
Abstract
Melatonin is synthesized from serotonin and it is excreted as sulphatoxymelatonin in urine. We aim to evaluate urinary sulphatoxymelatonin as a biomarker of brain serotonin status in a cohort of patients with mutations in genes related to serotonin biosynthesis. We analized urinary sulphatoxymelatonin from 65 healthy subjects and from 28 patients with genetic defects. A total of 18 patients were studied: 14 with autosomal dominant and recessive guanosine triphosphate cyclohydrolase-I deficiency; 3 with sepiapterin reductase deficiency; and 1 with aromatic L-amino acid decarboxylase deficiency. Further 11 patients were studied after receiving serotoninergic treatment (serotonin precursors, monoamine oxidase inhibitors, selective serotonin re-uptake inhibitors): 5 with aromatic L-amino acid decarboxylase deficiency; 1 with sepiapterin reductase deficiency; 3 with dihydropteridine reductase deficiency; and 2 with 6-pyruvoyltetrahydropterin synthase deficiency. Among the patients without therapy, 6 presented low urinary sulphatoxymelatonin values, while most of the patients with guanosine triphosphate cyclohydrolase-I deficiency showed normal values. 5 of 11 patients under treatment presented low urine sulphatoxymelatonin values. Thus, decreased excretion of sulphatoxymelatonin is frequently observed in cases with severe genetic disorders affecting serotonin biosynthesis. In conclusion, sulphatoxymelatonin can be a good biomarker to estimate serotonin status in the brain, especially for treatment monitoring purposes.
Similar content being viewed by others
Introduction
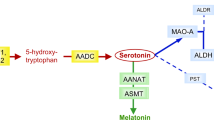
Melatonin (5-methoxy-N-acethyltriptamine) is secreted by the pineal gland and is synthesized from serotonin. Melatonin synthesis is regulated by two specific enzymes: serotonin-N-acetyl transferase (SNAT, EC 2.3.1.5), which is a rate-limiting enzyme, and 5-hydroxyindole-O-methyl transferase (HIOMT EC 2.1.1.4), which transfers a methyl group from S-adenosylmethionine to 2-hydroxyl of N-acetylserotonin (Fig. 1). Melatonin is released from the pineal gland and enters the circulation. Other melatonin sources are the retina, gut, skin, platelets and bone marrow, but their contribution to circulating melatonin is less relevant than that of pineal gland1. Melatonin is metabolized in the liver to 6-hydroxymelatonin by cytochrome CYP1A2 (EC 1.14.14.1), and it is excreted in urine as sulphatoxymelatonin (aMT6s) and, to a lower extent, as glucuronide conjugate1. Urine aMT6s excretion closely correlates to the plasma melatonin profile1,2 and is a good indicator of melatonin secretion from the pineal gland3. Thus, it has been suggested that the measurement of urinary aMT6s may be a good biomarker of serotonin status in the brain4. Yano et al. reported that blood melatonin and urine aMT6s levels may serve as biomarkers reflecting brain serotonin synthesis in subjects with phenylketonuria (PKU)4.

Melatonin pathway. Melatonin synthesis occurs in pineal gland (and other tissues) and its metabolism produces 6-sulphatoxymelatonin, the main urine melatonin metabolite. Serotonin and melatonin are marked in bold type. Synthesis and salvage pathways (grey arrows) of the tetrahydrobiopterin (BH4) are also represented. Enzymes are in cursive. AADC: aromatic L-amino acid decarboxylase; BH4: tetrahydrobiopterin; COMT: catechol O-methyltransferase; CYP1A2: cytochrome P450 isoform CYP1A2; DHPR: dihydropteridine reductase; GTP: guanosine triphosphate; GTPCH: GTP cyclohydrolase I; HIOMT: 5-hydroxyindole-O-methyltransferase; MAO: monoamine oxidase; OH-BH4: hydroxy-tetrahydrobiopterin; PCD: pterin-4a-carbinolamine dehydratase; PTPS: 6-pyruvoyl-tetrahydropterin synthase; q-BH2: quinonoid-dihydrobiopterin; SNAT: serotonin N-acetyltransferase; SR: sepiapterin reductase; TrpH: tryptophan hydroxylase; PLP: pyridoxal phosphate.
There are several genetic alterations affecting brain serotonin and dopamine biosynthesis (Fig. 1): aromatic L-amino acid decarboxylase (AADC) deficiency (OMIM#608643), pyridoxal phosphate (PLP) deficiency (pyridoxamine 5-phosphate oxidase deficiency, OMIM#610090), antiquitin deficiency (OMIM#266100) and tetrahydrobiopterin (BH4) disorders including the following deficiencies: dominant and recessive form of guanosine triphosphate cyclohydrolase-I (GTPCH-1, OMIM#128230 and OMIM#233910 respectively), 6-pyruvoyltetrahydropterin synthase (PTPS, OMIM#261640), sepiapterin reductase (SR, OMIM#612716), dihydropteridine reductase (DHPR, OMIM#261630) and primapterinuria (OMIM#264070). Other disorders such as tyrosine hydroxylase deficiency and the recently reported transportopathies5,6 impair dopamine biosynthesis in particular7. The clinical presentation of these disorders include symptoms related to autonomic dysfunction, which manifest as sweating, temperature dysregulation, hypersalivation, nasal congestion and psychiatric signs8 such as bad behaviour and autistic features. Movement disorders are caused mainly by dopamine dysregulation, which includes gait disturbances, dystonia, dyskinesia, Parkinsonism, tremor, oculogyric crises, palpebral ptosis and axial hypotonia7,9. However, the symptoms that are thought to be related to serotonin deficiency are more difficult to clinically assess.
To estimate serotonin deficiency in these conditions, the analysis of cerebrospinal fluid (CSF) 5-hydroxyindoleacetic acid (5HIAA) is the most appropriate tool10. Furthermore, most of the patients harbouring these disorders received different treatment approaches to restore normal serotonin (and dopamine) levels in the brain. To evaluate the biochemical response to treatment, patients often undergo a lumbar puncture for the monitoring of brain serotonin status by 5HIAA quantification.
To evaluate whether urine aMT6s is a reliable biomarker for serotonin status in the brain, we first established normal values for aMT6s in urine in a healthy population. In a second step, we analysed urine samples from patients with pathogenic mutations in genes involved in the serotonin biosynthetic pathway.
Methods
Subjects
The reference values (RVs) for urinary aMT6s were established in 65 healthy control subjects (36 females; age range 2–61 years; mean = 10.0) with similar age to our patient population. Lighting environment during the night when the morning urine was recovered was controlled (in darkness). The duration of sleep on that night was between 8–11 hours. Exclusion criteria were the presence of any pharmacological conditions affecting serotonin status, evidence of drug or alcohol abuse and a history of shift work or travel across two or more time zones in the preceding month. After establishing the RVs, urine aMT6s was studied in 28 patients with genetic defects affecting serotonin biosynthesis (age range 2–55 years; mean = 13.0). Details of these patients are described in Tables 1 and 2. Out of these 28 cases, patients 1–18 were patients without treatment (naive patients) or patients on L-dopa/carbidopa therapy (Table 1), and the rest of cases (18bis-28) were under serotoninergic treatment (Table 2) including serotonin precursors (5-hydroxytryptophan, (5HTP)), monoamine oxidase inhibitors (MAOIs), selective serotonin re-uptake inhibitors (SSRI), cofactors that enhance serotonin/melatonin biosynthesis (BH4, PLP and folinic acid) and melatonin treatment. Other treatments that were applied are also stated in Tables 1 and 2. Patient 18 was studied twice, at diagnosis (stated as patient 18 in Table 1) and after 3 months on MAOIs therapy (stated as patient 18bis in Table 2
).
Ethical issues
All samples from the patients were obtained in accordance with the 2013 revised Helsinki Declaration of 1964. For biochemical and genetic investigations, informed consent was collected from patients or their guardians. The Ethical Committee of Sant Joan de Déu Hospital approved the study.
Samples
For both the controls and the patients, the first morning urine samples were collected, centrifuged and stored at −20 °C until analysis. From the 28 patients, a total of 29 urine samples were collected, because, in one case with AADC deficiency, we analysed one urine sample at baseline conditions and again after therapy (patient 18). A total of 18 urine samples were taken from patients not treated with serotoninergic therapy (Table 1, patients 1–18). A further 11 urine samples were collected from subjects on different combinations of serotoninergic drugs (Table 2, patients 18bis-28).
In Tables 1 and 2, we also stated CSF 5HIAA values when available (13 out of 28 patients). Most of the CSF analyses were done during the treatment monitoring process (Table 2).
Urinary aMT6s analysis validation
Assay variables
-
To study urinary melatonin excretion changes related to the time of sampling, we analysed aMT6s values in the first and second morning urine samples in 10 healthy subjects (age range: 4–33 years) (Fig. 2A).
Figure 2 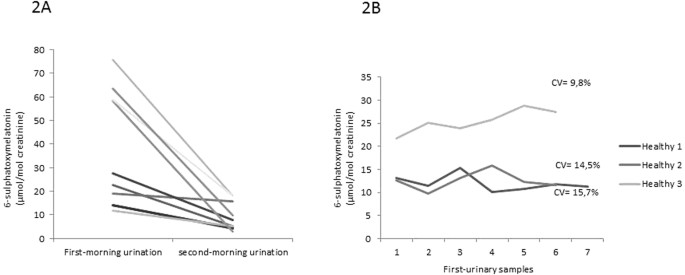
Assay variables. (A) Urine 6-sulphatoxymelatonin (aMT6s) values in the first and second morning urine samples in 10 healthy subjects. (B) Intra-individual variation of urinary aMT6s excretion in three healthy volunteers during 7 consecutive days.
-
To study the intra-individual variations of urinary aMT6s excretion, urinary aMT6s was analysed in three healthy volunteers during 7 consecutive days.
-
To evaluate assay imprecision, we calculated the within-run and between-run coefficients of variation (CV = standard deviation/mean aMT6s values*100) in 10 replicates.
-
After these metrological studies, RVs were established in the first morning urine samples, as reported by other groups4,11,12.

Urinary aMT6s was analysed by duplicate using a competitive ELISA kit (IBL; ref RE54021) and the optical density measured with a photometer (ATOM S.A. Barcelona, Spain) at 405 nm and 630 nm. Coefficients of variation of the duplicates were calculated. Creatinine concentration was determined by an automated spectrophotometric assay in the Architect c8000 analyser (Abbott). Results are expressed as µmol aMT6s/mol creatinine.
Statistical analysis
The Kolmogorov–Smirnow test was used to assess the distribution of the data. Because the data did not follow a Gaussian distribution, different non-parametric tests were applied. The Spearman simple correlation test was used to determine the correlations between urinary aMT6s and patient age. The Kruskal–Wallis and Mann–Whitney U tests were used to compare urinary aMT6s excretion among the different age groups and also between GTPCH naive patients and those under L-dopa/carbidopa treatment. Statistical calculations were performed using SPSS 23.0 software.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Results
Assay variables
The first morning urine sample showed the highest aMT6s concentrations in the 10 healthy cases, which remarkably dropped down in the second morning sample (Fig. 2A). The mean reduction from the first to the second urine samples was 68.7%.
The intra-individual variation studied in the three volunteers showed a CV value of 14.5%, 15.1% and 9.9%, respectively (Fig. 2B).
Regarding the imprecision assay studies, within-run CV was 17.0% for aMT6s = 11.5 ng/mL and between-run CV was 10.6% for aMT6s = 114.6 ng/mL.
Urinary aMT6s was analysed by duplicate. In the 80% studied samples, the variation of the duplicates was <10%, in the 10% studied samples was 10–15% and in the other 10% of studied samples the variation was >15%.
Establishment of reference values
In the healthy population (n = 65), a negative correlation was observed between urine aMT6s in the first morning urine sample and the age (r = −0.591; p < 0.001, Spearman test). RVs for our population were stratified according to age in three different groups as statistical differences were found (U = 28.8, p < 0.001 in groups 1–2; U = 9.0, p < 0.001 in groups 1–3; U = 34.0, p < 0.001 in groups 2–3; Table 3; Fig. 3).

Representation of urine 6-sulphatoxymelatonin (aMT6s) values from the reference population and patients with serotonin defects without serotoninergic treatment (A) and with serotoninergic treatment (B). Each disease is represented by different geometric shapes: square-AADC deficiency; cross-DHPR deficiency; inverted triangle-PTPS deficiency; triangle-adGTPCH deficiency; star-SR deficiency, and hexagon-arGTPCH deficiency. Each number matches with the patient’s number as shown in Tables 1 and 2. AADC: aromatic L-amino acid decarboxylase; DHPR: dihydropteridine reductase; GTPCH: guanosine triphosphate cyclohydrolase I; PTPS: 6-pyruvoyl-tetrahydropterin synthase; SR: sepiapterin reductase.
Patients not treated with serotoninergic treatment (Table 1)
Naive patients (patients 6, 8, 9, 12, 13, 17 and 18) and cases under L-dopa/carbidopa therapy (patients 1–5, 7, 10, 11, 14–16) were included (n = 18). Out of 18 cases, we studied 13 patients with autosomal dominant GTPCH (adGTPCH) deficiency (patients 1–13); 1 patient with autosomal recessive GTPCH (arGTPCH) deficiency (patient 14); 3 patients with SR deficiency (patients 15–17); and 1 patient with AADC deficiency (patient 18). We compared the results with those from our RVs (Table 1 and Fig. 3A). Regarding patients with adGTPCH deficiency (n = 13), only 2 presented decreased urine aMT6s values (patients 1 and 13; 34.9% and 69.8% of aMT6s reduction, respectively). No statistical differences were observed between adGTPCH patients and the control group. Patient with arGTPCH form (patient 14) had normal aMT6s values, whereas patients with SR and AADC deficiencies had very low urine aMT6s values compared to their age reference group.
No differences were observed in urinary aMT6s values when compared GTPCH naive patients (median = 13.4) and those under L-dopa/carbidopa therapy (median = 18.1; U = 20.5; p = 0.797).
Patients under serotoninergic treatment (Table 2)
We studied five patients with AADC deficiency (patients 18bis–22), one with SR deficiency (patient 23), three with DHPR deficiency (patients 24–26) and two with PTPS deficiency (patients 27–28). Details of the different treatments and results are stated in Table 2 and Fig. 3B. Three AADC deficiency cases (patients 18bis–20) presented low urine aMT6s values, while patient 21 showed normal aMT6 values. Patient 18bis was studied twice, at diagnosis (stated as patient 18) and after 3 months on MAOIs therapy (stated as patient 18bis). In this condition, aMT6s values increased from 1.1 to 9.2 µmol/mol creatinine, although they remained below the RVs (Table 1 and Table 2). Patient 22 was on melatonin therapy and therefore had a higher aMT6s concentration than RVs.
Regarding patients with pterin-related defects, some showed decreased aMT6s excretion despite receiving 5HTP treatment. Among them, the SR-deficient case (patient 23) and PTPS-deficient case (patient 28) showed remarkably reduced urinary aMT6s excretion (90.7% and 81.5%, respectively), while the DHPR-deficient cases (patients 24–26) and one PTPS-deficient case (patient 27) presented normal aMT6s values.
Discussion
This is the first report that assessed urinary aMT6s concentrations in genetic conditions that cause a severe effect on brain serotonin synthesis. Regarding assay variables, we corroborated that the first morning urine was the most suitable sample because the majority of melatonin is metabolized and excreted into urine as aMT6s4. Thus, any urine leak during the night would noticeably decrease aMT6s concentrations12. This fact would explain the unexpected low aMT6 value observed in two of our patients under serotoninergic treatment, who probably had troubles in urine sample collection (patient 18, who was 2 years old, and patient 23, because he had urinary continence problems).
Our analytical studies have shown that intra-individual variation only ranged from 9.9% to 15.1% (Fig. 2B). Furthermore, the metrological variations obtained were also reasonable for a proper interpretation of the results. Only 10% of patients presented CV of the duplicates higher than 15%, which was probably due to cross-reaction phenomena related to immune-based laboratory methods11. Thus, such samples were not considered in the study and should be reanalysed.
Regarding RVs, we established three different age groups (Table 3). Our results can be explained by the fact that melatonin synthesis reaches the highest rate at the age of 3–6 years, and later it decreases progressively by 80% until adult age1. These RVs are similar to others previously reported13, which supports the reproducibility of the ELISA method for aMT6s quantification in different populations.
Considering patients without serotoninergic treatment we studied 13 patients with adGTPCH deficiency, and 11 of them presented normal values of aMT6s. The adGTPCH deficiency is the mildest disease among those studied here, because the patients have a mild to moderate reduction in dopamine and serotonin biomarkers in CSF and were even normal in some cases. In fact, in patient 5 (the index case of one family14), CSF 5HIAA concentrations were normal at the time of diagnosis, which suggests minimal or no alteration in brain serotonin status. Moreover, most of our adGTPCH-deficient patients harboured the same mild mutation (p.Q89X) at the GCH1 gene as patient 5, and this fact would explain that adult cases from this cohort present a very mild (or even symptom-free) phenotype, as previously reported.
arGTPCH deficiency usually show PKU and has an early onset with a more severe clinical course than the adGTPCH deficiency15. Urine aMT6s levels were also normal in one case (patient 14) with arGTPCH deficiency, who showed normal phenylalanine levels and a phenotype resembling the dominant form of GTPCH deficiency, which suggested high GTPCH residual activity.
SR deficiency is inherited in an autosomal recessive manner. Patients present with a diurnally fluctuating motor disorder, and in most cases, it is associated with cognitive delay and severe neurologic dysfunction. The three patients reported here are siblings and they showed an important reduction of aMT6s levels (60.3%, 23.8% and 87.3%). In the index case (patient 15), the reduction of CSF 5HIAA at the time of diagnosis was also remarkable (Table 1). These three patients presented a mild phenotype with a late-onset presentation16. Moreover, they were under treatment with only L-dopa/carbidopa, as 5HTP was trialled some years ago, but the treatment was discontinued due to side effects (vomiting and diarrhoea). They presented a novel mutation in the SPR gene that affects splicing, which was reported as a mild change16. In SR deficiency, the dopamine and serotonin pathways are usually severely affected17, and the low levels of aMT6s could be a reflection of the impaired brain serotonin status.
Patient 18, with a severe form of AADC deficiency (at age of 1 year, she showed hypotonia, oculogyric crises and dystonia), presented, as expected, an extremely low value of urinary aMT6s, which was related to the concomitant dramatic reduction of the CSF 5HIAA values.
It has been reported that L-dopa therapy may be toxic to serotoninergic neurons in cell cultures by oxidative mechanisms producing highly reactive quinone species that reduce serotoninergic neurons18. These findings also have been observed “in vivo” by similar oxidative mechanisms producing a significant decrease in serotonin and 5HIAA metabolite19, as well as affecting the behaviour and cognitive functions in animal models19. However, no differences were observed when compared urinary aMT6s values between naive GTPCH patients and those under L-dopa/carbidopa treatment. It is interesting that carbidopa treatment (an inhibitor of peripheral AADC activity) does not seem to affect urine aMT6s excretion, emphasizing that the contribution of peripheral melatonin is less relevant than that of pineal gland1.
Regarding patients under serotoninergic treatment, three AADC-deficient patients showed low aMT6s concentrations despite serotoninergic treatment. In patient 18, urinary aMT6s excretion increased after 3 months of MAOIs therapy, which suggests that this therapy improves serotonin and melatonin status, although the aMT6s value was still below the normal values. Two patients (patients 19–20) with a severe phenotype, extremely low CSF 5HIAA levels at diagnosis, and who were under MAOIs and PLP therapy, showed reduced aMT6s urinary excretion, while patient 21 with a moderate phenotype, who was also on therapy with MAOIs and PLP, showed normal urinary aMT6s values. An explanation for these data is that AADC is the most severe condition affecting brain serotonin status20 with a very reduced capacity of serotonin and melatonin biosynthesis. Further investigations are required to determine whether long-term MAOIs treatment can normalize aMT6s excretion in AADC patients. Patient 22 with a mild phenotype, who was on MAOIs, SSRI and melatonin therapy, showed higher aMT6s values than the RVs.
Regarding patients with BH4-related defects, patient 23 with SR deficiency was diagnosed at 23 months of age and presented the typical phenotype with psychomotor retardation, hypotonia, ataxia and extrapyramidal signs16. CSF neurotransmitter studies showed a classical SR deficiency pattern with severely decreased 5HIAA values in CSF. We found very low urinary aMT6s values despite the patient being under 5HTP therapy, which should have increased serotonin and melatonin biosynthesis (Fig. 1). His parents reported night urinary incontinency problems, a fact that could explain the unexpectedly low aMT6s values of the patient.
All DHPR-deficient patients had normal urine aMT6s values. Although these patients were under different treatment regimens, all of them received 5HTP, the serotonin precursor. Patient 24 was also on long-lasting treatment with SSRI, folinic acid and BH4. At 12 months of age (when she was already under this treatment), she presented normal CSF 5HIAA values. At 5 years of age, she showed high urine aMT6s values, which suggested that the combination of all these drugs may have led to this result. In fact, it has been suggested that some SSRI drugs may increase plasma melatonin values21. Patients 25 and 26 undertook only 5HTP therapy, and both showed normal aMT6s excretion, which supports the hypothesis that this serotonin precursor is effective in increasing urinary aMT6s excretion. However, the reduced size of our series strongly advises the development of clinical trials to confirm or rule out this hypothesis.
PTPS-deficient patient 27 was diagnosed by a newborn screening program, and at 1 year of age (when she was already on treatment), she showed mildly reduced CSF 5HIAA values. Currently, this patient is receiving 5HTP, BH4, and calcium folinate as serotoninergic drugs. The last CSF analysis reported normal CSF neurotransmitter concentrations, which suggest adequate therapy; the urinary aMT6s values were also normal. Unexpectedly, patient 28 had very low aMT6s values despite being under 5HTP and BH4 treatment, and had underwent a previous CSF 5HIAA analysis that showed normal results. In this case, the doctor and the family confirmed that the urine was correctly sampled; one explanation we have, may be related to individual variations on CYP1A2 activity. CYP1A2 is one of the major isoforms of cytochrome P450 in the liver and metabolizes a number of clinical drugs and several important endogenous compounds, such as melatonin22. There is a large inter-individual variability in the expression and activity of CYP1A2, which is caused mainly by genetic (several polymorphisms) and environmental factors23. Other explanation woud be a possible cerebral folate deficiency, since HIOMT needs S-adenosylmethionine (SAM) as a cofactor for melatonin biosynthesis. However, CSF 5-methyltetrahydrofolate values were normal in this patient.
A limitation of this study is that aMT6s determination strictly depends on a proper sample collection, which may be difficult in severely handicapped infants. Moreover, in some cases, the inter-individual variations of CYP1A2 activity22,23 could contribute to unexpected variations in urinary aMT6s excretion. Another important aspect of melatonin fluctuation is that it is greatly influenced by dim light, whereas very bright light can block melatonin production11. Finally, the simultaneous measurement of both CSF 5HIAA and urinary aMT6s in larger series of patients should be done to stablish a correlation between these two variables.
In conclusion, we studied 28 patients with different genetic serotoninergic metabolism defects and found that decreased excretion of aMT6s is frequently observed in all patients with more severe disorders. No consistent alterations were documented in the adGTPCH deficiency, which is the mildest disease studied here. Sulphatoxymelatonin can be a non-invasive good biomarker to estimate serotonin status in the brain, especially for treatment monitoring purposes.
References
Claustrat, B. & Leston, J. Melatonin: Physiological effects in humans. Neurochirurgie 61, 77–84 (2015).
Arendt, J. et al. Some effects of melatonin and the control of its secretion in humans. Ciba Found. Symp. 117, 266–83 (1985).
Pääkkönen, T. et al. Urinary melatonin: A noninvasive method to follow human pineal function as studied in three experimental conditions. J. Pineal Res. 40, 110–115 (2006).
Yano, S., Moseley, K. & Azen, C. Large neutral amino acid supplementation increases melatonin synthesis in phenylketonuria: A new biomarker. J. Pediatr. 162, 999–1003 (2013).
Kurian, M. A. et al. Clinical and molecular characterisation of hereditary dopamine transporter deficiency syndrome: an observational cohort and experimental study. Lancet Neurol 10, 54–62 (2011).
Rilstone, J. J., Alkhater, R. A. & Minassian, B. A. Brain Dopamine–Serotonin Vesicular Transport Disease and Its Treatment. N Engl J Med 368, 543–50 (2013).
Pearl, P. L., Capp, P. K., Novotny, E. J. & Gibson, K. M. Inherited disorders of neurotransmitters in children and adults. Clin. Biochem. 38, 1051–1058 (2005).
Ng, J., Papandreou, A., Heales, S. J. & Kurian, M. A. Monoamine neurotransmitter disorders—clinical advances and future perspectives. Nat. Rev. Neurol. 11, 567–584 (2015).
Assmann, B., Surtees, R. & Hoffmann, G. F. Approach to the diagnosis of neurotransmitter diseases exemplified by the differential diagnosis of childhood-onset dystonia. Ann. Neurol. 54, 18–24 (2003).
Hyland, K. Clinical utility of monoamine neurotransmitter metabolite analysis in cerebrospinal fluid. Clin. Chem. 54, 633–641 (2008).
Benloucif, S. et al. Measuring melatonin in humans. J. Clin. Sleep Med. 4, 66–69 (2008).
Gholipour, T. et al. Decreased urinary level of melatonin as a marker of disease severity in patients with multiple sclerosis. Iran J Allergy Asthma Immunol 14, 91–97 (2015).
Rosen, R. et al. Urinary 6-sulfatoxymelatonin level in age-related macular degeneration patients. Mol. Vis. 15, 1673–9 (2009).
López-Laso, E. et al. Dopa-responsive infantile hypokinetic rigid syndrome due to dominant guanosine triphosphate cyclohydrolase 1 deficiency. J. Neurol. Sci. 256, 90–93 (2007).
Opladen, T., Hoffmann, G. F. & Blau, N. An international survey of patients with tetrahydrobiopterin deficiencies presenting with hyperphenylalaninaemia. J. Inherit. Metab. Dis. 35, 963–973 (2012).
Arrabal, L. et al. Genotype-phenotype correlations in sepiapterin reductase deficiency. A splicing defect accounts for a new phenotypic variant. Neurogenetics 12, 183–191 (2011).
Friedman, J. et al. Sepiapterin reductase deficiency: A Treatable Mimic of Cerebral Palsy. Ann. Neurol. 71, 520–530 (2012).
Stansley, B. J. & Yamamoto, B. K. L-dopa-induced dopamine synthesis and oxidative stress in serotonergic cells. Neuropharmacology 67, 243–251 (2013).
Stansley, B. J. & Yamamoto, B. K. Behavioral impairments and serotonin reductions in rats after chronic L-dopa. Pshychopharmacology 232, 3203–3213 (2015).
Pons, R. et al. Aromatic L-amino acid decarboxylase deficiency: clinical features, treatment, and prognosis. Neurology 62, 1058–1065 (2004).
Kirecci, S. L. et al. Relationship between plasma melatonin levels and the efficacy of selective serotonin reuptake inhibitors treatment on premature ejaculation. Int. J. Urol. 21, 917–920 (2014).
Zhou, S.-F., Wang, B., Yang, L.-P. & Liu, J.-P. Structure, function, regulation and polymorphism and the clinical significance of human cytochrome P450 1A2. Drug Metab. Rev. 42, 268–354 (2010).
Jin, T. et al. Genetic polymorphism analysis of the drug-metabolizing enzyme CYP2C9 in a Chinese Tibetan population. Gene 567, 196–200 (2015).
Acknowledgements
The authors want to thank the collaborators from the various hospitals in Spain, Germany and Greece that sent us their urine samples for analysis. This work is funded by the "Plan Estatal de I+D+I and Instituto de Salud Carlos III-Subdirección General de Evaluación y Fomento de la Investigación Sanitaria", project PI15/01082, project PI14/0003 and the European Regional Development Fund (FEDER). Instituto de Salud Carlos III and the FEDER Funding Program from the European Union. CIBERER U-703. [Artuch]. Instituto de Salud Carlos III and the FEDER Funding Program from the European Union. CIBERER U-703. [Molero-Luis]. Instituto de Salud Carlos III and the FEDER Funding Program from the European Union. PI15/01082 [Ormazabal]. This work was supported by grants from the Instituto de Salud Carlos III. PI14/00032. [Garcia-Cazorla].
Author information
Authors and Affiliations
Contributions
M.B. and M.M.L. contributed to conception and design, acquisition, analysis and interpretation of data, drafted the initial manuscript, and approved the final manuscript as submitted. A.O. contributed to analysis and interpretation of data, reviewed and revised the manuscript, and approved the final manuscript as submitted. L.A., J.D.L.H., J.A.F.R., L.G.G.S., S.I.M., R.D., J.C., T.O., B.Z., R.P., A.G.C., F.S. and E.L.L. contributed to acquisition of data, reviewed and revised the manuscript, and approved the final manuscript as submitted. R.A. contributed to conception and design, analysis and interpretation of data, reviewed and supervised the manuscript, and approved the final manuscript as submitted. Every one of the authors has participated sufficiently in the study, meeting the appropriate authorship criteria, and each has seen, reviewed and approved this version of the manuscript and takes full responsibility for it. We all agree to its submission for publication. Nobody who qualifies for authorship has been omitted from the list of authors. All the authors have complete access to the study data. Considering that the work has been performed with the collaboration of different Hospitals of Spain, and also from Greece and Germany, we believe that we are 17 authors who have contributed and participated in this multidisciplinary work.
Corresponding author
Ethics declarations
Competing Interests
Yes there is potential Competing Interest. MedDay Pharmaceuticals gave economical support for the purchase of melatonin ELISA kits.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Batllori, M., Molero-Luis, M., Arrabal, L. et al. Urinary sulphatoxymelatonin as a biomarker of serotonin status in biogenic amine-deficient patients. Sci Rep 7, 14675 (2017). https://doi.org/10.1038/s41598-017-15063-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-15063-8
This article is cited by
-
Insights into the expanding phenotypic spectrum of inherited disorders of biogenic amines
Nature Communications (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
