novel discoveries of metabolic biomarkers and implications of their causal relationship to CHD pathogenesis6,7,8. Previous metabolomics studies concerning CHD, with few exceptions, were assessed on targeted metabolomics platforms6,7,9,10,11. These studies examined a predefined group of potential metabolites that was mostly culled from diabetes metabolomics studies6,7,9,10. Meanwhile, the nontargeted metabolomics approach provides a global fingerprint of information through the simultaneous measurement of as many metabolites as possible and may better contribute to discovery-driven research on coronary atheroma8,12. Our study used an ultra-high performance liquid chromatography coupled with the quadruple time-of-flight mass spectrometry (UHPLC-QTOF/MS)-based nontargeted metabolomics approach to explore the molecular perturbation profile of severe CHD patients compared with angiographically normal controls. We aimed to extend the current knowledge beyond previously reported targeted metabolite changes by examining the global plasma metabolomics profile of coronary heart disease and searching for new metabolites that are potentially involved in CHD pathogenesis.
Methods
Participant recruitment
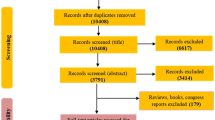
Blood samples of 980 patients who underwent coronary angiography to evaluate CHD at FuWai Hospital between June 2011 and March 2015 were collected for our study. We excluded participants who had organic heart diseases (rheumatic heart disease, etc.), liver/renal dysfunctions, a severe infection or malignant tumours and hyperthyroidism or any other autoimmune disease, resulting in a cohort of 795 eligible participants. To explore alterations in the plasma metabolites that may be associated with CHD, two independent case-control groups were constructed based on strict coronary angiographic evidence: one group consisted of 150 CHD subjects (at least 1 major coronary artery with ≥ 80% stenosis and at least one stent implanted), and one group consisted of 150 age- and gender-matched control subjects (coronary arteries with ≤ 20% stenosis). The nontargeted plasma metabolomics profiling and coronary angiography data from these 300 subjects were analysed. Information regarding the traditional risk factors (TRFs) of CHD was collected from a clinical database and analysed by the study physicians. The Ethics Committee of Fuwai Hospital approved this study, which complies with the Declaration of Helsinki. Before collection of blood samples, all study participants provided written informed consent. The study strategy is shown in Fig. 1.

Flowchart of study strategy.
Serum collection and preparation
Fasting plasma samples were collected in heparinized tubes before cardiac catheterization, chilled to 4 °C, centrifuged within 1 hour of collection (10 min at 2,300 rpm), separated into aliquots, and stored at −80 °C. The serum samples were then thawed at 4 °C, and 100 μL of serum from each sample was extracted with 400 μL of methanol using the Bravo liquid handling system (Agilent Technologies, USA). After vortexing for 2 min and incubating for 1 hour at −20 °C to precipitate the proteins, the mixture was then centrifuged at 12,000 rpm for 15 min at 4 °C. The supernatants were transferred to test vials and stored at −80 °C until the UHPLC-QTOF/MS analysis.
By mixing equal amounts of all 300 plasma samples, quality control (QC) samples were created to monitor the stability between the analytical batches and ensure data normalization. These QC samples were processed in the same manner as the testing samples.
UHPLC-QTOF/MS analysis
The plasma samples were randomly injected for UHPLC-QTOF/MS analysis in both the electrospray positive and negative modes. Blank samples (75% ACN in water) and QC samples were injected between every eight samples during the acquisition. A UHPLC system (1290 series, Agilent Technologies, USA) coupled to a quadruple time-of-flight (QTOF) mass spectrometer (Agilent 6550 iFunnel QTOF, Agilent Technologies, USA) was employed for the primary metabolic detection and determination. Tandem mass spectrometry (MS/MS) was performed using another QTOF mass spectrometer (Triple TOF 5600+, AB SCIEX, USA), which was better at performing a qualitative metabolite analysis. The QC samples were used for the MS/MS data acquisition. Four mass range segments, 50–300 Da, 290–600 Da, 590–900 Da, and 890–1200 Da, were used to expand the coverage of the MS/MS spectra. The acquired MS/MS spectra were matched with the in-house MS/MS spectral library (Shanghai Institute of Organic Chemistry), which has been verified by the metabolite standards for metabolite identification (details are described in the Supplementary Information).
Data pre-processing and annotation
ProteoWizard was employed to transform the MS raw data (.d) files to the mzXML format. The R package XCMS (version 3.2) was used to sequentially process the data. A data matrix that consisted of the retention time (RT), mass-to-charge ratio (m/z) values, and peak intensity was generated. The R package CAMERA was employed for peak annotation after XCMS data processing13. To obtain consistent variables, the metabolic matrix was further reduced by removing peaks with more than 80% missing values. A LOESS regression model based on the intensity of the metabolites in the QC samples was built to normalize the metabolite peaks in the test samples14.
Statistical analysis
Continuous variables were compared using Student’s t-test, while categorical variables were compared using analysis of variance (ANOVA). For the comparison of CHD and control subjects, we ran multiple linear regression analyses that were adjusted for the TRFs of CHD (age, sex, body mass index (BMI), hypertension, diabetes, hyperlipidaemia, family history of CHD and smoking) and for the metabolite batch. The beta (β) value and 95% confidence interval (95% CI) were estimated. We also used a conservative Bonferroni correction to mitigate type I error in multiple testing, resulting in a significant threshold of 8.2 × 10−5 (0.05/611). To further quantify the independent correlations between significant metabolites and CHD, a logistic regression model was constructed. All of the analyses were performed using SAS version 9.4. P values < 0.05 were considered to be statistically significant.
Data availability
The datasets generated and/or analysed in the current study are available from the corresponding author upon reasonable request.
Results
Demographic of the study population
The baseline characteristics and TRFs of the 150 CHD cases and 150 age- and gender-matched controls are displayed in Table 1. The average age of CHD patients was 55.24 ± 10.16 years and that of control subjects was 55.23 ± 10.23 years. Additionally, 50% of patients were female. As expected, CHD patients had significantly higher incidences of hypertension, diabetes, and hyperlipidaemia and more likely had a family history of CHD and smoker status.
Metabolic perturbation profile of CHD
The levels of 611 fasting plasma metabolites (466 in the positive mode (ESI+) and 145 in the negative mode (ESI−)) were obtained. After adjusting for the TRFs of CHD (age, sex, BMI, hypertension, diabetes, hyperlipidaemia, family history of CHD, and smoking) and for the metabolite batch using multiple linear regression models, 105 of the 611 metabolites were significantly different between the CHD cases and controls. As depicted in Fig. 2, the 105 metabolites (84 of which are known and 21 of which are unknown) could be classified into several principal classes: 51 are phospholipids, 10 are carnitines, 8 are free fatty acids, 5 are bile acid metabolites, 3 are amino acids and 7 are separate particles. The β value and 95% CI were collected, and these results are shown in Supplementary Table S1.

Metabolic perturbation profile of CHD. A total of 105 metabolites were significantly altered in CHD cases compared to angiographically normal controls. Each metabolite super-pathway is represented in a different colour.
Metabolites exhibiting the strongest association with CHD
To avoid type I error in multiple testing, a total of 6 metabolites out of the 105 differential metabolites mentioned above were identified with a Bonferroni-corrected cut-off of 8.2 × 10−5 (0.05/611), including palmitic acid (β = 0.205; p < 0.0001), linoleic acid (β = 0.133; p < 0.0001), 4-pyridoxic acid (β = 0.142; p < 0.0001), phosphatidylglycerol (20:3/2:0) (β = 0.287; p < 0.0001), carnitine (14:1) (β = 0.332; p < 0.0001), and lithocholic acid (β = 0.224; p < 0.0001). Their positive independent correlations with CHD were further quantified using a logistic regression model: palmitic acid (odds ratio [OR]: 7.24; 95% CI: (2.39, 21.912); P = 0.0001), linoleic acid (OR: 6.13; 95% CI: (2.367, 15.893); P = 0.0005), 4-pyridoxic acid (OR: 4.879; 95% CI: (2.158, 11.035); P = 0.0002), phosphatidylglycerol (20:3/2:0) (OR: 3.938; 95% CI: (1.891, 8.204); P = 0.0001), carnitine (14:1) (OR: 3.379; 95% CI: (1.913, 5.967); P < 0.0001), and lithocholic acid (OR: 2.791; 95% CI: (1.74, 4.477); P = 0.0003) (Table 2).
Discussion
Using a nontargeted metabolomics approach, we explored the molecular perturbation profile of patients with severe CHD compared with angiographically normal controls. We identified 105 metabolites that were distinctly altered in plasma samples, including 51 phospholipids, 10 carnitines, 8 free fatty acids, 5 bile acid metabolites, 3 amino acids and 7 separate particles. Of these, six metabolites were discovered as having the strongest correlations with CHD after adjusting for multiple testing (the adjusted ORs ranged from 2.79 to 7.24); to the best of our knowledge, the correlations of CHD with 4-pyridoxic acid, lithocholic acid and phosphatidylglycerol (20:3/2:0) were first reported in this study. The links between metabolic dysregulation and CHD that were detected in our analysis not only verified the well-established implications about lipids but also identified novel metabolic targets that are potentially involved in CHD pathogenesis. Here, we theoretically estimated the assumed causality between significant metabolic perturbations and CHD and provided insight on the internal mechanisms that may be involved.
Palmitic acid
Of the six identified metabolites, palmitic acid had the strongest positive correlation with CHD (OR: 7.24; 95% CI: (2.39, 21.912); P = 0.0001), which was consistent with previous findings15,16. As the most abundant saturated fatty acid (SFA), palmitic acid (16:0) accounts for nearly 50% of the total daily SFA intake. Although the association between SFA intake and CHD risk is debated17, strong positive associations have been identified between CHD development and palmitic acid intake18,19. A recently updated meta-analysis of clinical trials reported that palmitic acid (16:0) significantly increased total cholesterol levels and low-density lipoprotein (LDL) cholesterol levels20. Furthermore, an in vitro experiment confirmed that palmitic acid could induce vascular endothelial dysfunction by triggering inflammatory signals and provided novel insight on the mechanisms underlying the correlation between palmitic acid and atherosclerosis21.
Linoleic acid
Linoleic acid (LA) levels were elevated in severe CHD patients compared with control subjects in our study. Each integer increase in the LA level is associated with a 6.13-fold increase (95% CI: (2.367, 15.893); P = 0.0005) in CHD risk. As the most abundant polyunsaturated fatty acid (PUFA), omega 6 (n-6) LA is usually confused with general PUFAs or with omega 3 (n-3) PUFAs when explaining clinical findings of its relationship with the risk of cardiovascular disease. Meanwhile, a comprehensive randomized controlled trial discovered that the well-known atheroprotective properties of PUFA intake are specifically attributable to n-3 PUFAs and are not relevant to n-6 LA22. Moreover, they found that increased LA intake was associated with higher risks of all-cause death, CHD, and cardiovascular disease23. This may explain the positive connection between the plasma LA level and CHD found in our study. The mechanism linking an increased LA level to cardiovascular pathogenesis has also been confirmed. Researchers found that an increased level of linoleic acid resulted in increased oxidized LA metabolites (OXLAMs), which can induce the formation of macrophage foam cells and endothelial cell dysfunction24,25. Another study suggests that LA may also activate the NF-κB pathway and TNF-mediated endothelial cell dysfunction, which play critical roles in the pathogenesis of atherosclerosis26.
Pyridoxic acid
The third metabolite found to be associated with CHD in this study is pyridoxic acid, which is the end product of vitamin B6 catabolism. Three forms of Vitamin B6 are typically detected in the plasma: pyridoxal 5′-phosphate (PLP), pyridoxal (PL), and 4-pyridoxic acid (4-PA). PLP is the biochemically active form of Vitamin B6 and is an essential coenzyme in almost all living systems27. Pyridoxic acid has received little attention in cardiovascular research. Meanwhile, a number of previous studies have confirmed that lower circulating PLP concentrations or lowering vitamin B6 intake is associated with an increased risk of CHD, hypertension, diabetes or stroke28,29,30,31. What is often overlooked is that low circulating PLP levels have been observed in combination with an increase in plasma 4-PA levels under inflammatory conditions32,33,34. To our knowledge, our study was the first to confirm a positive association between the plasma 4-PA level and CHD risk (OR: 4.879; 95% CI: (2.158, 11.035); P = 0.0002). Several hypotheses have been suggested to explain the inverse changes shown in B6 vitamins during inflammation-related pathogenesis. Some studies have suggested the mobilization of PLP from the plasma to the site of inflammation (erythrocytes or affected tissues) for use of PLP-dependent enzymes34,35. Other studies have indicated a higher catabolic rate of PLP under oxidative stress and activated cellular immunity conditions33,36. A study also proposed the PA:(PL + PLP) ratio (PAr) as an indicator of vitamin B6 catabolism and confirmed that the PAr is a predictor for all-cause mortality37. A possible inhibitory effect of 4-PA on vitamin B6 metabolism has also been discussed. Experimental data have shown the inhibitory effect of 4-PA on PL kinase37,38. Additionally, an observational study showed that the plasma 4-PA level was a stronger covariate with homocysteine (an independent CHD risk factor) than the plasma PLP level39. In conclusion, it is likely that CHD pathogenesis may cause an increase in the 4-PA level, but a perturbed 4-PA status may contribute to the development of more severe inflammation as well.
Phosphatidylglycerol (20:3/2:0)
A strong correlation of phosphatidylglycerol (20:3/2:0) with CHD was identified in our study. Since hundreds of different glycerophospholipids have been observed in plasma lipoproteins (Lp (a)) and atherosclerotic plaques, our finding was not unexpected40. However, recent studies have suggested that the function of glycerophospholipids might different depending on the cell type that is activated, the oxidation/inflammation status, the subclass of glycerophospholipid or the species of the bound fatty acids16,40. Some glycerophospholipids may act as antioxidants41,42, whereas others may induce pro-atherogenic effects on endothelial cells43,44. Regarding phosphatidylglycerol (PGs), which a phosphoglycerol moiety occupies a glycerol substitution site, little evidence has been shown on their relationship with CHD, and we were the first to reveal a positive relationship between PG(20:3/2:0) and CHD (OR: 3.938; 95% CI: (1.891, 8.204); P = 0.0001) in human plasma. Several previous studies have provided evidence that PGs are significantly increased in atherosclerotic rats45,46. Elevated levels of PGs were also found in patients with type 2 diabetes and prediabetes47. PGs are precursors for another glycerophospholipid, cardiolipin (CL). Thus, the association of oxidant stress and mitochondrial dysfunction with CL may suggest a pro-atherogenic role for PGs48. Meanwhile, the involvement of PGs in endogenous antioxidant defence mechanisms has also been proposed, as PGs are the second-most abundant phospholipid in lung surfactant and play an anti-inflammatory role during infection49.
Acylcarnitine (14:1)
Almost all metabolomics studies concerning CHD thus far have mentioned acylcarnitines6,9,10. We identified acylcarnitine (14:1), a medium-chain dicarboxylated acylcarnitine, as being an important metabolite associated with CHD (OR: 3.379; 95% CI: (1.913, 5.967); P < 0.0001). As expected, the medium-chain acylcarnitine cluster has been reported as being associated with cardiovascular diseases in multiple studies of different designs50. The well-known metabolic function of acylcarnitines is to transport long-chain fatty acids into the mitochondria for β-oxidation, which is how the heart acquires most of its energy. Therefore, the accumulation of acylcarnitines, especially those of medium-chain species, has been attributed to incomplete fatty acid oxidation and energy metabolism in CHD51. Studies have also suggested the elevation of acylcarnitines as being a signature for inadequate β-oxidation in macrophage lipid metabolism52, which is a key process underlying atherosclerosis. Moreover, L-carnitine and acylcarnitines have been shown to contribute to the development of atherosclerosis by increasing the levels of the independent CHD risk factor trimethylamine N-oxide (TMAO) in experimental animals53. Another study also showed that decreasing the levels of L-carnitine and its derivatives can attenuate atherosclerosis pathogenesis54.
Lithocholic acid
Lithocholic acid (LCA) belongs to the bile acid family, which is composed of molecules that are essential for the absorption of dietary fats and the regulation of cholesterol homeostasis. Our study was the first to report a relationship between elevated plasma lithocholic acid levels and CHD (OR: 2.791; 95% CI: (1.74, 4.477); P = 0.0003). Faecal metabolomics studies have shown that CHD patients are unable to excrete adequate amounts of bile acids to remove excess serum cholesterol55. This can lead to an accumulation of plasma LCA, which we detected in CHD patients in our study. Researchers who observed that elevated lithocholic acid levels could induce hepatic inflammation in cholestasis also uncovered the vascular inflammatory role of circulating lithocholic acid. Experimental evidence showed that elevated LCA levels might cause endothelial dysfunction via the production of reactive oxygen species (ROS) by the induction of the NF-κB and p38 MAPK signalling pathways, which suggested a potential pro-atherogenic role for LCA56. However, several studies have provided conflicting data on the relationship between LCA and coronary atheroma57, which reminds us that this field of study is worthy of further investigation.
Strengths of the current study
We employed a nontargeted metabolomics approach that not only confirmed previous findings from targeted assays but also revealed a much wider metabolite spectrum that is related to CHD. We mainly explored the CHD metabolomics profile in Chinese populations. There are very large differences in both the prevalence and levels of risk factors for coronary heart disease among different ethnic groups58,59, and previous CHD metabolomics studies were predominately conducted on Caucasian populations6,7,8,9,10,11. Thus, it is important to develop ethnicity-specific CHD metabolomics analyses to more accurately determine the underlying metabolic perturbations involved in CHD pathogenesis. In addition, we optimized the high-resolution mass spectrometry (HRMS) process by combining two QTOF mass spectrometers, the Agilent 6550 iFunnel QTOF and Triple TOF 5600+. The Agilent 6550 iFunnel QTOF is better for metabolite detection and determination, while the Triple TOF 5600+ is better for qualitative metabolite analyses. Furthermore, the study cohort was screened with very restrictive exclusion criteria and strictly grouped based on angiographic evidence to reduce variations in the levels of the plasma metabolites.
Limitations of the current study
First, the current study is a case-control study that predominately reveals the association between metabolite signatures and CHD status. However, the causal relationship and the internal mechanisms of metabolite signatures and CHD status are still unclear. Therefore, a prospective nontargeted study is recommended in the future. Second, our results lack the verification of a targeted analysis. An independent cohort is required to further replicate and confirm our findings. Moreover, as the metabolic fingerprinting assessment was conducted at a single time point, this study is unable to trace back to the disturbed metabolic flux in CHD patients. Additional isotope tracer techniques, especially those used in animal models, are recommended to complement the techniques used in the current study.
Conclusions
Overall, our study examined the global plasma metabolomics profile related to CHD by employing a nontargeted UHPLC-QTOF/MS metabolomics approach. We not only confirmed common alterations in plasma metabolism but also identified 3 novel metabolites (4-pyridoxic acid, PG (20:3/2:0) and lithocholic acid) that exhibited strong correlations with CHD. These findings provide further insight into the pathogenic mechanisms of CHD and may lead to improved diagnostic and therapeutic options for patients with CHD. Further targeted studies based on the metabolic perturbations that we reported are warranted.
References
National Cholesterol Education Program Expert Panel on Detection, E. & Treatment of High Blood Cholesterol in, A. Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 106, 3143–3421 (2002).
Grundy, S. M. Obesity, metabolic syndrome, and cardiovascular disease. J Clin Endocrinol Metab 89, 2595–2600, https://doi.org/10.1210/jc.2004-0372 (2004).
von Eckardstein, A. & Assmann, G. Plasma homocysteine levels and mortality in patients with coronary artery disease. N Engl J Med 337, 1632–1633 (1997).
Ballantyne, C. M. et al. Influence of low high-density lipoprotein cholesterol and elevated triglyceride on coronary heart disease events and response to simvastatin therapy in 4S. Circulation 104, 3046–3051 (2001).
Rubins, H. B. Triglycerides and coronary heart disease: implications of recent clinical trials. J Cardiovasc Risk 7, 339–345 (2000).
Shah, S. H. et al. Association of a peripheral blood metabolic profile with coronary artery disease and risk of subsequent cardiovascular events. Circ Cardiovasc Genet 3, 207–214, https://doi.org/10.1161/CIRCGENETICS.109.852814 (2010).
Magnusson, M. et al. A diabetes-predictive amino acid score and future cardiovascular disease. Eur Heart J 34, 1982–1989, https://doi.org/10.1093/eurheartj/ehs424 (2013).
Wang, Z. et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472, 57–63, https://doi.org/10.1038/nature09922 (2011).
Shah, S. H. et al. Baseline metabolomic profiles predict cardiovascular events in patients at risk for coronary artery disease. Am Heart J 163, 844–850 e841, https://doi.org/10.1016/j.ahj.2012.02.005 (2012).
Shah, A. A. et al. Metabolic profiles predict adverse events after coronary artery bypass grafting. J Thorac Cardiovasc Surg 143, 873–878, https://doi.org/10.1016/j.jtcvs.2011.09.070 (2012).
Tang, W. H., Wang, Z., Cho, L., Brennan, D. M. & Hazen, S. L. Diminished global arginine bioavailability and increased arginine catabolism as metabolic profile of increased cardiovascular risk. J Am Coll Cardiol 53, 2061–2067, https://doi.org/10.1016/j.jacc.2009.02.036 (2009).
Sevin, D. C., Kuehne, A., Zamboni, N. & Sauer, U. Biological insights through nontargeted metabolomics. Current opinion in biotechnology 34, 1–8, https://doi.org/10.1016/j.copbio.2014.10.001 (2015).
Kuhl, C., Tautenhahn, R., Bottcher, C., Larson, T. R. & Neumann, S. CAMERA: an integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Analytical chemistry 84, 283–289, https://doi.org/10.1021/ac202450g (2012).
Dunn, W. B. et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nature protocols 6, 1060–1083, https://doi.org/10.1038/nprot.2011.335 (2011).
Zong, G. et al. Intake of individual saturated fatty acids and risk of coronary heart disease in US men and women: two prospective longitudinal cohort studies. Bmj 355, i5796, https://doi.org/10.1136/bmj.i5796 (2016).
Park, J. Y., Lee, S. H., Shin, M. J. & Hwang, G. S. Alteration in metabolic signature and lipid metabolism in patients with angina pectoris and myocardial infarction. PloS one 10, e0135228, https://doi.org/10.1371/journal.pone.0135228 (2015).
Fattore, E. & Fanelli, R. Palm oil and palmitic acid: a review on cardiovascular effects and carcinogenicity. International journal of food sciences and nutrition 64, 648–659, https://doi.org/10.3109/09637486.2013.768213 (2013).
Hu, F. B. et al. Dietary saturated fats and their food sources in relation to the risk of coronary heart disease in women. The American journal of clinical nutrition 70, 1001–1008 (1999).
Praagman, J. et al. Dietary Saturated Fatty Acids and Coronary Heart Disease Risk in a Dutch Middle-Aged and Elderly Population. Arteriosclerosis, thrombosis, and vascular biology 36, 2011–2018, https://doi.org/10.1161/ATVBAHA.116.307578 (2016).
Mensink, R. P., Zock, P. L., Kester, A. D. & Katan, M. B. Effects of dietary fatty acids and carbohydrates on the ratio of serum total to HDL cholesterol and on serum lipids and apolipoproteins: a meta-analysis of 60 controlled trials. The American journal of clinical nutrition 77, 1146–1155 (2003).
Ishida, T., Naoe, S., Nakakuki, M., Kawano, H. & Imada, K. Eicosapentaenoic Acid Prevents Saturated Fatty Acid-Induced Vascular Endothelial Dysfunction: Involvement of Long-Chain Acyl-CoA Synthetase. Journal of atherosclerosis and thrombosis 22, 1172–1185, https://doi.org/10.5551/jat.28167 (2015).
Ramsden, C. E., Hibbeln, J. R., Majchrzak, S. F. & Davis, J. M. n-6 fatty acid-specific and mixed polyunsaturate dietary interventions have different effects on CHD risk: a meta-analysis of randomised controlled trials. The British journal of nutrition 104, 1586–1600, https://doi.org/10.1017/S0007114510004010 (2010).
Ramsden, C. E. et al. Use of dietary linoleic acid for secondary prevention of coronary heart disease and death: evaluation of recovered data from the Sydney Diet Heart Study and updated meta-analysis. Bmj 346, e8707, https://doi.org/10.1136/bmj.e8707 (2013).
Vangaveti, V., Baune, B. T. & Kennedy, R. L. Hydroxyoctadecadienoic acids: novel regulators of macrophage differentiation and atherogenesis. Therapeutic advances in endocrinology and metabolism 1, 51–60, https://doi.org/10.1177/2042018810375656 (2010).
Wang, L. et al. Triglyceride-rich lipoprotein lipolysis releases neutral and oxidized FFAs that induce endothelial cell inflammation. Journal of lipid research 50, 204–213, https://doi.org/10.1194/jlr.M700505-JLR200 (2009).
Toborek, M. et al. Linoleic acid and TNF-alpha cross-amplify oxidative injury and dysfunction of endothelial cells. Journal of lipid research 37, 123–135 (1996).
Merrill, A. H. Jr. & Henderson, J. M. Diseases associated with defects in vitamin B6 metabolism or utilization. Annual review of nutrition 7, 137–156, https://doi.org/10.1146/annurev.nu.07.070187.001033 (1987).
Lin, P. T. et al. Low pyridoxal 5′-phosphate is associated with increased risk of coronary artery disease. Nutrition 22, 1146–1151, https://doi.org/10.1016/j.nut.2006.08.013 (2006).
Lin, P. T., Cheng, C. H., Wei, J. C. & Huang, Y. C. Low plasma pyridoxal 5′-phosphate concentration and MTHFR 677C– > T genotypes are associated with increased risk of hypertension. International journal for vitamin and nutrition research. Internationale Zeitschrift fur Vitamin- und Ernahrungsforschung. Journal international de vitaminologie et de nutrition 78, 33–40, https://doi.org/10.1024/0300-9831.78.1.33 (2008).
Shen, J., Lai, C. Q., Mattei, J., Ordovas, J. M. & Tucker, K. L. Association of vitamin B-6 status with inflammation, oxidative stress, and chronic inflammatory conditions: the Boston Puerto Rican Health Study. The American journal of clinical nutrition 91, 337–342, https://doi.org/10.3945/ajcn.2009.28571 (2010).
Kelly, P. J. et al. Low vitamin B6 but not homocyst(e)ine is associated with increased risk of stroke and transient ischemic attack in the era of folic acid grain fortification. Stroke 34, e51–54, https://doi.org/10.1161/01.STR.0000071109.23410.AB (2003).
Bates, C. J., Pentieva, K. D., Prentice, A., Mansoor, M. A. & Finch, S. Plasma pyridoxal phosphate and pyridoxic acid and their relationship to plasma homocysteine in a representative sample of British men and women aged 65 years and over. The British journal of nutrition 81, 191–201 (1999).
Ulvik, A., Midttun, O., Pedersen, E. R., Nygard, O. & Ueland, P. M. Association of plasma B-6 vitamers with systemic markers of inflammation before and after pyridoxine treatment in patients with stable angina pectoris. The American journal of clinical nutrition 95, 1072–1078, https://doi.org/10.3945/ajcn.111.029751 (2012).
Chiang, E. P. et al. Inflammation causes tissue-specific depletion of vitamin B6. Arthritis research & therapy 7, R1254–1262, https://doi.org/10.1186/ar1821 (2005).
Midttun, O. et al. Low plasma vitamin B-6 status affects metabolism through the kynurenine pathway in cardiovascular patients with systemic inflammation. The Journal of nutrition 141, 611–617, https://doi.org/10.3945/jn.110.133082 (2011).
Ulvik, A. et al. Evidence for increased catabolism of vitamin B-6 during systemic inflammation. The American journal of clinical nutrition 100, 250–255, https://doi.org/10.3945/ajcn.114.083196 (2014).
Ulvik, A. et al. Vitamin B-6 catabolism and long-term mortality risk in patients with coronary artery disease. The American journal of clinical nutrition 103, 1417–1425, https://doi.org/10.3945/ajcn.115.126342 (2016).
Gaut, Z. N. & Solomon, H. M. Phosphorylation of pyridoxine by human blood platelets–effects of structure analogs and metabolic inhibitors. Biochemical pharmacology 21, 2395–2400 (1972).
Bates, C. J., Mansoor, M. A., Gregory, J., Pentiev, K. & Prentice, A. Correlates of plasma homocysteine, cysteine and cysteinyl-glycine in respondents in the British National Diet and Nutrition Survey of young people aged 4-18 years, and a comparison with the survey of people aged 65 years and over. The British journal of nutrition 87, 71–79 (2002).
Meikle, P. J. et al. Plasma lipidomic analysis of stable and unstable coronary artery disease. Arteriosclerosis, thrombosis, and vascular biology 31, 2723–2732, https://doi.org/10.1161/ATVBAHA.111.234096 (2011).
Rosenblat, M., Vaya, J., Shih, D. & Aviram, M. Paraoxonase 1 (PON1) enhances HDL-mediated macrophage cholesterol efflux via the ABCA1 transporter in association with increased HDL binding to the cells: a possible role for lysophosphatidylcholine. Atherosclerosis 179, 69–77, https://doi.org/10.1016/j.atherosclerosis.2004.10.028 (2005).
Wallner, S. & Schmitz, G. Plasmalogens the neglected regulatory and scavenging lipid species. Chemistry and physics of lipids 164, 573–589, https://doi.org/10.1016/j.chemphyslip.2011.06.008 (2011).
Oka, H. et al. Lysophosphatidylcholine induces urokinase-type plasminogen activator and its receptor in human macrophages partly through redox-sensitive pathway. Arteriosclerosis, thrombosis, and vascular biology 20, 244–250 (2000).
Kume, N. & Gimbrone, M. A. Jr. Lysophosphatidylcholine transcriptionally induces growth factor gene expression in cultured human endothelial cells. The Journal of clinical investigation 93, 907–911, https://doi.org/10.1172/JCI117047 (1994).
Dang, V. T., Huang, A., Zhong, L. H., Shi, Y. & Werstuck, G. H. Comprehensive Plasma Metabolomic Analyses of Atherosclerotic Progression Reveal Alterations in Glycerophospholipid and Sphingolipid Metabolism in Apolipoprotein E-deficient Mice. Scientific reports 6, 35037, https://doi.org/10.1038/srep35037 (2016).
Jia, P. et al. The anti-atherosclerotic effect of tanshinol borneol ester using fecal metabolomics based on liquid chromatography-mass spectrometry. The Analyst 141, 1112–1120, https://doi.org/10.1039/c5an01970b (2016).
Meikle, P. J. et al. Plasma lipid profiling shows similar associations with prediabetes and type 2 diabetes. PloS one 8, e74341, https://doi.org/10.1371/journal.pone.0074341 (2013).
Zhong, H., Lu, J., Xia, L., Zhu, M. & Yin, H. Formation of electrophilic oxidation products from mitochondrial cardiolipin in vitro and in vivo in the context of apoptosis and atherosclerosis. Redox biology 2, 878–883, https://doi.org/10.1016/j.redox.2014.04.003 (2014).
Numata, M., Chu, H. W., Dakhama, A. & Voelker, D. R. Pulmonary surfactant phosphatidylglycerol inhibits respiratory syncytial virus-induced inflammation and infection. Proceedings of the National Academy of Sciences of the United States of America 107, 320–325, https://doi.org/10.1073/pnas.0909361107 (2010).
Shah, S. H., Kraus, W. E. & Newgard, C. B. Metabolomic profiling for the identification of novel biomarkers and mechanisms related to common cardiovascular diseases: form and function. Circulation 126, 1110–1120, https://doi.org/10.1161/CIRCULATIONAHA.111.060368 (2012).
Koves, T. R. et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell metabolism 7, 45–56, https://doi.org/10.1016/j.cmet.2007.10.013 (2008).
Blair, H. C., Sepulveda, J. & Papachristou, D. J. Nature and nurture in atherosclerosis: The roles of acylcarnitine and cell membrane-fatty acid intermediates. Vascular pharmacology 78, 17–23, https://doi.org/10.1016/j.vph.2015.06.012 (2016).
Koeth, R. A. et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nature medicine 19, 576–585, https://doi.org/10.1038/nm.3145 (2013).
Vilskersts, R. et al. Methyl-gamma-butyrobetaine decreases levels of acylcarnitines and attenuates the development of atherosclerosis. Vascular pharmacology 72, 101–107, https://doi.org/10.1016/j.vph.2015.05.005 (2015).
Charach, G. et al. Decreased fecal bile acid output in patients with coronary atherosclerosis. Journal of medicine 29, 125–136 (1998).
Qin, P., Tang, X., Elloso, M. M. & Harnish, D. C. Bile acids induce adhesion molecule expression in endothelial cells through activation of reactive oxygen species, NF-kappaB, andp38. American journal of physiology. Heart and circulatory physiology 291, H741–747, https://doi.org/10.1152/ajpheart.01182.2005 (2006).
Duboc, H. et al. Crosstalk between the hepatologist and the cardiologist: a future place for the lithocholic acid as a coronary atheroma risk factor? Hepatology 56, 2426, https://doi.org/10.1002/hep.25839 (2012).
Budoff, M. J. et al. Ethnic differences of the presence and severity of coronary atherosclerosis. Atherosclerosis 187, 343–350, https://doi.org/10.1016/j.atherosclerosis.2005.09.013 (2006).
Kuller, L. H. Ethnic differences in atherosclerosis, cardiovascular disease and lipid metabolism. Curr Opin Lipidol 15, 109–113 (2004).
Acknowledgements
The authors would like to thank all of the participants in our study cohort.
Author information
Authors and Affiliations
Contributions
Li Yp., Wang Y., Chen Jz. and Dou Kf. conceived and designed the study. Li Yp., He Y. and Chen Cz. collected the plasma samples. Li Yp. and Zhang D. conducted the metabolomics analysis and wrote part of the Methods and Supplementary Information sections. Zhao Yy., Bai Yx. and Wang Y. performed the data analysis. Li Yp. wrote the main manuscript. Li Yp. and Song Cx prepared all of the tables and figures. Dou Kf., Pu Jl. and Yang Yj. approved the final version of the manuscript for publication. All of the authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Y., Zhang, D., He, Y. et al. Investigation of novel metabolites potentially involved in the pathogenesis of coronary heart disease using a UHPLC-QTOF/MS-based metabolomics approach. Sci Rep 7, 15357 (2017). https://doi.org/10.1038/s41598-017-15737-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-15737-3
This article is cited by
-
Combined metabolomics and machine learning algorithms to explore metabolic biomarkers for diagnosis of acute myocardial ischemia
International Journal of Legal Medicine (2023)
-
Forensic identification of sudden cardiac death: a new approach combining metabolomics and machine learning
Analytical and Bioanalytical Chemistry (2023)
-
A HILIC-MS/MS method development and validation for the quantitation of 13 acylcarnitines in human serum
Analytical and Bioanalytical Chemistry (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
