
Abstract
Clostridium difficile infection (CDI) is typically associated with disturbed gut microbiota and changes related to decreased colonization resistance against C. difficile are well described. However, nothing is known about possible effects of C. difficile on gut microbiota restoration during or after CDI. In this study, we have mimicked such a situation by using C. difficile conditioned medium of six different C. difficile strains belonging to PCR ribotypes 027 and 014/020 for cultivation of fecal microbiota. A marked decrease of microbial diversity was observed in conditioned medium of both tested ribotypes. The majority of differences occurred within the phylum Firmicutes, with a general decrease of gut commensals with putative protective functions (i.e. Lactobacillus, Clostridium_XIVa) and an increase in opportunistic pathogens (i.e. Enterococcus). Bacterial populations in conditioned medium differed between the two C. difficile ribotypes, 027 and 014/020 and are likely associated with nutrient availability. Fecal microbiota cultivated in medium conditioned by E. coli, Salmonella Enteritidis or Staphylococcus epidermidis grouped together and was clearly different from microbiota cultivated in C. difficile conditioned medium suggesting that C. difficile effects are specific. Our results show that the changes observed in microbiota of CDI patients are partially directly influenced by C. difficile.
Similar content being viewed by others


Introduction
Clostridium difficile is recognized as the main cause of infectious diarrhoea associated with hospitalization and is also becoming an important cause of intestinal infections in the community1,2. Crucial for the development of C. difficile infection (CDI) is the disturbance of the normal gut microbiota, which is usually due to treatment with antibiotics1,2. Several in vivo and in vitro models have been used to study interactions between gut microbiota and C. difficile 3,4. In addition, different patient populations have been studied in respect to changes of gut microbiota associated with C. difficile colonization5,6. Individuals colonized with C. difficile have less diverse gut microbiota, with the general decrease of anaerobic bacteria (e.g. Bacteroides, Bifidobacterium) and an increased proportion of facultative anaerobes (e.g. Enterobacteriaceae, Enterococcus)5,6. Not only changes within particular bacterial groups, but also specific microbial patterns are associated with C. difficile colonization7. However, recent findings suggest that CDI development is not associated with specific gut microbial composition, but rather with the presence of a limited set of bacterial species and their functional capacities8,9,10,11,12.
Gut microbiota could interact with C. difficile through several mechanisms13,14, including nutrient competition, regulation of sporulation and vegetative growth and modulation of immune response. The best studied mechanism by which gut commensal bacteria (e.g. Clostridium scindens) confer colonization resistance is modification of host-derived bile salts12,15,16,17. Depletion of bile salts metabolites (i.e. secondary bile acid deoxycholate), due to absent bacterial groups in disturbed microbiota, can permit C. difficile growth12. Specimens from CDI patients contain significantly lower levels of secondary bile acids compared to samples from healthy controls18. On the other hand, the reduction of several bacterial taxa after antibiotic treatment leads to an increased proportion of primary bile acid taurocholate that favours C. difficile germination19,20. Another possible mechanism of colonization resistance is through production of the short chain fatty acid (SCFA), butyrate. Butyrate plays a central role in maintaining gut homeostasis21. Butyrate-producing bacteria are found within the Lachnospiraceae and Ruminococcaceae families; taxa that are greatly reduced in stool specimens from CDI patients22. In nutrient competition, sialic acid is one of the important factors. Gut commensal bacteria (e.g. Bacteroides thetaiotaomicron) release sialic acids from mucus. Several bacterial groups that would normally metabolize free sialic acid are not present anymore upon antibiotic treatment and this allows C. difficile to use this nutrient source and expand its population in the gut23. The gut symbiont B. thetaiotaomicron also produces high levels of succinate, another key nutrient promoting C. difficile expansion after antibiotic treatment24. C. difficile growth is also favoured in the abundance of some other carbon sources (i.e. mannitol, sorbitol) in altered gut microbiota25. Furthermore, the development of CDI is favoured because microbiota changes are associated with unbalanced immune responses after antibiotic treatment26.
While mechanisms through which gut microbiota influences C. difficile are being partially elucidated, not much is known about the possible effect of C. difficile on gut microbiota. During CDI, C. difficile colonizes the intestine with dysbiotic microbiota and could represent a significant proportion of the bacterial population. In a mouse model, C. difficile was shown to be associated with other species in mucus-associated bacterial communities27. Moreover, in a gnotobiotic mouse model with simplified 12 species murine microbiota (Oligo-MM12), colonization with C. difficile resulted in significant compositional perturbation28. Another recent study demonstrated that epidemic C. difficile isolates successfully compete for nutrients even in the presence of a complex gut microbiota29.
It is very likely that C. difficile overgrowth in the gut niche affects the reconstitution of gut microbiota during and after the CDI. To address the possible impact of C. difficile on the growth of common gut bacteria, we analyzed the differences in bacterial community structure of fecal microbiota after in vitro growth in C. difficile conditioned medium. Representative strains from two PCR ribotypes were used. PCR ribotype 027 was selected because it was previously associated with more disturbed microbiota than others7,30,31, while PCR ribotype 014/020 was selected as the most commonly isolated ribotype on global scale32. To characterize the specific changes in the structure of the fecal microbiota community cultivated in C. difficile conditioned medium, several data analysis approaches were used, including the LEfSe algorithm (in the mothur software) and three different machine learning methods. LEfSe uses linear discriminant analysis (LDA) to find operational taxonomic units (OTUs) that differ significantly in abundance between samples33. The machine learning method ReliefF (WEKA software) ranks the OTUs in such a way, that the highest ranking OTUs would best differentiate between the ribotypes (and controls)34. The machine learning methods jRip (WEKA software) and predictive clustering trees (PCT, CLUS software) provide interpretable models (rules and trees), that can be used to predict target values (such as ribotype and relative OTU abundances) and explain the predictions through the model structure35,36,37.
Results
C. difficile conditioned medium decreases the diversity of fecal microbiota in vitro
Two growth media were tested and showed major differences supporting the growth of diverse microbial communities. The stability of the community composition grown in Wilkins Chalgren Anaerobe Broth (WCAB) was better as compared to that grown in Anaerobe Basal Broth (ABB) after a 5 day incubation period (Fig. S1, Table S1). In experiments with the ABB medium, significant changes were observed already between controls and original uncultivated pooled fecal samples (Fig. S1). Hence, this medium was not included in further analyses.
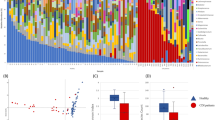
When comparing the growth between unconditioned control WCAB growth medium and C. difficile conditioned WCAB growth medium, an effect on fecal microbiota diversity was detected. We compared the diversity for each day (3 and 5) separately and for each strain separately, to the diversity of the unconditioned control growth medium. The Shannon diversity index values of communities grown in conditioned medium as compared to those grown in control medium were significantly lower for three out of six tested strains (t-test: p = 0.003–0.047) only on day 3 and not on day 5 (Fig. 1). The decrease of the Shannon diversity index in conditioned medium was not ribotype associated, as decreased diversity was observed with strains that belonged to ribotype 027 (1 strain) and ribotype 014/020 (2 strains).

Species diversity (Shannon index with corresponding standard deviation) in WCAB (Wilkins Chalgren Anaerobe Broth) control medium and in WCAB conditioned medium of six different C. difficile strains (1–6), belonging to two different ribotypes (014/020, 027) on day 3 and day 5.
C. difficile conditioned medium affects specific groups within fecal microbiota and changes are ribotype associated
In the first step all samples were grouped either by hierarchical clustering or by learning a PCT (Fig. 2). Both methods distributed our samples and ranked the growth conditions in the same way; the conditioned medium had the highest impact on the grouping, followed by day of incubation and finally by ribotype (014/020 vs. 027). Significant differentiation between ribotypes was noticed with NMDS analysis (R, vegan package; AMOVA: p < 0.001) and within the PCT only after 5 days of incubation (Fig. 2a, panel C; Fig. 3). Differences among the three strains within each ribotype were not statistically significant.

Clustering of samples according to (a) machine learning (PCT, CLUS software) and (b) mothur analysis (tree viewed by MEGA software, v.7.0.26). Samples clustered in three groups, i.e. group of WCAB (Wilkins Chalgren Anaerobe Broth) control medium samples (segment A), group of WCAB conditioned medium samples after three days of incubation (segment B) and group of WCAB conditioned medium samples after five days of incubation (segment C). A predictive clustering tree (PCT) predicts the OTU composition of a sample from the environmental conditions and the ribotype. The leaves of the tree, corresponding to clusters of samples with similar OTU composition, predict the relative abundances of the OTUs, while the path to each leaf from the root of the tree explains the cluster in terms of the environmental conditions. During PCT construction, tests for the internal nodes of the tree are chosen to maximize the similarity in OTU composition for the samples that go to the same branch and maximize the differences between the samples that go to the left and the right branch, respectively (segments A, B, C). The segment C shows the difference in OTU composition between the fifth and the sixth leaf node from the left. The fifth node represents OTUs associated with ribotype 027 (blue box), the sixth node represents OTUs associated with ribotype 014/020 (red box), after five day incubation period in WCAB growth medium. The differences in values of relative abundances between the two ribotypes for OTUs are presented in Fig. 4b.

Non-Metric Multidimensional Scaling (NMDS) analysis for samples cultured in WCAB (Wilkins Chalgren Anaerobe Broth) after a five day incubation period (R, vegan package). Black lines separate two C. difficile ribotypes, whereas colored lines indicate different C. difficile strains.
Changes in OTUs were analysed with LEfSe and the machine learning algorithms and are presented for different pairs of sample types (014/020 and control; 027 and control; 014/020 and 027) (Tables 1 and S2; Figs. 2, 4 and S2).

OTUs associated with WCAB (Wilkins Chalgren Anaerobe Broth) conditioned medium of ribotype 027 ( ) or ribotype 014/020 (
) or ribotype 014/020 ( ), on day 5, selected by using (a) the mothur software (LEfSe test) and (b) machine learning (PCT, CLUS software). Blue font marks OTUs increased in ribotype 027 conditioned medium, red font marks OTUs increased in ribotype 014/020 conditioned medium, detected with both data analysis approaches, respectively. For the mothur analysis, only the LDA scores of significant OTUs are shown. For the machine learning analysis, the values represent the difference of OTU relative abundances between the left and right branch of the segment C in the PCT (shown in Fig. 2).
), on day 5, selected by using (a) the mothur software (LEfSe test) and (b) machine learning (PCT, CLUS software). Blue font marks OTUs increased in ribotype 027 conditioned medium, red font marks OTUs increased in ribotype 014/020 conditioned medium, detected with both data analysis approaches, respectively. For the mothur analysis, only the LDA scores of significant OTUs are shown. For the machine learning analysis, the values represent the difference of OTU relative abundances between the left and right branch of the segment C in the PCT (shown in Fig. 2).
When using LEfSe algorithm, OTUs that significantly decreased in C. difficile conditioned medium according to the control were Escherichia_Shigella, Clostridium_sensu_stricto, Veillonella, Streptococcus, Dorea, Bacteroides, Lactobacillus and an unclassified member of Ruminococcaceae (Table 1) on day 3. On day 5, both ribotypes were also associated with a decrease in Blautia (Table 1). The same genera, but different OTUs (e.g. Dorea, Bacteroides) could be also increased in conditioned medium (Table 1). Bacterial groups with increased proportions in conditioned medium of both ribotypes included Enterococcus and an unclassified member of Burkholderiales (Table 1). Additionally, ribotype 014/020 was associated with an increase in Sutterella.
Among the machine learning approaches, we first used PCTs and learned to predict OTU composition from the incubation period and type of sample (control, ribotype 014/020 and ribotype 027). OTUs that contribute most to the differentiation between microbial communities in conditioned medium on day 5 are given in Fig. 2a (panel C). The top 10 OTUs, ranked in terms of their difference in relative abundance between the two ribotypes are shown.
For the second machine learning task, we used the relative OTU abundances to predict the type of the sample (conditioned medium of specific ribotype). For this purpose, we used the RIPPER algorithm for learning classification rules, i.e. its implementation jRip in the WEKA software. The OTUs identified by jRip as relevant for distinguishing between control and C. difficile conditioned medium samples were Escherichia_Shigella, Phascolarctobacterium and Clostridium_sensu_stricto (Fig. S2).
Rather than a very short list of OTUs, that appear in the jRip rules and are sufficient for predicting the type of sample, feature ranking (ReliefF) in the WEKA software returns a ranking of the complete list of OTUs in terms of their importance. With this third machine learning task, we ranked OTUs according to their relevance for distinguishing between control and C. difficile conditioned medium samples. Most of the top 20 relevant OTUs identified by WEKA ReliefF were also identified by the mothur analysis. These included Escherichia_Shigella, Bacteroides, Clostridium_XIVa, Enterococcus, Clostridium_sensu_stricto, Streptococcus, Coprococcus, Lactobacillus and an unclassified member of Ruminococcaceae (Table 1).
Several OTUs also significantly differed between the two ribotypes. LEfSe and PCT have agreed that the following OTUs are associated with ribotype 027: Escherichia_Shigella, Phascolarctobacterium, Clostridium_XIVa, Dorea and Clostridium_XVIII (Fig. 4). On the other hand, ribotype 014/020 was associated with an increased proportion of Bacteroides, Clostridium_XIVa, Flavonifractor, Coprococcus and an unclassified taxon from Ruminococcaceae (Fig. 4). Each approach also detected some additional OTUs that differed between the two ribotypes.
Changes in fecal microbiota after growth in C. difficile conditioned medium are associated with nutrient availability
To partially address the possible mechanisms underlying the observed changes in fecal microbiota cultivated in C. difficile conditioned media the growth of selected representatives in pure culture in conditioned medium was tested (Fig. S3). Of all tested strains the growth in conditioned medium was mostly affected for Bifidobacterium longum, E. coli and Ruminococcus gnavus and less so for Bacteroides vulgatus and Enterococcus faecium. To rule out direct inhibitory effect of C. difficile, the overlapping drop assay was performed on solid media. No direct inhibition, except partially for R. gnavus, was observed (Fig. S4).
Changes in fecal microbiota after growth in conditioned medium are C. difficile specific
As the original pooled fecal material used in the experiments described in the previous sections was no longer available, new fecal starting material was prepared for subsequent experiments. The first fecal sample was prepared by pooling specimens from healthy donors before antibiotic therapy and the second fecal sample by pooling specimens from the same individuals after antibiotic therapy.
Changes in both types of fecal microbiota (healthy and post-antibiotic) after cultivation in C. difficile conditioned medium and conditioned media of other bacteria (E. coli, S. Enteritidis, Staphylococcus epidermidis) were compared to cultivation in un-conditioned medium.
Changes detected in fecal microbiota composition showed significant differences after growth in all conditioned media (Fig. 5; Fig. S5). The deepest branching divided samples of C. difficile conditioned media from all other samples (Fig. 5). Controls from unconditioned medium were more similar to conditioned medium of all other bacteria than to conditioned medium of C. difficile. Within C. difficile samples, further clustering was associated with fecal microbiota type (healthy or post-antibiotic) and further with C. difficile ribotype. Also, in the group of samples from all other bacteria, the first branching was microbiota type associated (healthy or post-antibiotic). Subsequently, all three typical enteric strains clustered together, while fecal microbiota from S. epidermidis conditioned medium clustered separately.

Hierarchical clustering of samples before antibiotic therapy (mixH2) and after antibiotic therapy (mixD2) cultured in WCAB (Wilkins Chalgren Anaerobe Broth) conditioned media of four different bacteria (Clostridium difficile, 1998 and WF270; E. coli, EH7 and EO157; Salmonella Enteritidis, SALM; Staphylococcus epidermidis, STAPH), obtained by MEGA software (v.7.0.26).
Discussion
C. difficile colonizes the gut only in the presence of non-mature or non-functional microbiota. The expanded C. difficile population could subsequently shape the gut microenvironment and affect gut microbiota reestablishment, but this possibility is poorly explored. In the present study, we have mimicked the conditions of a C. difficile influenced environment by comparing fecal microbiota community structure after cultivation in C. difficile conditioned and fresh growth medium.
Possible mechanisms of interactions between pathobionts and gut microbiota include nutrient depletion, direct inhibition, and effect via immune defenses38. Effects observed in experiments with conditioned medium could be the result of changes in nutrients or inhibitory substances. In co-culture of C. difficile and selected bacterial species, no direct inhibition was detected, except for R. gnavus, while all tested strains showed various degrees of growth decrease in C. difficile conditioned medium (Fig. S3). Hence, we concluded that poor growth in C. difficile conditioned medium is more likely due to changed nutrient availability than to inhibitory substances potentially produced by C. difficile and the same mechanisms could also be effective in vivo during CDI.
Growth medium conditioned by any bacterial species has modified composition and is likely nutrient depleted and will therefore affect the composition of microbiota after cultivation. To test whether changes in microbiota grown in C. difficile conditioned medium are specific, we compared two different types of pooled fecal microbiota (pre- and post-antibiotics) after cultivation in C. difficile conditioned medium with conditioned media of other bacteria. E.coli and Salmonella were selected as representatives of gut pathogens/commensals, whereas S. epidermidis was selected as an example of a bacterium not associated with the gut environment. Interestingly, microbiota in E. coli and Salmonella conditioned medium were more similar to microbiota in Staphylococcus conditioned medium than to C. difficile. The specific nature of changes was to some extent confirmed also in gnotobiotic mouse model with simplified 12 species murine microbiota (Oligo-MM12), where colonization with C. difficile (but not with C. scindens) resulted in significant compositional perturbation as compared to uninfected control animals28. The exact changes were not described in detail, as the main focus of the study was bile acid metabolism in association with colonization resistance.
Cultivation of pooled fecal sample in C. difficile conditioned medium decreased microbial diversity and changed OTU distribution. The decreased diversity is in accordance with the majority of studies on human fecal samples, where lower microbial diversity in C. difficile positive samples was detected by culture or molecular approaches7,30,39,40,41,42,43,44,45,46,47,48. Although the association of C. difficile colonization with a decrease in gut microbiota diversity is well known, our results additionally show, that it could be explained as a cause, as well as a consequence, of CDI. CDI is initially caused by antibiotics or other factors and allows C. difficile overgrowth. Our results suggest that subsequently decreased microbiota diversity is to some extent maintained by C. difficile during CDI.
In some studies, ribotype 027 has been reported to be associated with less diverse microbiota7,30,31. However, in our in vitro model, only fecal microbiota composition, but not microbial diversity, differed between ribotypes 027 and 014/020.
Overall, both analysis approaches, mothur and machine learning, showed that similar genera were affected in conditioned media of both C. difficile ribotypes. The most prominently influenced OTUs have well defined roles within gut microbiota functions and/or have been previously associated with presence or absence of C. difficile (Dorea, Enterococcus, Clostridium_XIVa, Veillonella, Escherichia_Shigella and Lactobacillus).
The microbiota in C. difficile conditioned medium generally contained significantly over-represented opportunistic pathogens, particularly Enterococcus members. This is in agreement with several other studies that found association between C. difficile colonization/infection and enterococci44,49 or vancomycin-resistant enterococci (VRE)50,51,52,53. VRE colonization was also described as a risk factor for CDI recurrence51,52. Another opportunistic pathogen, Escherichia_Shigella, was found in reduced proportion in C. difficile conditioned medium. Reduced growth was also observed for pure cultures of E. coli in C. difficile conditioned medium, while E. faecium was affected to a much lesser degree (Fig. S3).
On the other hand, in C. difficile conditioned medium we found a significant under-representation of gut commensals with putative protective functions, particularly lactate (Lactobacillus) and butyrate (Clostridium_XIVa) producers. Some previous studies already reported on lower lactobacilli counts in C. difficile positive samples39,42. Our findings suggest that lactobacilli, an important bacterial group for balanced microbiota, regenerate poorly in C. difficile presence. Poor growth of lactobacilli in the presence of C. difficile could also explain why some probiotics have low efficiency in the prevention of CDI in several clinical trials54,55. Nevertheless, the probiotic strain Lactobacillus plantarum 299v 56,57, a mixture of Lactobacillus probiotic strains58 or lactobacilli mixed with other probiotic strains59 could have strong potential as adjunctive therapies to antibiotics for the prevention of CDI. Due to a decreased abundance of several butyrate-producing bacteria, including Clostridium_XIVa members, in C. difficile positive samples43 and their re-establishment after fecal microbiota transplantation60,61, there is growing evidence about the important role of those bacteria in colonization resistance to CDI.
Furthermore, in C. difficile conditioned media, we found a decreased proportion of some other putative butyrate producing bacteria, belonging to the family Lachnospiraceae, i.e. Blautia and Dorea. This correlates with studies on humans and animal models showing association of both genera with colonization resistance to CDI12,62. In addition to their different relative abundance between C. difficile conditioned medium (any ribotype) and control medium, both genera (together with Bacteroides), were among those that differentiated between ribotype 027 and ribotype 014/020 exposed fecal microbiota in our in vitro model. Some Dorea OTUs showed relative increase and other decrease between the C. difficile conditioned and control media. This could correspond to different functions associated with the genus. Although Dorea is a genus with recognised positive effects on gut and human health, increased proportions of Dorea have previously also been associated with colorectal adenomas63 and the irritable bowel syndrome64.
Another affected genus was Veillonella, which showed decrease in relative abundance in C. difficile conditioned medium. To date, the reports on Veillonella and CDI are somewhat conflicting. A recent study by Antharam and co-workers (2016) linked two Veillonella species with specific changes in the gut metabolome that correlated strongly with CDI9. Patients with recurrent CDI possessed an increased proportion of Veillonella 62, while oncological patients colonized with C. difficile had decreased levels of Veillonella 65.
In summary, we could show that C. difficile causes specific changes in fecal microbiota and that nutrient availability is a more likely mechanism than direct inhibitory effects. Changes impact common gut microbiota members associated with colonization resistance or are associated with an increased proportion of opportunistic microorganisms. The two tested C. difficile ribotypes differ in their impact on fecal microbiota composition. Our results suggest that decreased microbiota diversity initially caused by antibiotics and predisposing to CDI, is to some extent maintained by C. difficile during and after the infection.
Methods
Conditioned medium preparation
Six different C. difficile strains, belonging to two PCR ribotypes (014/020, 027), were selected from our C. difficile strain collection (strain designations ZZV10-2514, ZZV11-3188, ZZV11-3298, ZZV11-3304, ZZV12-4777, ZZV14-5907). Selected strains originated from humans and were isolated between the years 2010 and 2014. All strains were incubated in anaerobic workstation at 37 °C (Don Whitley Scientific) for 48 hours in Wilkins Chalgren Anaerobe Broth (WCAB) (Oxoid) and Anaerobe Basal Broth (ABB) (Oxoid) to obtain C. difficile suspensions with up to 1010 CFU/ml (WCAB) and up to 109 CFU/ml (ABB), respectively. After centrifugation at 10,000 rpm for 5 min, supernatants were filtered (0.2 μm, cellulose acetate, Sarstedt) to prepare conditioned medium, which was subsequently used for fecal microbiota culturing.
In vitro model of C. difficile conditioned medium and fecal microbiota
C. difficile negative fecal samples were randomly selected from samples sent for C. difficile testing (male: n = 5; female: n = 5, all aged under 65 years). Specimens were pooled and diluted in glycerol to form 10% slurry and immediately frozen at −80 °C until further processing. Fecal slurry was added to growth media (ABB, WCAB) (1:100, v:v) and incubated overnight to prepare fecal inoculum for in vitro cultivation. This overnight fecal inoculum (50 μl) was added into ABB or WCAB conditioned medium (4,950 μl) in a 6-well plate. Cultures were gently mixed and then incubated anaerobically at 37 °C for five days. Samples (5 ml) for total bacterial DNA extraction were taken after 3 and 5 days of incubation. For each sample with a specific combination of growth medium, C. difficile strain and incubation time, three parallels were tested. Fecal inoculum grown in freshly prepared and prereduced ABB and WCAB growth media was used as a control. All incubations and sample handling were performed anaerobically at 37 °C in an anaerobic workstation (10% CO2, 10% H2, 80% N2) (Don Whitley Scientific).
Cultivation of fecal microbiota in C. difficile conditioned medium in comparison to media conditioned by other bacteria
This set of experiments was performed as described above but using only WCAB medium, different inoculum of pooled fecal microbiota, only two C. difficile strains and additional other bacteria.
Besides two C. difficile strains (ZZV11-3298, ZZV14-5907) we tested Salmonella Enteritidis strain (ATCC 13076), Staphylococcus epidermidis strain (ATCC 12228) and two E. coli strains. One of them was O157 strain isolated in diagnostic laboratory (ZZV17-8567) and an isolate obtained from fecal sample used also for inoculum preparation (ZZV17-8330). Strains were incubated anaerobically or aerobically and conditioned medium was prepared as described above.
Two different fecal inoculums were prepared by pooling fecal samples of three donors (male: n = 1, female: n = 2, all aged under 65 years). Inoculum designated ‘mixH2’ was prepared from samples obtained before antibiotic therapy (clarithromycin, penicillin V or amoxicillin). Inoculum designated ‘mixD2’ was prepared from samples obtained from same individuals three days after completed antibiotic therapy.
Cultures were gently mixed and incubated anaerobically at 37 °C for three days. For each sample with a specific combination of growth medium, bacterial strain and type of microbiota, three parallels were tested.
Cultivation of pure cultures of selected bacteria in C. difficile conditioned media or in co-culture with C. difficile
Bifidobacterium longum (ZZV17-8523), Bacteroides vulgatus (ZZV17-8401), Enterococcus faecium (ZZV17-8382), E. coli (ZZV17-8330) and Ruminococcus gnavus (ZZV11-4196) were grown anaerobically at 37 °C in fresh WCAB medium and in conditioned WCAB medium of two C. difficile strains (ZZV11-3298, ZZV14-5907). For each condition, the experiment was done in triplicates. Growth was monitored by OD620 measurement (Synergy2, BioTek Instruments) at different time points (0 hr, 4 hr, 8 hr, 12 hr, 24 hr, 48 hr and 72 hr).
The same isolates were co-cultured with the same two C. difficile strains on Wilkins Chalgren Anaerobe Agar (WCAA). We used the overlapping drop technique as described by Parker and Simmons (1959)66 with minor modifications. Pairs of overlapping drops of overnight WCAB broth cultures were placed on WCAA solid medium. Drops of 7 µl were deposited on WCAA plate: the second drop of each pair was added as soon as the first had dried and overlapped the first by approximately 1/3 of its area. Plates were examined for inhibition extending beyond the area of overlap after 24 hours of anaerobic incubation at 37 °C.
Isolation of the total bacterial DNA and high-throughput 16S rDNA amplicon sequencing
Samples were centrifuged at 10,000 rpm for 10 min and the pellet was used for total bacterial DNA isolation (QIAamp Fast Stool DNA Mini Kit, Qiagen) after mechanical disruption (speed 7,000 for 70 s) with the SeptiFast Lyse Kit (Roche) on MagNA Lyser (Roche). Bacterial community composition was determined by paired-end sequencing on an Illumina MiSeq platform, targeting the V3-V4 hypervariable region of the 16 S rRNA gene. Library preparation was carried out using the 341 F (5′-CCTACGGGNGGCWGCAG-3′) – 805 R (5′-GACTACHVGGGTATCTAATCC-3′) set of primers. Sequencing yielded an average depth of 140,000 sequences per sample. A template-free sample processed in the same way as the real samples and sequenced on the same run was included as negative sequencing control.
Data analysis
MiSeq output data was analysed with statistical tools included in the mothur software and different machine learning approaches.
Analysis in mothur (v.1.36.1)67 was done according to the MiSeq standard operating procedure (SOP) for Illumina paired end reads68. The following criteria were included in the analysis: i) sequences were not allowed any ambiguous bases and the maximum homopolymer length was set to 8 base pairs; ii) sequences were aligned against the Silva reference alignment; iii) chimeras were searched with the UCHIME algorithm; iv) classification of sequences was performed using the RDP training set (v.9) with 0.80 threshold value; v) OTUs (operational taxonomic units) were constructed with 0.03 cutoff value; vi) OTUs with relative abundance lower than 0.001% were removed from contingency table. Alpha sample diversity was calculated with the Shannon diversity index. Beta diversity was estimated with the AMOVA test, which was performed using Bray Curtis distances69. OTUs that differed between treatments were selected with respect to linear discriminant analysis (LDA) effect size (LEfSe) method33. Non-Metric Multidimensional Scaling (NMDS) analysis was done in R with the vegan package using the Bray Curtis distance matrix as input. Hierarchical clustering was obtained with MEGA software (v.7.0.26)70 using the Bray Curtis distance matrix as input.
We applied three types of machine learning methods to analyze the collected data. In machine learning terminology, each data sample is called an example and corresponds to a row in the data table. The variables, also called attributes, correspond to columns in the data table. We distinguish between descriptive variables (inputs) and target variables (outputs). In the first machine learning task in this study, we used incubation period and type of sample (control, ribotype 014/020 and ribotype 027) as descriptive variables and the bacterial community composition in terms of the relative abundance of OTUs as target variables. In the second and third tasks, we used the relative abundances of OTUs as description of the type of sample as the target variable.
The different types of machine learning methods we applied were: learning predictive clustering trees (PCTs)35, learning classification rules (RIPPER)36 and feature ranking (ReliefF)34. In particular, we used the software packages CLUS (available at: http://sourceforge.net/projects/clus/)35 for learning PCTs and WEKA v3.8 (Waikato Environment for Knowledge Analysis) for learning rules (jRip implementation of RIPPER) and feature ranking37.
References
Rupnik, M., Wilcox, M. & Gerding, D. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nature Reviews Microbiology 7, 526–536, https://doi.org/10.1038/nrmicro2164 (2009).
Leffler, D. & Lamont, J. Clostridium difficile Infection. New England Journal of Medicine 372, 1539–1548, https://doi.org/10.1056/NEJMra1403772 (2015).
Best, E. L., Freeman, J. & Wilcox, M. H. Models for the study of Clostridium difficile infection. Gut Microbes 3, 145–167, https://doi.org/10.4161/gmic.19526 (2012).
Leslie, J. & Young, V. A whole new ball game: Stem cell-derived epithelia in the study of host microbe interactions. Anaerobe 37, 25–28, https://doi.org/10.1016/j.anaerobe.2015.10.016 (2016).
Zalig, S. & Rupnik, M. Clostridium difficile infection and gut microbiota. Seminars in Colon and Rectal. Surgery 25, 4, https://doi.org/10.1053/j.scrs.2014.05.005 (2014).
Theriot, C. M. & Young, V. B. Microbial and metabolic interactions between the gastrointestinal tract and Clostridium difficile infection. Gut Microbes 5, 86–95, https://doi.org/10.4161/gmic.27131 (2014).
Skraban, J. et al. Gut microbiota patterns associated with colonization of different Clostridium difficile ribotypes. PLoS One 8, e58005, https://doi.org/10.1371/journal.pone.0058005 (2013).
Moya, A. & Ferrer, M. Functional Redundancy-Induced Stability of Gut Microbiota Subjected to Disturbance. Trends Microbiol 24, 402–413, https://doi.org/10.1016/j.tim.2016.02.002 (2016).
Antharam, V. et al. An Integrated Metabolomic and Microbiome Analysis Identified Specific Gut Microbiota Associated with Fecal Cholesterol and Coprostanol in Clostridium difficile Infection. Plos One 11, https://doi.org/10.1371/journal.pone.0148824 (2016).
Rojo, D. et al. Clostridium difficile heterogeneously impacts intestinal community architecture but drives stable metabolome responses. Isme Journal 9, 2206–2220, https://doi.org/10.1038/ismej.2015.32 (2015).
Knecht, H. et al. Effects of beta-Lactam Antibiotics and Fluoroquinolones on Human Gut Microbiota in Relation to Clostridium difficile Associated Diarrhea. Plos One 9, https://doi.org/10.1371/journal.pone.0089417 (2014).
Buffie, C. et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517, 205–U207, https://doi.org/10.1038/nature13828 (2015).
Rupnik, M. Toward a True Bacteriotherapy for Clostridium difficile Infection. New England Journal of Medicine 372, 1566–1568, https://doi.org/10.1056/NEJMcibr1500270 (2015).
Britton, R. & Young, V. Interaction between the intestinal microbiota and host in Clostridium difficile colonization resistance. Trends in Microbiology 20, 313–319, https://doi.org/10.1016/j.tim.2012.04.001 (2012).
Greathouse, K., Harris, C. & Bultman, S. Dysfunctional Families: Clostridium scindens and Secondary Bile Acids Inhibit the Growth of Clostridium difficile. Cell Metabolism 21, 9–10, https://doi.org/10.1016/j.cmet.2014.12.016 (2015).
Weingarden, A. et al. Changes in Colonic Bile Acid Composition following Fecal Microbiota Transplantation Are Sufficient to Control Clostridium difficile Germination and Growth. Plos One 11, https://doi.org/10.1371/journal.pone.0147210 (2016).
Winston, J. & Theriot, C. Impact of microbial derived secondary bile acids on colonization resistance against Clostridium difficile in the gastrointestinal tract. Anaerobe 41, 44–50, https://doi.org/10.1016/j.anaerobe.2016.05.003 (2016).
Allegretti, J. R. et al. Recurrent Clostridium difficile infection associates with distinct bile acid and microbiome profiles. Aliment Pharmacol Ther 43, 1142–1153, https://doi.org/10.1111/apt.13616 (2016).
Sorg, J. & Sonenshein, A. Bile salts and glycine as cogerminants for Clostridium difficile spores. Journal of Bacteriology 190, 2505–2512, https://doi.org/10.1128/JB.01765-07 (2008).
Vincent, C. & Manges, A. Antimicrobial Use, Human Gut Microbiota and Clostridium difficile Colonization and Infection. Antibiotics-Basel 4, 230–253, https://doi.org/10.3390/antibiotics4030230 (2015).
Wong, J. M., de Souza, R., Kendall, C. W., Emam, A. & Jenkins, D. J. Colonic health: fermentation and short chain fatty acids. J Clin Gastroenterol 40, 235–243 (2006).
Ross, C. L., Spinler, J. K. & Savidge, T. C. Structural and functional changes within the gut microbiota and susceptibility to Clostridium difficile infection. Anaerobe 41, 37–43, https://doi.org/10.1016/j.anaerobe.2016.05.006 (2016).
Ng, K. M. et al. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 502, 96–99, https://doi.org/10.1038/nature12503 (2013).
Ferreyra, J. A. et al. Gut microbiota-produced succinate promotes C. difficile infection after antibiotic treatment or motility disturbance. Cell Host Microbe 16, 770–777, https://doi.org/10.1016/j.chom.2014.11.003 (2014).
Theriot, C. M. et al. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat Commun 5, 3114, https://doi.org/10.1038/ncomms4114 (2014).
Buonomo, E. L. & Petri, W. A. The microbiota and immune response during Clostridium difficile infection. Anaerobe 41, 79–84, https://doi.org/10.1016/j.anaerobe.2016.05.009 (2016).
Semenyuk, E. G. et al. Analysis of Bacterial Communities during Clostridium difficile Infection in the Mouse. Infect Immun 83, 4383–4391, https://doi.org/10.1128/IAI.00145-15 (2015).
Studer, N. et al. Functional Intestinal Bile Acid 7α-Dehydroxylation by Clostridium scindens Associated with Protection from Clostridium difficile Infection in a Gnotobiotic Mouse Model. Front Cell Infect Microbiol 6, 191, https://doi.org/10.3389/fcimb.2016.00191 (2016).
Robinson, C., Auchtung, J., Collins, J. & Britton, R. Epidemic Clostridium difficile Strains Demonstrate Increased Competitive Fitness Compared to Nonepidemic Isolates. Infection and Immunity 82, 2815–2825, https://doi.org/10.1128/IAI.01524-14 (2014).
Rea, M. C. et al. Clostridium difficile carriage in elderly subjects and associated changes in the intestinal microbiota. J Clin Microbiol 50, 867–875, https://doi.org/10.1128/JCM.05176-11 (2012).
Lawley, T. D. et al. Targeted restoration of the intestinal microbiota with a simple, defined bacteriotherapy resolves relapsing Clostridium difficile disease in mice. PLoS Pathog 8, e1002995, https://doi.org/10.1371/journal.ppat.1002995 (2012).
Martin, J., Monaghan, T. & Wilcox, M. Clostridium difficile infection: epidemiology, diagnosis and understanding transmission. Nature Reviews Gastroenterology & Hepatology 13, 206–216, https://doi.org/10.1038/nrgastro.2016.25 (2016).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol 12, R60, https://doi.org/10.1186/gb-2011-12-6-r60 (2011).
Robnik-Sikonja, M. & Kononenko, I. Theoretical and empirical analysis of ReliefF and RReliefF. Machine Learning 53, 23–69, https://doi.org/10.1023/A:1025667309714 (2003).
Blockeel, H. & De Raedt, L. Top-down induction of first-order logical decision trees. Artificial Intelligence 101, 285–297, https://doi.org/10.1016/S0004-3702(98)00034-4 (1998).
Cohen, W. W. In Proceedings of the 12th International Conference on Machine Learning. (eds Prieditis, A. & Russell, S.) 115–123 (ACM Press New York).
Hall, M. et al. The WEKA data mining software: an update. ACM SIGKDD Explorations Newsletter 11, 9, https://doi.org/10.1145/1656274.1656278 (2009).
Stecher, B. & Hardt, W. Mechanisms controlling pathogen colonization of the gut. Current Opinion in Microbiology 14, 82–91, https://doi.org/10.1016/j.mib.2010.10.003 (2011).
Hopkins, M. & MacFarlane, G. Changes in predominant bacterial populations in human faeces with age and with Clostridium difficile infection. Journal of Medical Microbiology 51, 448–454 (2002).
Chang, J. et al. Decreased diversity of the fecal microbiome in recurrent Clostridium difficile-associated diarrhea. Journal of Infectious Diseases 197, 435–438, https://doi.org/10.1086/525047 (2008).
De La Cochetiere, M. et al. Effect of antibiotic therapy on human fecal microbiota and the relation to the development of Clostridium difficile. Microbial Ecology 56, 395–402, https://doi.org/10.1007/s00248-007-9356-5 (2008).
Sepp, E. et al. Intestinal lactoflora in Estonian and Norwegian patients with antibiotic associated diarrhea. Anaerobe 17, 407–409, https://doi.org/10.1016/j.anaerobe.2011.04.012 (2011).
Antharam, V. et al. Intestinal Dysbiosis and Depletion of Butyrogenic Bacteria in Clostridium difficile Infection and Nosocomial Diarrhea. Journal of Clinical Microbiology 51, 2884–2892, https://doi.org/10.1128/JCM.00845-13 (2013).
Vincent, C. et al. Reductions in intestinal Clostridiales precede the development of nosocomial Clostridium difficile infection. Microbiome 1, https://doi.org/10.1186/2049-2618-1-18 (2013).
Perez-Cobas, A. et al. Structural and functional changes in the gut microbiota associated to Clostridium difficile infection. Frontiers in Microbiology 5, https://doi.org/10.3389/fmicb.2014.00335 (2014).
Zhang, L. et al. Insight into alteration of gut microbiota in Clostridium difficile infection and asymptomatic C. difficile colonization. Anaerobe 34, 1–7, https://doi.org/10.1016/j.anaerobe.2015.03.008 (2015).
Gu, S. et al. Identification of key taxa that favor intestinal colonization of Clostridium difficile in an adult Chinese population. Microbes and Infection 18, 30–38, https://doi.org/10.1016/j.micinf.2015.09.008 (2016).
Milani, C. et al. Gut microbiota composition and Clostridium difficile infection in hospitalized elderly individuals: a metagenomic study. Scientific Reports 6, https://doi.org/10.1038/srep25945 (2016).
Ozaki, E. et al. Clostridium difficile colonization in healthy adults: transient colonization and correlation with enterococcal colonization. Journal of Medical Microbiology 53, 167–172, https://doi.org/10.1099/jmm.0.05376-0 (2004).
Fujitani, S., George, W., Morgan, M., Nichols, S. & Murthy, A. Implications for vancomycin-resistant Enterococcus colonization associated with Clostridium difficile infections. American Journal of Infection Control 39, 188–193, https://doi.org/10.1016/j.ajic.2010.10.024 (2011).
Choi, H., Kim, K., Lee, S. & Lee, S. Risk Factors for Recurrence of Clostridium difficile Infection: Effect of Vancomycin-resistant Enterococci Colonization. Journal of Korean Medical Science 26, 859–864, https://doi.org/10.3346/jkms.2011.26.7.859 (2011).
Stripling, J. et al. Loss of Vancomycin-Resistant Enterococcus Fecal Dominance in an Organ Transplant Patient With Clostridium difficile Colitis After Fecal Microbiota Transplant. Open Forum Infectious Diseases 2, https://doi.org/10.1093/ofid/ofv078 (2015).
McKinley, L. et al. Vancomycin-resistant Enterococcus co-colonization rates with methicillin-resistant Staphylococcus aureus and Clostridium difficile in critically ill veterans. Am J Infect Control 44, 1047–1049, https://doi.org/10.1016/j.ajic.2016.02.005 (2016).
Allen, S. et al. Lactobacilli and bifidobacteria in the prevention of antibiotic-associated diarrhoea and Clostridium difficile diarrhoea in older inpatients (PLACIDE): a randomised, double-blind, placebo-controlled, multicentre trial. Lancet 382, 1249–1257, https://doi.org/10.1016/S0140-6736(13)61218-0 (2013).
Evans, C. & Johnson, S. Prevention of Clostridium difficile Infection With Probiotics. Clinical Infectious Diseases 60, S122–S128, https://doi.org/10.1093/cid/civ138 (2015).
Klarin, B. et al. Lactobacillus plantarum 299v reduces colonisation of Clostridium difficile in critically ill patients treated with antibiotics. Acta Anaesthesiologica Scandinavica 52, 1096–1102, https://doi.org/10.1111/j.1399-6576.2008.01748.x (2008).
Kujawa-Szewieczek, A. et al. The Effect of Lactobacillus plantarum 299v on the Incidence of Clostridium difficile Infection in High Risk Patients Treated withAntibiotics. Nutrients 7, 10179–10188, https://doi.org/10.3390/nu7125526 (2015).
Maziade, P., Pereira, P. & Goldstein, E. A Decade of Experience in Primary Prevention of Clostridium difficile Infection at a Community Hospital Using the Probiotic Combination Lactobacillus acidophilus CL1285, Lactobacillus casei LBC80R, and Lactobacillus rhamnosus CLR2 (Bio-K+). Clinical Infectious Diseases 60, S144–S147, https://doi.org/10.1093/cid/civ178 (2015).
Bakken, J. Staggered and Tapered Antibiotic Withdrawal With Administration of Kefir for Recurrent Clostridium difficile Infection. Clinical Infectious Diseases 59, 858–861, https://doi.org/10.1093/cid/ciu429 (2014).
van Nood, E. et al. Duodenal Infusion of Donor Feces for Recurrent Clostridium difficile. New England Journal of Medicine 368, 407–415, https://doi.org/10.1056/NEJMoa1205037 (2013).
Jalanka, J. et al. Long-term effects on luminal and mucosal microbiota and commonly acquired taxa in faecal microbiota transplantation for recurrent Clostridium difficile infection. Bmc Medicine 14, https://doi.org/10.1186/s12916-016-0698-z (2016).
Khanna, S. et al. Gut microbiome predictors of treatment response and recurrence in primary Clostridium difficile infection. Alimentary Pharmacology & Therapeutics 44, 715–727, https://doi.org/10.1111/apt.13750 (2016).
Shen, X. J. et al. Molecular characterization of mucosal adherent bacteria and associations with colorectal adenomas. Gut Microbes 1, 138–147, https://doi.org/10.4161/gmic.1.3.12360 (2010).
Rajilic-Stojanovic, M. et al. Global and Deep Molecular Analysis of Microbiota Signatures in Fecal Samples From Patients With Irritable Bowel Syndrome. Gastroenterology 141, 1792–1801, https://doi.org/10.1053/j.gastro.2011.07.043 (2011).
Zwielehner, J. et al. Changes in Human Fecal Microbiota Due to Chemotherapy Analyzed by TaqMan-PCR, 454 Sequencing and PCR-DGGE Fingerprinting. Plos One 6, https://doi.org/10.1371/journal.pone.0028654 (2011).
Parker, M. T. & Simmons, L. E. The inhibition of Corynebacterium diphtheriae and other gram-positive organisms by Staphylococcus aureus. J Gen Microbiol 21, 457–476, https://doi.org/10.1099/00221287-21-2-457 (1959).
Schloss, P. et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Applied and Environmental Microbiology 75, 7537–7541, https://doi.org/10.1128/AEM.01541-09 (2009).
Kozich, J., Westcott, S., Baxter, N., Highlander, S. & Schloss, P. Development of a Dual-Index Sequencing Strategy and Curation Pipeline for Analyzing Amplicon Sequence Data on the MiSeq Illumina Sequencing Platform. Applied and Environmental Microbiology 79, 5112–5120, https://doi.org/10.1128/AEM.01043-13 (2013).
Schloss, P. Evaluating different approaches that test whether microbial communities have the same structure. Isme Journal 2, 265–275, https://doi.org/10.1038/ismej.2008.5 (2008).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Molecular Biology and Evolution 33, 1870–1874, https://doi.org/10.1093/molbev/msw054 (2016).
Acknowledgements
The authors acknowledge the financial support from the Slovenian Research Agency (research core funding No. P3-0387, P2-0103, L2-7509), as well as the European Commission, via the grant ICT-2013-612944 MAESTRA. SH, AM and MB are supported by the Slovenian Research Agency Grants (Young Researcher).
Author information
Authors and Affiliations
Contributions
S.H. and M.R. prepared study design. S.H. performed the majority of experiments. A.M. contributed to the sequencing. S.H., A.M., M.B. and S.D. performed the data analysis. M.R. contributed to the data analysis. All authors contributed to the text preparation and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Horvat, S., Mahnic, A., Breskvar, M. et al. Evaluating the effect of Clostridium difficile conditioned medium on fecal microbiota community structure. Sci Rep 7, 16448 (2017). https://doi.org/10.1038/s41598-017-15434-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-15434-1
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
