
Abstract
The major isoform of the GABAA receptor is α1β2γ2. The binding sites for the agonist GABA are located at the β2+/α1− subunit interfaces and the modulatory site for benzodiazepines at α1+/γ2−. In the absence of α1 subunits, a receptor was formed that was gated by GABA and modulated by diazepam similarly. This indicates that alternative subunits can take over the role of the α1 subunits. Point mutations were introduced in β2 or γ2 subunits at positions homologous to α1− benzodiazepine binding and GABA binding positions, respectively. From this mutation work we conclude that the site for GABA is located at a β2+/β2− subunit interface and that the diazepam site is located at the β2+/γ2− subunit interface. Computational docking leads to a structural hypothesis attributing this non-canonical interaction to a binding mode nearly identical with the one at the α1+/γ2− interface. Thus, the β2 subunit can take over the role of the α1 subunit for the formation of both sites, its minus side for the GABA binding site and its plus side for the diazepam binding site.
Similar content being viewed by others

Introduction
γ-Aminobutyric acid type A (GABAA) receptors are the major inhibitory neurotransmitter receptors in the mammalian central nervous system. The GABAA receptor is a pentameric protein complex, whose subunits are drawn from the following different isoforms: α(1–6), β(1–4), γ(1–3), δ, ε, θ, π and ρ(1–3). The five subunits form a chloride selective ion channel1,2,3. The most common isoform of this receptor consists of two α1, two β2 and one γ2 subunit(s)4,5,6 arranged α1γ2β2α1β2 counterclockwise when viewed from the extracellular space7,8,9. These receptors have two agonist GABA binding sites and one benzodiazepine binding site10. By using in vitro mutagenesis the binding sites for the agonist GABA were located to the β2+/α1− subunit interfaces11,12, and the modulatory site for benzodiazepines was at the α1+/γ2− subunit interface13. Thus, the α1 subunit is commonly accepted to contribute to the formation of both sites.
The GABAA receptors can be activated by the agonist GABA and modulated by many drugs14. Among these drugs are the benzodiazepines, such as diazepam, that have sedative, anxiolytic, anticonvulsant, hypnotic, and muscle relaxant properties15. Coexpression of different combinations of recombinant subunits has generated GABAA receptors with distinct pharmacological and electrophysiological properties.
As early as 1990, we observed that β2γ2 GABAA receptors, lacking the α1 subunit, were activated by GABA and potentiated by diazepam16. Later this observation was confirmed by several groups for GABA and diazepam17,18,19 or other modulators20. Expression was also documented for β1γ2 21,22 and β3γ2 22,23 GABAA receptors. In the present study, we tried to understand this apparent contradiction and decided to investigate whether alternative GABA and benzodiazepine-binding subunit interfaces exist. Site-directed mutagenesis was combined with two-electrode voltage clamp in Xenopus oocytes. Our findings suggest that the β2 subunit may replace the α1 subunit for the formation of either site. We have previously utilized experimentally guided computational docking that led to a diazepam bound structure model at the α1+/γ2− interface24. Computational docking at the β2+/γ2− interface yielded structural models which strongly suggest that diazepam can interact with this site in a binding mode nearly identical with the one observed at the canonical α1+/γ2− site, thus explaining the similar apparent potency.
Results
Functional expression of β2γ2 GABAA receptors in Xenopus oocytes
We initially determined whether varying the subunit ratio led to a different extent of expression of β2γ2 GABAA receptors. We injected β2 and γ2 cRNAs at the three ratios 1:1 (1 fMol each/oocyte), 2:1 (2 fMol and 1 fMol/oocyte) and 1:3 (1 fMol and 3 fMol/oocyte) into oocytes and measured the maximum current amplitudes elicited by 10 mM GABA. 5–7 days after microinjection of RNA, the β2γ2 GABAA receptors formed by the 1:3 cRNA injection ratio gave the highest maximal current amplitude (131 ± 19 nA, n = 13). In contrast receptors formed from 1:1 and 2:1 cRNA ratios resulted in current amplitudes less than 100 nA. Thus, we used the 1:3 cRNA ratio coding for wild type or mutant β2 or γ2 subunits for all following experiments. Possibly the subunit arrangement is affected by the injection ratio as it has been documented in the case of β3γ2 GABAA receptors25. Oocytes injected with 1 fMol coding for the β2 subunit only, or with 3 fMol coding for the γ2 subunit only, both did not result in current expression.
β2γ2 receptors respond to GABA
5–7 days after injection, Xenopus oocytes expressing β2γ2 GABAA receptors were investigated for the presence of currents elicited by 10 mM GABA. Figure 1a shows original current traces obtained from oocytes clamped at −80 mV. Figure 1b shows an averaged concentration-response curve for β2γ2 GABAA receptors. The curve was characterized by an EC50 of 75 ± 5 µM and a Hill coefficient of 1.0 ± 0.1 (n = 5). This EC50 is similar to that reported for α1β2γ2, which amounts to 51 ± 15 µM8.

Concentration response curve for GABA at β2γ2 GABAA receptors. Receptors were expressed in Xenopus oocytes and exposed to subsequently higher concentrations of GABA and the elicited current amplitude was determined. Individual curves were first normalized to the fitted maximal current amplitude and subsequently averaged. Data are expressed as mean ± S.E.M., n = 5 from two batches of oocytes. (a) Original current traces. GABA applications are indicated by a bar. The numbers indicate the concentration of GABA in μM. (b) Averaged concentration-response curve for β2γ2 GABAA receptors. The dotted line shows for comparison corresponding data on α1β2γ2 GABAA receptors.
β2γ2 receptors respond to diazepam
Diazepam is a positive allosteric modulator of certain GABAA receptors enhancing the GABA-induced chloride ion influx. We examined the current potentiation by 1 µM diazepam using a GABA concentration that elicited about 5% of the respective maximal current amplitude. Figure 2a shows original current traces from an experiment were oocytes were exposed to either GABA alone or in combination with increasing concentrations of diazepam. Figure 2b shows an averaged concentration-response curve. The curve was characterized by an EC50 of 69 ± 14 nM and a Hill coefficient of 0.6 ± 0.1 (n = 3). The EC50 is similar to that reported earlier for α1β2γ2 with 92 ± 6 nM8, while the Hill coefficient is, for reasons we do not understand, significantly lower than 1. No evidence for a possible receptor heterogeneity that could explain this finding was found (see below). Potentiation by 1 µM diazepam amounted to 216 ± 30% (n = 15).

Concentration response curve for diazepam at β2γ2 GABAA receptors. Receptors were expressed in Xenopus oocytes and exposed to either GABA alone or GABA in the presence of subsequently higher concentrations of diazepam and the elicited current amplitude was determined. At each concentration of diazepam current potentiation was calculated. Individual curves for potentation were first normalized to the fitted maximal current amplitude and subsequently averaged. Data are expressed as mean ± S.E.M., n = 3 from two batches of oocytes. (a) Original current traces. (b) Averaged concentration-response curve for β2γ2 GABAA receptors. The dotted line shows for comparison corresponding data on α1β2γ2 GABAA receptors.
Selection of point mutations
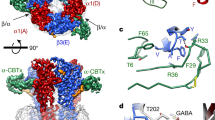
Obviously the α1 subunit is dispensable for the formation of a GABAA receptor responsive to both channel agonist GABA and diazepam as shown above. We aimed to localize both binding sites in β2γ2 receptors. For this purpose we selected some point mutations that have been described to affect either the response to GABA or that to diazepam in α1β2γ2 11,12,26,27,28,29 (Table 1). A total of nine different point mutations were introduced into either the β2 subunit or γ2 subunit. In the β2 subunit we generated one mutation at the minus side (β2Y62L) and four mutations at the plus side (β2T202A, β2T202S, β2Y205S, β2Y205Q) and in the γ2 subunit one mutation on the minus side (γ2F77Y) and three mutations on the plus side (γ2S217A, γ2Y220S and γ2Y220Q). A sequence alignment of the corresponding regions in α1, β2 and γ2 is shown in Fig. 3. Table 1 summarizes the consequences of the mutations in α1β2γ2 receptors. In addition the effect of homologous mutations in other subunits is listed.

Sequence alignment of α1, β2 and γ2 subunits of the rat GABAA receptor. Mutated residues of β2 and γ2 subunits are indicated with numbers. Homologous positions of the α1 subunit are also highlighted.
All mutated subunits were expressed in Xenopus oocytes in combination with wild-type subunits to result in β2γ2 GABAA receptors. Functional properties were determined by using two-electrode voltage clamp.
Functional expression of receptors was verified using 50 µM etomidate as agonist since mutations affecting the response to GABA are not very likely to influence the response to this agent. Amino acid residues affecting the latter property have been described to be located within the membrane embedded part of the receptor30. The mutation β2Y62L interfered very strongly with channel activation by etomidate. As the mutation is located far away from the suspected etomidate site31, this indicates that this mutation also affects gating by etomidate (see below). Unfortunately, β2T202Aγ2, β2Y205Sγ2 and β2Y205Qγ2 all resulted in no or very little expression (Table 2), so that these receptors could not be further investigated.
Effect of a mutation at the plus side of β2
β2T202S was the only investigated point mutation at the plus side of β2 that did not disrupt β2γ2 receptor assembly as evidenced by the large response to etomidate (Table 2). In α1β2γ2 receptors this mutation led to a 20-fold decrease in GABA sensitivity12. In β2γ2 receptors the same mutation led to a 42-fold decrease in GABA sensitivity (Fig. 4, Table 2). The EC50 for GABA dependent channel gating was 3130 ± 260 µM and a Hill coeffient of 0.8 ± 0.1 (n = 4). Potentiation by 1 µM diazepam was not significantly affected by the mutation (Fig. 5, Table 2).

Influence of different point mutations on the concentration dependence of GABA. Wild type or mutant β2γ2 GABAA receptors were expressed in Xenopus oocytes and exposed to subsequently higher concentrations of GABA and the elicited current amplitude was determined. Individual curves for each subunit combination were first normalized to the fitted maximal current amplitude and subsequently averaged. Averaged concentration-response curve are shown. Data are expressed as mean ± S.E.M., n = 3–7 from two batches of oocytes.

Influence of different point mutations on the potentiation by diazepam. Receptors were expressed in Xenopus oocytes and first exposed to 7 µM GABA alone or the same concentration of GABA in the presence of 1 µM diazepam and the elicited current amplitude was determined. Current potentiation by diazepam was calculated and averaged for each subunit combination. Data are expressed as mean ± S.E.M., n = 4–15 from two batches of oocytes.
Effect of a mutation at the minus side of β2
In α1β2γ2 receptors the mutation β2Y62L leads to a 30-fold decrease in GABA sensitivity with no effect on the antagonist affinity as compared to wild-type α1β2γ2 GABAA receptors11. The homologous mutation in the α1 subunit, α1F64L caused a 200-fold drop in both properties11. Here, we found that combination of β2Y62L with the γ2 subunit showed 2-fold smaller current amplitude (Table 2) as compared to wild-type β2γ2 receptors, indicating that this mutation might somehow disturb efficient assembly of functional channels and/or that gating is affected. In addition the mutation β2Y62L led to a 3-fold decrease in the sensitivity for GABA with an EC50 of 228 ± 50 µM and a Hill coefficient of 1.0 ± 0.1 (n = 7) (Fig. 4, Table 2). Again, potentiation by 1 µM diazepam was not significantly affected by the mutation (Fig. 5, Table 2).
The notion that gating is affected by the mutation is strongly supported by the fact that 50 µM etomidate elicits about 4-fold smaller currents than saturating concentrations of GABA (Table 2). In wild type β2γ2 receptors this current was about 7-fold larger than the one elicited by GABA. Similarly, in mutated α1β2Y62Lγ2 receptors this current was about 30-fold smaller than the one elicited by GABA. As the site for etomidate is located far away from the mutated residue, this is a strong indication that β2Y62 is not only involved in binding of GABA, but also in gating.
Effect of mutations at the plus side of γ2
Among the investigated mutations, γ2S217A and γ2Y220Q led to sizeable expression in combination with β2 and were further studied. To our knowledge nothing is known about both mutations. The homologous mutation to γ2S217A in the β2 subunit, β2T202A, led to a drastic loss in GABA sensitivity12. The homologous mutation to γ2Y220Q in the α1 subunit, α1Y209Q, disrupted the site for diazepam, while leaving GABA sensitivity unaffected26. This residue was also identified with photoaffinity labeling by the benzodiazepine binding site ligand Ro15–451332. In β2γ2S217A and β2γ2Y220Q the sensitivity to GABA was decreased 2-fold and 5-fold, respectively with EC50s of 133 ± 20 µM (n = 3) and 367 ± 139 µM (n = 3), and Hill coefficients of 0.8 ± 0.1 and 0.7 ± 0.1, respectively (Fig. 4, Table 2). Potentiation by 1 µM diazepam was not significantly affected by both mutations (Fig. 5, Table 2).
Effect of a mutation at the minus side of γ2
We studied the mutation γ2F77Y. In α1β2γ2 receptors this mutation abolishes the binding site for diazepam, while leaving GABA sensitivity unaffected29. Similar findings were made in β2γ2 receptors. The EC50 for GABA was 72 ± 15 µM (n = 5) and the Hill coefficient 0.9 ± 0.1 (Fig. 4, Table 2). Modulation by 1 µM diazepam was nearly lost (Fig. 5, Table 2).
Summary of the findings
No appreciable currents could be elicited upon expression of β2 or γ2 subunits alone and the receptors β2T202Aγ2, β2Y205Sγ2, β2γ2Y220S and β2Y205Qγ2. Modulation by diazepam was nearly lost in β2γ2F77Y receptors, whereas the reponse to GABA remained unaffected. Activation by GABA was strongly affected in β2T202Sγ2 receptors and weakly affected in β2Y62Lγ2, β2γ2S217A and β2γ2Y220Q receptors.
Computational Docking
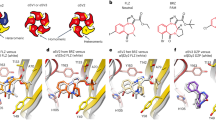
We performed computational docking of diazepam utilizing a homology model of the β2+/γ2− interface based on the β3 crystal structure 4COF33 as specified in the Methods section. The overal sequence similarity between β2+ and α1+ is high, especially in loops B and C where several aromatic and polar amino acids are conserved. We have shown previously that loop C residues are engaged in key interactions with diazepam24,26. The docking as specified in the Methods section provided for sidechain flexibility (loops D, G, E, B and C,) as well as a limited degree of backbone flexibility in the loop C tip, very similar to the approach used in our previous docking studies at the canonical high affinity α1+/γ2− site24. Computational docking usually generates correct binding poses, but they are not always correctly ranked by the different scoring functions34. We therefore analyzed the top 100 poses of the docking run based on multiple different criteria: Only poses that display interactions with γ2F77 were considered, to limit poses to those that reflect experimental findings. Poses were then filtered by similarity to the binding mode in the high affinity α1+/γ2− site24, where similarity was judged on ligand binding mode and major interactions with the pocket. Lastly, two different scoring functions were employed to identify the best candidate poses based on consensus scoring. Overall, six poses were identified that show high similarity with the high affinity binding mode at α1+/γ2−. Among these, two were found in rank one and two positions in the ChemScore ranking, and five were among top 30 ChemScored. Similarly, one of the six candidates was found in the rank two position of the GoldScore Fitness ranking, and a total of three were among the GoldScore top 30. Thus, consensus scoring leads to a binding mode model that features a binding mode very similar to the one that is observed at the canonical high affinity site.
Due to the sterically demanding sidechain β2F200, we find that sidechain rotamers adjust differently to ligand binding compared to the high affinity pocket, but gross binding mode and key interactions are highly similar (Fig. 6) where one of the representative poses is shown in comparison with the pose depicted in our previous study24.

Structural hypothesis for diazepam binding at the extracellular β2+/γ2 interface. Panel (a) shows the reference binding pose from our previous studies17 at the α1+ (orange)/ γ2− (cyan) interface. Panel (b) shows the most closely corresponding binding pose from the computational docking at the β2+ (red)/γ2− (cyan) interface. The homologous key amino acids in the binding pockets, as well as diazepam, are rendered in stick representation. While sidechain rotamers show some differences, ligand position, binding mode and key interations are very similar.
In the proposed binding mode the pendant phenyl ring is in close contact to loop A, which bears in the diazepam sensitive α isoforms a histidine, and in α4 and α6 an arginine which is known to interfere with diazepam binding35. Several H101X mutations were investigated in the past, and it was demonstrated that F (Phe), Y (Tyr) or Q (Gln) have only a small impact on flunitrazepam modulation36. The homologous position in β2 is the hydrophobic Leu99, which is sterically similar to Gln, and as hydrophobic as Phe. Thus, the proposed binding mode is compatible with previous mutagenesis studies and with the present observations.
Discussion
Several early studies reported responsiveness of β2γ2 GABAA receptors expressed in heterologous systems to both GABA and diazepam16,17,18,19 or to GABA in βxγ2 GABAA receptors20,21,22,23. Later, the two binding sites for GABA in α1β2γ2 GABAA receptors were localized to the two β2+/α1− subunit interfaces11,12 and the diazepam binding site to α1+/γ2− subunit interface13. Thus, the α1 subunit seemed to be required for the formation of both sites. The major point of this study was to localize the subunit interfaces that harbor the alternative GABA and benzodiazepine binding sites in β2γ2 GABAA receptors lacking the α1 subunit.
After having shown that β2γ2 GABAA receptors responded similarly to GABA and diazepam as α1β2γ2 GABAA receptors, we used point mutations abrogating one of the two in α1β2γ2. Unfortunately, some of the chosen mutations interfered with receptor expression or gating, presumably by negatively affecting assembly or by leading to mainly inactive channels.
The γ2+/β2− subunit interface may be excluded for both sites
There are four possible subunit interfaces in β2γ2 GABAA receptors: β2+/β2−, β2+/γ2−, γ2+/β2− and γ2+/γ2−. Of these, the γ2+/β2− subunit interface also occurs in the α1β2γ2 receptors. Mutation β2T202A dramatically impaired GABA activation in these receptors12 and mutation α1Y209Q led to loss of flumazenil sensitivity26. Obviously the γ2+/β2− subunit interface can not take over the formation of both sites. In addition three mutations located at this interface in β2γ2 receptors, β2Y62Lγ2, γ2S217Aβ2 and γ2Y220Qβ2, had no strong impact on the responses to GABA or diazepam (Table 2). Taken together, we can exclude that the γ2+/β2− subunit interface is the location of GABA and benzodiazepine binding sites.
Localization of the diazepam binding subunit interface
Mutations β2T202S12 and γ2F77Y29 have been described to disrupt GABA and diazepam binding sites in α1β2γ2 receptors, respectively. Mutations β2T202S and γ2F77Y similarly strongly affect the response of β2γ2 receptors to GABA and diazepam. Mutations β2T202S and γ2F77Y had little impact on diazepam and GABA sites, respectively. This implies a role of β2+ and γ2− in the formation of GABA site and diazepam site, respectively. Thus, the GABA binding site must be located at β2+/β2− or β2+/γ2− subunit interfaces and that for diazepam at β2+/γ2− or γ2+/γ2−.
The mutation α1Y209Q abrogates the diazepam site in α1β2γ2 receptors26. Therefore, it may be expected that the homologous mutation in the γ2 subunit, γ2Y220Q, affects the apparent affinity for diazepam. Similarly, the mutation α1T206A decreases the affinity for diazepam in α1β2γ2 receptors26. Thus, it may be expected that the homologous mutation in the γ2 subunit, γ2S217A, affects the apparent affinity for diazepam. Both mutations failed to affect the response to diazepam, arguing strongly against involvement of the γ2+ subunit interface in the diazepam site. For these reasons, we locate this site to the β2+/γ2− subunit interface (Fig. 7). The failure of the mutation β2T202S to affect the response to diazepam may be explained by the fact that a homologous, similar mutation α1T206C did not affect the response to diazepam in α1β2γ2 receptors27. The high affinity interaction of diazepam with the β2+/γ2− subunit interface is also supported by the docking experiments (Fig. 6), which strongly suggest that diazepam binds in this non-canonical site in a fashion very similar to the one that is observed in the high affinity site.

Schematic representation of β2γ2 GABAA receptors. The location of amino acid residues of interest is indicated. Point mutations resulting in disrupting assembly are not shown. The binding site for GABA is concluded to locate at the β2+/ β2− interface, that for diazepam at the β2+/γ2− interface. Please note that the subunit arrangement was not addressed in this study.
Putative localization of the GABA binding subunit interface
Thus, we are left with the β2+/β2− and the γ2+/γ2− subunit interfaces. The mutation β2T202S at the β2+ has very strong effect on the EC50 for GABA shifting the concentration response curve 42-fold, while effects of mutations at the γ2+ side are much smaller. Therefore we localize the GABA binding site at the β2+/β2− subunit interface (Fig. 7). However, we can not fully exclude an additional site at a γ2+ side. As β2+/γ2− has been excluded, this additional GABA site would have to be at the γ2+/γ2− interface. It should be noted that a binding site for GABA has also been described at the β3+/δ− subunit interface37.
Functional expression
We observed that individual β2 or γ2 subunits did not form functional receptors on the surface of oocytes. Within 1 day after injection, the α1β2γ2 receptors expressed currents in µA range38. In contrast, we observed in this work that the β2γ2 receptors need longer period for channel expession (5–7 days) and the maximal current amplitudes elicited by 10 mM GABA amount to only 100–200 nA. If silent receptors and different single channel open frequency between receptors are ignored, β2γ2 receptors form less efficiently than α1β2γ2 receptors. A previous study described a important role of α1 subunits for receptor trafficking and assembly in α1β2γ2 receptors39. Thus, in the presence of large amounts of α1, β2γ2 may not be formed. Under special circumstances where α subunit expression is low, the formation of a limited amount of β2γ2 receptors may occur. Recent single cell RT-PCR data indicate that cells devoid of mRNA coding for α subunits are not present in the hypothalamus, where diversity of neurons is huge. However, the endocrine system may have receptors without α subunits. Chromaffine cells (at least in certain developmental stages) have only mRNA coding for β3 and ε subunits (personal communication, I. Adameyko).
While so far it is not considered a candidate receptor to exist in the adult mammalian nervous system, the possible existence of such receptors has also not been specifically excluded. Given that fact that in the developing mammalian brain expression of all three γ isoforms is higher than in the postnatal brain, non-canonical receptor arrangements should be considered. In this vein, it is very important to realize that a high affinity benzodiazepine binding site at the β2+/γ2− interface implies that such receptors cannot be distinguished from α+/γ2− “canonical” receptors in radioligand and PET studies where benzodiazepine ligands are used as presumably selective probes for α+/γ2− canonical benzodiazepine-sites.
What may be the biological relevance of our observations? Ralvenius et al.40 studied mice carrying a point mutation in all those four alpha subunits that can form diazepam sensitive GABAA receptors. At 10 mg/kg diazepam these mice were completely protected from diazepam-induced muscle relaxation and motor impairment. However, they showed a trend towards reduced locomotor activity that was quite prominent at higher doses. At least part of this response could be due to β2γ2 GABAA receptors. We have not tested whether β1 or β3 (that may form β1γ2 and β3γ2) behave as β2. As their loop C differs (see Supplementary Fig. S1), it is conceivable that the diazepam site described here does not exist or has different properties in these receptors.
Summary
While in α1β2γ2 receptors diazepam binds to the α1+/γ2− subunit interface and GABA to β2+/α1−, in β2γ2 receptors diazepam binds to the β2+/γ2− subunit interface and GABA to β2+/β2− (Fig. 7). Thus, the β2 subunit can take over the role of the α1 subunit for the formation of both sites, its minus side for the GABA binding site and its plus side for the diazepam binding site.
Methods
Construction of mutated receptor subunits
The point mutations β2Y62Lγ2, β2T202Aγ2, β2T202Sγ2, β2Y205Sγ2, β2Y205Qγ2, β2γ2F77Y, β2γ2S217A, β2γ2Y220S and β2γ2Y220Q were prepared using the QuickChangeTM mutagenesis kit (Stratagene, Agilent Technologies, Basel, Switzerland).
Expression in Xenopus oocytes
Animal experiments were carried out in strict accordance to the Swiss ethical guidelines, and have been approved by the local committee of the Canton Bern Kantonstierarzt, Kantonaler Veterinärdienst Bern (BE85/15). Surgery of Xenopus laevis to obtain the oocytes was done under anesthesia, and all efforts were made to diminish animal suffering. Oocytes were prepared, injected and defolliculated as described previously41,42. Polyadenylated cRNA coding for the subunits of GABAA receptors were prepared in vitro with the mMESSAGE mMACHINE kit (Ambion, Austin, TX, USA). Oocytes were injected with 50 nl of solution containing cRNA coding for wild type or mutants β2 (1 fMol) or γ2 (3 fMol) subunits and then incubated in modified Barth’s solution (10 mM HEPES, pH 7.5, 88 mM NaCl, 1 mM KCl, 2.4 mM NaHCO3, 0.82 mM MgSO4, 0.34 mM Ca(NO3)2, 0.41 mM CaCl2, 100 units/ml penicillin, 100 µg/ml streptomycin) at 18 °C for 5–7 days before measurements.
Functional characterization in Xenopus oocytes
Electrophysiological experiments were performed using an Oocyte Clamp OC-725 (Warner Instrument Corp., Hamden, USA) two-electrode voltage clamp amplifier. Currents were digitized at 5 kHz with MacLab/200 (AD Instruments, Spechbach, Germany).
The holding potential was −80 mV. The perfusion medium contained 90 mM NaCl, 1 mM KCl, 1 mM MgCl2, 1 mM CaCl2 and 5 mM Na-HEPES (pH 7.4). The perfusion solution (6 ml/min) was applied through a glass capillary with an inner diameter of 1.35 mm, the mouth of which was placed about 0.5 mm from the surface of the oocyte. Individual concentration response curves for GABA were fitted with the equation I(c) = Imax/[1 + (EC50/c)n], where c is the concentration of GABA, EC50 the concentration of GABA eliciting half-maximal current amplitude, Imax is the maximal current amplitude, I is the current amplitude, and n is the Hill coefficient. Maximal current amplitudes (Imax) were obtained from the fits of the concentration-response curves. The individual curves were fitted and standardized to Imax and subsequently averaged. For all receptors studied, potentiation was measured at a GABA concentration eliciting 1–5% of the maximal GABA current amplitude. GABA was applied twice alone for 20 s, and then in combination with diazepam for 20 s. The duration of washout periods was 4 min in between agonist or agonist/drug applications to prevent receptor desensitization. At the beginning of the experiments, GABA applications were repeated when the elicited current amplitude altered by >5%. Potentiation was calculated by the following equation: (IModulator + GABA/IGABA−1) * 100%. Concentration dependent potentiation was fitted with the equation I(c) = Imax/[1 + (EC50/c)n], where c is the concentration of diazepam, EC50 the concentration of diazepam eliciting half-maximal current amplitude, Imax is the maximal current amplitude, I is the current amplitude, and n is the Hill coefficient. Maximal current amplitudes (Imax) were obtained from the fits of the concentration-response curves. The individual curves were fitted and standardized to Imax and subsequently averaged.
All data are from at least two different batches of oocytes. Data represent mean ± S.E.M as indicated in each case. An unpaired t-test was used to compare two means. ***p < 0.001.
Computational Modelling and Docking
Homology models of the β2+/γ2− interface were generated based on the 4COF GABAA receptors human β3 homopentamer structure24. Due to the high homology of the β2 and β3 subunits, no insertions or deletions requiring gaps occur in the extracellular domain, while an alignment of the γ2 subunit as described previously43 was used to account for the lower homology in the loop F region. Computational docking was subsequently performed using the GOLD software v1.6.244, where the binding site was defined to be at the interface between loops A – G of the subunits. Sidechains β2Y157, β2T160, β2Y159, β2T161, β2F200, β2T202, and β2Y205 as well as γ2Y58, γ2F77 and γ2T142 were kept flexible, soft potential were applied to the tip of loop C (β2V198 - β2G203) to allow some degree of backbone flexibility, default settings for the docking run were used and the top 100 ranked poses were retained for subsequent analysis. Ligand interactions of poses were computed using the MOE program (MOE (The Molecular Operating Environment), Version 2011.10, Chemical Computing Group Inc., Montreal), and poses featuring interactions with γ2F77 were inspected using two scoring functions (ChemScore Fitness as implemented in GOLD, GoldScore Fitness). “Hits” were defined as poses among top 30 ranked in both scoring functions (consensus score hits) and featuring interactions with γ2F77 and the resulting hit poses were subsequently compared to our poses from previous work at the canonical binding site24.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Macdonald, R. L. & Olsen, R. W. GABAA receptor channels. Annu. Rev. Neurosci. 17, 569–602 (1994).
Olsen, R. W. & Sieghart, W. International Union of Pharmacology. LXX. Subtypes of γ-aminobutyric acid A receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol. Rev. 60, 243–260 (2008).
Sigel, E. & Steinmann, M. E. Structure, function, and modulation of GABAA receptors. J. Biol. Chem. 287, 40224–40231 (2012).
Chang, Y., Wang, R., Barot, S. & Weiss, D. S. Stoichiometry of a recombinant GABAA receptor. J. Neurosci. 16, 5415–5424 (1996).
Farrar, S. J., Whiting, P. J., Bonnert, T. P. & McKernan, R. M. Stoichiometry of a ligand-gated ion channel determined by fluorescence energy transfer. J. Biol. Chem. 274, 10100–10104 (1999).
Tretter, V., Ehya, N., Fuchs, K. & Sieghart, W. Stoichiometry and assembly of a recombinant GABAA receptor subtype. J. Neurosci. 17, 2728–2737 (1997).
Baumann, S. W., Baur, R. & Sigel, E. Subunit arrangement of γ-aminobutyric acid type A receptors. J. Biol. Chem. 276, 36275–36280 (2001).
Baumann, S. W., Baur, R. & Sigel, E. Forced subunit assembly in α1β2γ2 GABAA receptors. Insight into the absolute arrangement. J. Biol. Chem. 277, 46020–46025 (2002).
Baur, R., Minier, F. & Sigel, E. A GABAA receptor of defined subunit composition and positioning: concatenation of five subunits. FEBS Lett. 580, 1616–1620 (2006).
Sigel, E., Stephenson, F. A., Mamalaki, C. & Barnard, E. A. A γ-aminobutyric acid/benzodiazepine receptor complex of bovine cerebral cortex. J. Biol. Chem. 258, 6965–6971 (1983).
Sigel, E., Baur, R., Kellenberger, S. & Malherbe, P. Point mutations affecting antagonist affinity and agonist dependent gating of GABAA receptor channels. EMBO J. 11, 2017–2023 (1992).
Amin, J. & Weiss, D. S. GABAA receptor needs two homologous domains of the β-subunit for activation by GABA but not by pentobarbital. Nature 366, 565–569 (1993).
Sigel, E. & Buhr, A. The benzodiazepine binding site of GABAA receptors. Trends Pharmacol. Sci. 18, 425–429 (1997).
Sieghart, W. Allosteric modulation of GABAA receptors via multiple drug-binding sites. Adv. Pharmacol. (San Diego, Calif.) 72, 53–96 (2015).
Woods, J. H., Katz, J. L. & Winger, G. Benzodiazepines: use, abuse, and consequences. Pharmacol. Rev. 44, 151–347 (1992).
Sigel, E., Baur, R., Trube, G., Mohler, H. & Malherbe, P. The effect of subunit composition of rat brain GABAA receptors on channel function. Neuron 5, 703–711 (1990).
Im, H. K., Im, W. B., Hamilton, B. J., Carter, D. B. & Vonvoigtlander, P. F. Potentiation of γ-aminobutyric acid-induced chloride currents by various benzodiazepine site agonists with the α1γ2, β2γ2 and α1β2γ2 subtypes of cloned γ-aminobutyric acid type A receptors. Mol. Pharmacol. 44, 866–870 (1993).
Whittemore, E. R., Yang, W., Drewe, J. A. & Woodward, R. M. Pharmacology of the human γ-aminobutyric acid A receptor α4 subunit expressed in Xenopus laevis oocytes. Mol. Pharmacol. 50, 1364–1375 (1996).
Amin, J., Brooks-Kayal, A. & Weiss, D. S. Two tyrosine residues on the α subunit are crucial for benzodiazepine binding and allosteric modulation of γ-aminobutyric acid A receptors. Mol. Pharmacol. 51, 833–841 (1997).
Sanna, E. et al. Characterization of the electrophysiological and pharmacological effects of 4-iodo-2,6-diisopropylphenol, a propofol analogue devoid of sedative-anaesthetic properties. Br. J. Pharmacol. 126, 1444–1454 (1999).
Knoflach, F. et al. Pharmacological and electrophysiological properties of recombinant GABAA receptors comprising thealpha3, beta1 and gamma2 subunits. Eur. J. Neurosci. 4, 1–9 (1992).
Miko, A., Werby, E., Sun, H., Healey, J. & Zhang, L. A TM2 Residue in the β1 subunit determines spontaneous opening of homomeric and heteromeric γ-aminobutyric acid-gated ion channels. J. Biol. Chem. 279, 22833–22840 (2004).
Taylor, P. M. et al. Identification of amino acid residues within GABAA receptor β subunits that mediate both homomeric and heteromeric receptor expression. J. Neurosci. 19, 6360–6371 (1999).
Middendorp, S. J., Puthenkalam, R., Baur, R., Ernst, M. & Sigel, E. Accelerated discovery of novel benzodiazepine ligands by experiment-guided virtual screening. ACS Chem. Biol. 9, 1854–1859 (2014).
Chua, H. C., Absalom, N. L., Hanrahan, J. R., Viswas, R. & Chebib, M. The Direct Actions of GABA, 2′-Methoxy-6-Methylflavone and general anaesthetics at β3γ2L GABAA receptors: evidence for receptors with different subunit stoichiometries. PLoS One 10, e0141359, https://doi.org/10.1371/journal.pone.0141359 (2015).
Buhr, A., Schaerer, M. T., Baur, R. & Sigel, E. Residues at positions 206 and 209 of the α1 subunit of γ-aminobutyric acid A receptors influence affinities for benzodiazepine binding site ligands. Mol. Pharmacol. 52, 676–682 (1997).
Tan, K. R., Baur, R., Charon, S., Goeldner, M. & Sigel, E. Relative positioning of diazepam in the benzodiazepine-binding-pocket of GABA receptors. J. Neurochem. 111 (2009).
Schaerer, M. T., Buhr, A., Baur, R. & Sigel, E. Amino acid residue 200 on the α1 subunit of GABAA receptors affects the interaction with selected benzodiazepine binding site ligands. Eur. J. Pharmacol. 354, 283–287 (1998).
Buhr, A., Baur, R. & Sigel, E. Subtle changes in residue 77 of the γ subunit of α1β2γ2 GABAA receptors drastically alter the affinity for ligands of the benzodiazepine binding site. J. Biol. Chem. 272, 11799–11804 (1997).
Belelli, D., Lambert, J. J., Peters, J. A., Wafford, K. & Whiting, P. J. The interaction of the general anesthetic etomidate with the γ-aminobutyric acid type A receptor is influenced by a single amino acid. Proc. Natl. Acad. Sci. USA 94, 11031–11036 (1997).
Chiara, D. C. et al. Mapping general anesthetic binding site(s) in human α1β3 γ-aminobutyric acid type A receptors with [3H]TDBzl-etomidate, a photoreactive etomidate analogue. Biochemistry 51, 836–847 (2012).
Sawyer, G. W., Chiara, D. C., Olsen, R. W. & Cohen, J. B. Identification of the bovine gamma-aminobutyric acid type A receptor alpha subunit residues photolabeled by the imidazobenzodiazepine [3H]Ro15-4513. J. Biol. Chem. 277, 50036–50045 (2002).
Miller, P. S. & Aricescu, A. R. Crystal structure of a human GABAA receptor. Nature 512, 270–275 (2014).
Moitessier, N., Englebienne, P., Lee, D., Lawandi, J. & Corbeil, C. R. Towards the development of universal, fast and highly accurate docking/scoring methods: a long way to go. Br. J. Pharmacol. 153(Suppl 1), S7–26 (2008).
Wieland, H. A., Lüddens, H. & Seeburg, P. H. A single histidine in GABAA receptors is essential for benzodiazepine agonist binding. J. Biol. Chem. 267, 1426–1429 (1992).
Dunn, S. M. J., Davies, M., Muntoni, A. L. & Lambert, J. J. Mutagenesis of the rat α1 subunit of the γ-aminobutyric acidA receptor reveals the importance of residue 101 in determining the allosteric effects of benzodiazepine site ligands. Mol. Pharmacol. 56, 768–774 (1999).
Lee, H. J. et al. A pharmacological characterization of GABA, THIP and DS2 at binary α4β3 and β3δ receptors: GABA activates β3δ receptors via the β3(+) δ(−) interface. Brain Res. 1644, 222–30 (2016).
Baur, R. & Sigel, E. Low expression in Xenopus oocytes and unusual functional properties of α1β2γ2 GABAA receptors with non-conventional subunit arrangement. PLoS One 12 (2017).
Wong, L. W., Tae, H. S. & Cromer, B. A. Assembly, trafficking and function of α1β2γ2 GABAA receptors are regulated by N-terminal regions, in a subunit-specific manner. J. Neurochem. 134, 819–832 (2015).
Ralvenius, W. T., Benke, D., Acuña, M. A., Rudolph, U. & Zeilhofer, H. U. Analgesia and unwanted benzodiazepine effects in point-mutated mice expressing only one benzodiazepine-sensitive GABAA receptor subtype. Nat. Commun. 6, 6803 (2015).
Sigel, E. Properties of single sodium channelstranslated by Xenopus oocytes after injection with messenger ribonucleic acid. J. Physio. 386, (73–90 (1987).
Sigel, E. & Minier, F. The Xenopus oocyte: system for the study of functional expression and modulation of proteins. Mol. Nutr. Food. Res. 49, 228–234 (2005).
Puthenkalam, R. et al. Structural studies of GABAA receptor binding sites: which experimental structure tells us what? Front. Mol. Neurosci. 9, 44 (2016).
Verdonk, M. L., Cole, J. C., Hartshorn, M. J., Murray, C. W. & Taylor, R. D. Improved protein-ligand docking using GOLD. Proteins 52, 609–623 (2003).
Acknowledgements
This work was supported by Swiss National Science Foundation Grant 315230_156929/1 and by the Austrian Science Fund Grant P27746.
Author information
Authors and Affiliations
Contributions
N.W., M.C.M., X.S. and R.B. performed molecular biology and electrophysiological experiments, X.S. and M.E. performed computational studies, N.W. and E.S. designed the experiments, analysed the data and wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wongsamitkul, N., Maldifassi, M.C., Simeone, X. et al. α subunits in GABAA receptors are dispensable for GABA and diazepam action. Sci Rep 7, 15498 (2017). https://doi.org/10.1038/s41598-017-15628-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-15628-7
This article is cited by
-
Differential assembly diversifies GABAA receptor structures and signalling
Nature (2022)
-
Topiramate potential neurotoxicity and mitigating role of ginger oil in mice brain
Environmental Science and Pollution Research (2022)
-
Structure of a human synaptic GABAA receptor
Nature (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
